

Abstract
Summary Background Data:
Duodenogastric–esophageal reflux disease is directly linked to Barrett's esophagus and to the development of esophageal adenocarcinoma. Despite this link, little is known about the mutagenic potential of refluxed material on the esophageal mucosa. We hypothesize that the reflux of gastric and duodenal content causes mutations in esophageal mucosa in vivo.
Methods:
Seven Sprague Dawley/Big Blue F1 lacI transgenic rats underwent esophagoduodenostomy (ED) to surgically create duodeno–gastric–esophageal reflux. Fourteen nonoperated rats served as negative (n = 7) and as positive (n = 7/methyl-N-amyl-nitrosamine [MNAN] intraperitoneally) controls. The animals were killed 16 weeks after operation or injection, the entire esophageal mucosa was harvested, and mutation frequency was determined through standard Big Blue Mutagenesis Assay.
Results:
Gross esophagitis was evident in all operated animals. The frequency of lacI mutations in esophageal mucosal cells of animals with ED was significantly higher, nearly 1.5-fold, than that of nonoperated animals. Nitrosamine administration resulted in a nearly 20-fold increase of lacI mutation frequency. Thirteen mutations were successfully sequenced, 46% occurred at CpG dinucleotide sites and 61% were either C to T or G to A transitions.
Conclusions:
The data provide preliminary evidence of the mutagenic potential of bile reflux on esophageal epithelium. The specific mutations are markedly higher than would be expected by chance and are similar to that found in p53 mutations of human esophageal adenocarcinoma, providing a link to human esophageal cancer.
The frequency and spectrum of mutations of the lac-I gene were investigated in a well-described rat model of esophageal carcinoma. Surgical creation of duodenoesophageal reflux resulted in a significant increase in lac-I mutation frequency, which, on analysis, had a spectrum of mutations remarkably similar to those seen in human esophageal adenocarcinoma.
Bile salts, or more accurately, duodenal content, have been implicated in the pathophysiology of esophageal mucosal injury for decades.1,2 Early studies using 24-hour ambulatory pH monitoring showed a higher prevalence of “alkaline” reflux in patients with esophagitis and Barrett's esophagus.3 These early observations were confirmed by ambulatory spectrophotometric monitoring of esophageal bilirubin exposure4 and esophageal aspiration studies,5 both of which showed a high esophageal exposure to duodenal content and bile salts, particularly in patients with Barrett's epithelium. Recent studies have shown significant effects of bile acids on cellular physiology. Bile salts have been shown to activate protein kinase C in isolated rat hepatocytes,6 to activate nuclear transcription factors,7 and nuclear receptors for bile acids have been identified.8 Parks et al have shown that physiological concentrations of free and conjugated chenodeoxycholic acid, lithocholic acid, and deoxycholic acid activate the farnesoid X receptor, a heretofore orphan nuclear receptor.8 This provides evidence that bile acids may modulate nuclear activity. These findings, in concert with the strong link between gastroesophageal reflux disease (GERD) and esophageal adenocarcinoma,9 suggests that bile salts may play a role in the pathophysiology of esophageal carcinogenesis.
Few studies examine the genetic effects of bile salts, and virtually no studies do so in vivo. It has been demonstrated that bile salts can promote colon, liver, stomach, and pancreas carcinogenesis in rats or hamsters. The mechanism of the cocarcinogenic effect is still unclear but may involve an increase in cell turnover. Transgenic rodents for measuring mutation frequency in vivo have recently been developed and are commercially available (Big Blue Transgenic Rodent Mutagenesis System; Stratagene Inc., La Jolla, CA).10,11 These animals (both rats and mice) provide a significant advance in the ability to measure tissue-specific mutation frequency following chemical treatment, and in combination with the rat model of esophageal carcinoma,12 provides a powerful tool to test the hypotheses that duodenal content is important in the pathophysiology of esophageal adenocarcinoma.
METHODS
Experimental Preparation
Fisher 344 lacI transgenic rats (Stratagene Inc.) were crossed with outbred Sprague Dawley rats to create an F1 strain. Pilot studies using homozygous Big Blue rats resulted in excess mortality. At 10 weeks of age, animals were randomly assigned to 1 of 3 groups: 1) control: n = 7 no operation; 2) positive control: n = 7 no operation, 25 mg/kg n-methyl-amyl-nitrosamine; or 3) experimental: n = 7 esophagoduodenostomy.
Surgical diversion of duodenal secretions into the esophagus was induced by end-to-side esophagoduodenostomy (ED) in the experimental group (Fig. 1). Each animal was then exposed to the chemical carcinogen or duodenogastric reflux for 16 weeks and euthanized. The 16-week time interval was chosen to investigate early genetic events in the process of carcinogenesis, before the development of frank carcinoma. It is known that this experimental preparation results in a 40% to 50% prevalence of frank cancers in the 30- to 50-week timeframe. Animals that received nitrosamines were housed continuously in a fume exhaust hood to remove the volatile carcinogens. They were injected intraperitoneally with 25 mg/kg (50% LD50) weekly for 10 weeks. After killing, the esophagus, duodenum, and liver were removed and esophageal mucosa removed from the underlying musculature.
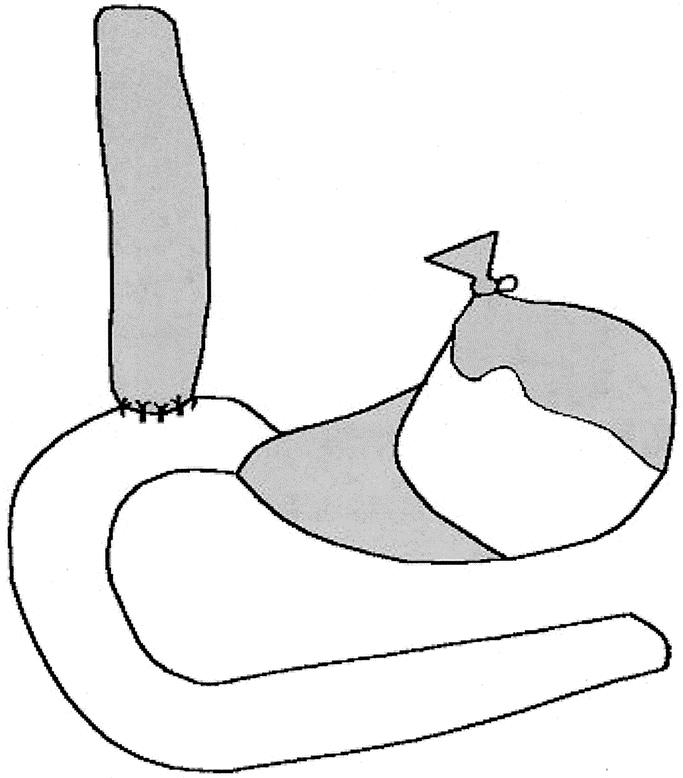
FIGURE 1. Surgical preparation consisting of esophagoduodenostomy with gastric preservation. Shaded areas represent 1) esophagus, 2) squamous forestomach, and 3) gastric antrum.
DNA Extraction, Packaging, and Plating
Sixteen weeks after the treatment, the rats were euthanized by intraperitoneal injection of pentobarbital. The esophagus was immediately resected and the mucosa stripped of the surrounding muscular layer and connective tissue and put into liquid nitrogen until further use. The esophageal mucosa was ground in a mortar on dry ice and incubated overnight with DNA lysis buffer (10 mm TrisHCL, pH 8.0, 0.1 M EDTA, 0.5% SDS) at 56°C. After phenol–chloroform extraction, DNA was ethanol-precipitated at room temperature and resuspended in TE buffer.
Transgenic Mutagenesis Assay
The Lac Operon of Escherichia coli
The lac operon codes for a set of regulated genes coordinately involved in bacterial lactose metabolism.13 LacI codes for a repressor protein that is assembled into a tetrameric conformation, which binds to the lacO operator sequence, preventing transcription of the genes coding for β-galactosidase. In the absence of an inducer, the lacI repressor product is bound to the operator and transcription is blocked, thus inhibiting β-galactosidase activity. β-galactosidase activity can be measured in E. coli by plating on media containing the chromogenic substrate 5-bromo-4-chloro-3-indolyl-β-galactopyranoside (X-gal), a substrate for β-galactosidase.
Packaging of the LacI Gene
Genomic DNA was packaged into lambda phage using Transpack Packaging Reagent (Stratagene Inc.). The packaged phage were then adsorbed to E. coli host strain SCS-8 cells for 15 minutes at 37°C. The 8-phage system provides a vehicle for the transfer of the vector between the eukaryotic rodent cell and E. coli. Lambda phage protein extracts are commercially available and recognize and excise the integrated vector at its cos sites. The sequence is then incorporated into l phage-based infectious viral particles. To minimize DNA cleavage and background β-galactosidase activity, the packaging extracts are deficient in DNA restriction enzymes and β-galactosidase activity. The viral particles are then preadsorbed to bacteria, plated on agar, and incubated overnight in the presence of X-gal. The cell-phage solution was then plated in NZCYM agar with X-gal and incubated overnight at 37°C.
Determination of Mutation Frequency
Mutant strains appear as blue plaques among the predominant colorless nonmutant plaques. Plates are examined for the presence of blue mutant plaques in the presence of colorless nonmutant plaques. Density of the plaques is limited to 15,000 plaque-forming units per 25 cm2 plate. The mutation ratio is calculated as the number of blue plaques divided by the total plaque count.
Sequence Analysis of LacI Mutants
Several blue (mutant) plaques were selected from the mutagenesis assay of random animals and resuspended in SM buffer. The phage solution was adsorbed to E. coli SCS-8 host cells and then replated at low density to allow isolation of a single plaque. The isolated blue plaques were then amplified through standard polymerase chain reaction (PCR) and DNA extracted. The lacI gene was then sequenced through automated sequencing apparatus to determine the exact mutation present. Primer 2 for the forward PCR (5′ ATA GAC ACC ATC GAA TG) and primer 12 for the reverse PCR (5′ AGA ACT TAA TGG GCC CGC) were used. It has been shown that more than 90% of mutations are found within this range.
Statistics
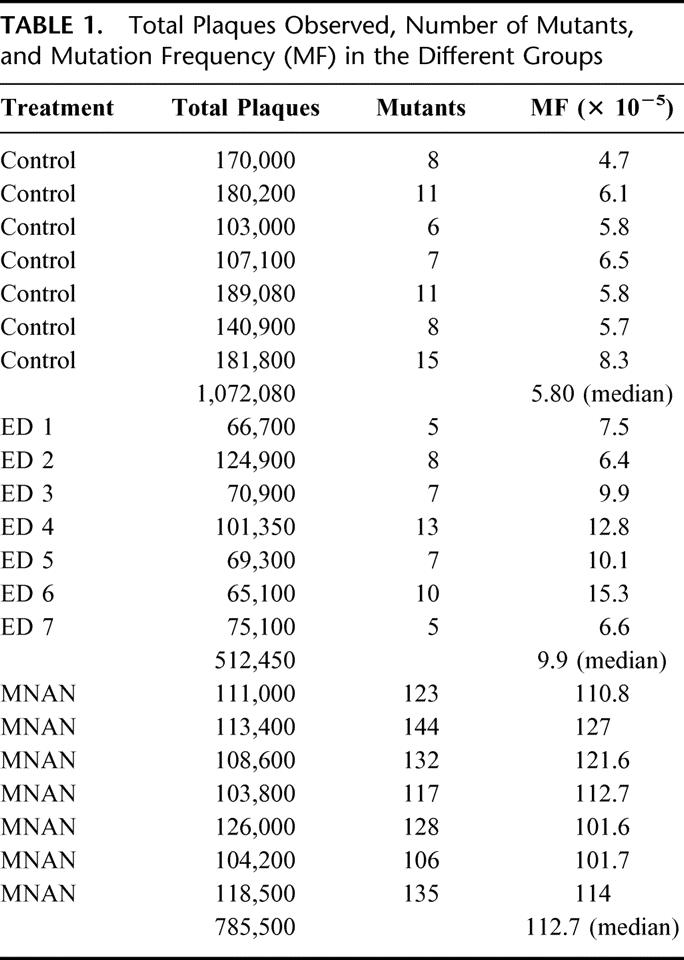
Statistical analyses of the influence of viability in the number of plaques screened per packaging reaction, the number of packaging reactions for each animal, and the number of animals per exposure group have been published.14,15 Our experimental design included 7 animals per group, 2 tissues per animal (esophagus and duodenum), and several packaging reactions per tissue. Approximately 100,000 plaques were recovered from each tissue so that hundreds of thousands of plaques were screened per tissue (512,450–1,072,080; Table 1). Published analyses suggest that this design will result in a 93% probability of detecting a 50% increase in mutant fraction (P = 0.05).14 Data are presented as median ± interquartile range (IQR). Mutation ratios in the control and experimental groups were compared by Mann-Whitney U test.
TABLE 1. Total Plaques Observed, Number of Mutants, and Mutation Frequency (MF) in the Different Groups
RESULTS
Gross esophagitis was evident in all operated animals. Table 1 shows the number of plaques screened, the number of mutants observed, and the mutation frequency for each group. Median mutation frequency was 5.8 × 10−5 (IQR 5.7–6.5) in animals without treatment, and 113 × 10−5 (IQR 101–127) in animals receiving weekly methyl-N-amyl-nitrosamine (MNAN), an approximately 20-fold increase. The difference was highly significant (P <0.001) and validates the assay system. Surgical creation of duodenoesophageal reflux increased the mutation frequency approximately 1.5-fold compared with the control group (median 9.9 × 10−5, IQR 6.6–12.8, P <0.007). Figure 2 illustrates the median mutation frequency in the 3 different groups.
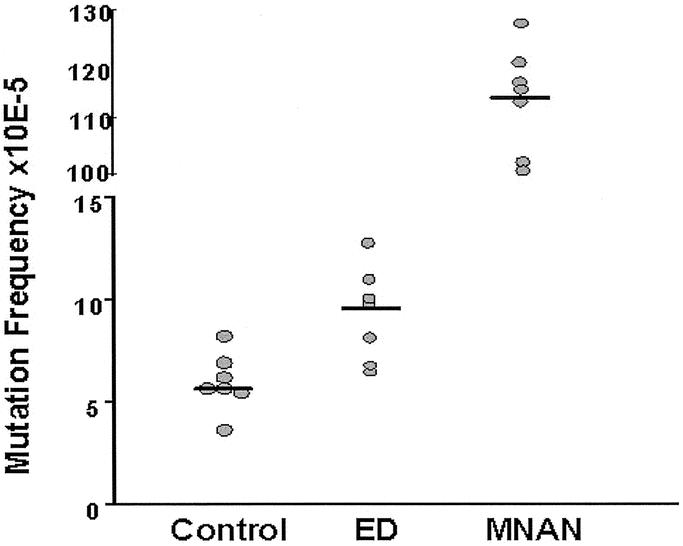
FIGURE 2. Individual values for the mutation frequency in each animal of the control and experimental groups. The bar indicates the median value. ED, esophagoduodenostomy; MNAN, n-methyl-amyl-nitrosamine.
Twenty-two mutant plaques from 6 animals in the duodenoesophageal reflux group were selected for sequencing. Five of these were not confirmed on replating, 2 were mutations outside of the range of the primers used, and 2 were not sequenced as a result of technical errors. The remaining 13 were successfully sequenced. Table 2 shows the specific point mutations identified. Of note, 46% of the mutations occurred at CpG dinucleotide sites and 61% were either C to T or G to A transitions (Fig. 3A). Both of these frequencies are markedly higher than would be expected by chance.
TABLE 2. Specific Mutations Found in 13 Plaques Isolated From 6 Animals

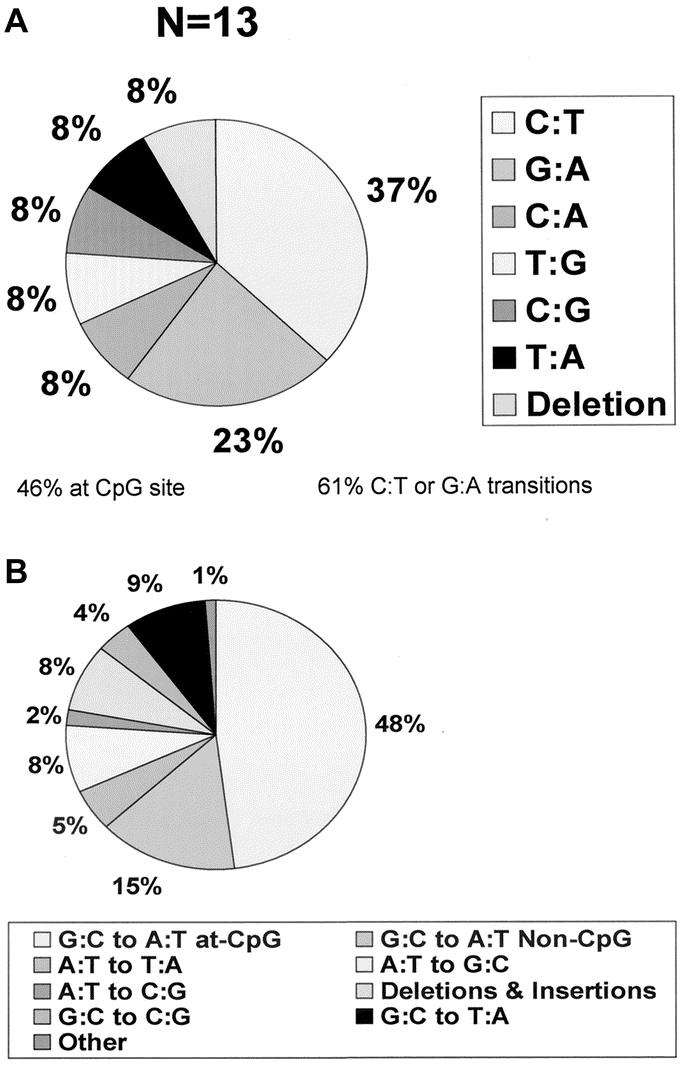
FIGURE 3. (A) Spectrum of mutations found in the esophageal mucosa of experimental animals exposed to duodenogastroesophageal reflux. (B) p53 mutational spectrum in human colon adenocarcinoma.19 The prominence of CpG dinucleotide mutations and C:T or G:A transitions is evident in both.
DISCUSSION
This study presents the first evidence of esophageal molecular changes induced in vivo by mixed duodenogastric reflux (DGR). We not only identified a 1.5-fold increase in esophageal mucosal mutation frequency in animals exposed to DGR, but also found that 46% of the mutations occurred at CpG dinucleotides and that 61% were C to T or G to A transversions. These changes are similar to the mutation pattern seen in human esophageal adenocarcinoma and provide preliminary evidence that DGR has a carcinogenic role in the development of adenocarcinoma of the esophagus.
The identification of carcinogens involved in human cancer is often based on the pattern of mutation observed, coupled with epidemiologic evidence of increased exposure to an environmental or endogenous carcinogens.16 Because mutations seldom occur randomly, the pattern of mutation in a gene of interest is related to the underlying cause of the mutation. This “fingerprint” of a carcinogen can be exploited epidemiologically to imply cause and effect between a carcinogen and a specific tumor.17 Identification of the specific types of mutations caused by ultraviolet radiation, nitrosamines, and aflatoxins has revealed a characteristic mutational spectrum for each of these agents. For example, benzopyrene often causes G to T transversions18 and melphalan causes A to T transversions.19 In contrast, the binding of alkylating agents to guanine results in alterations of hydrogen-binding properties and G to A transitions, whereas ionizing radiation often causes deletion of DNA sequences. Using this rationale, Hsu et al. and Otzurk et al. have linked the mutational spectra observed in hepatocellular carcinoma to exposure to aflatoxin B.20,21 Similar studies have linked the mutational spectra in the p53 gene caused by ultraviolet radiation to human squamous cell skin cancer, and those caused by exposure to tobacco and alcohol to squamous carcinoma of the lung and esophagus.22
Mutations in the p53 tumor suppressor gene have been widely studied and reported for a large number of tumor types, including esophageal carcinoma. One of the more striking observations in the study of the mutational spectra of the p53 gene is the high prevalence of mutation at CpG dinucleotides found in human gastrointestinal adenocarcinoma. C to T and/or G to A transitions at CpG dinucleotides have been found to represent 60% of all p53 mutations in esophageal, 48% in colon, 41% in gastric, and 35% in pancreatic adenocarcinoma (Fig. 3). This suggests a different etiologic origin of these tumors compared with squamous cell carcinomas of the esophagus, lung, and upper airway, which have a 9% to 18% prevalence of CpG transitions and are known to be associated with tobacco and alcohol.22,23 The etiologic significance of mutation at CpG dinucleotides in adenocarcinomas of the gastrointestinal tract is emphasized by the fact that CpG dinucleotides represent only 1% of all dinucleotides in the human genome, and that C to T or G to A mutations at CpG are only 2 of 96 possible single-base substitutions at all dinucleotides.
CpG dinucleotides are also the target sequence for 5-methylcytosine DNA methylation in mammals. 5-methylcytosine is known to undergo spontaneous deamination to yield thymine, which, if not repaired, would represent a C to T transition. It is interesting that tumors with the highest frequency of CpG transitions arise in those parts of the gastrointestinal tract where exposure to bile occurs (colon, stomach, and adenocarcinoma of the esophagus in association with reflux-induced Barrett's esophagus). The pancreas has a low bile exposure and pancreatic carcinomas have a low (near-baseline) CpG transition frequency. The high prevalence of CpG transition mutations in the lacI gene in our experimental model provides the first molecular biologic support for a causal relationship between bile exposure and high CpG transition frequency.
Published data on the mutagenesis of bile salts are few and conflicting. Studies of the mutagenicity of bile acids in the Ames salmonella–microsome assay suggest that bile acids are mutagenic when given with the addition of suboptimal levels of a variety of other known carcinogens.24 Wantanabe has shown a synergistic effect of human bile on the induction of chromosome abnormalities in mitomycin C-treated lymphocytes in culture.25 These in vitro studies must be considered in light of limitations of the bacterial assay systems used, as well as the physical chemistry of bile salts. Negative results may in part be the result of the fact that the physical behavior of bile salts depends on their degree of hydroxylation (pH) and the type and presence of conjugation of the basic steroid nucleus. Most in vitro assay systems occur at neutral pH, when conjugated bile salts are in their ionic form and may not have access into the cell and nucleus.
Although the Big Blue mutagenesis assay does have limitations, it is the best in vivo measure of mutagenesis presently available. One of its limitations is the fact that the lacI transgene is prokaryotic and not expressed in the rat genome. Thus, the mutation frequency may not be representative of genes that are normally transcribed. The accuracy of the magnitude of the mutation frequency is, however, not central to our findings, only the relative increase present with duodenogastric reflux. On the positive side, it is a well-described system documented by a large amount of literature, allows testing for mutation in all cell and tissue types, does not require tissue culture, and is appropriate for the generation of the large data series necessary to achieve significance. Validation testing has suggested that the model accurately reflects both the mutagenic potential and the expected type of mutations of other well-studied agents, including 4-acety-laminofluorene, a nonmutagenic chemical which is a potent hepatic cell proliferator (no increase in prevalence of mutations); benzo(a)pyrene, a potent alkylating mutagen and carcinogen (significant increases in mutant frequencies in all tissues tested); and N-ethyl N-nitrosourea, a direct acting alkylating agent and a potent germ cell mutagen (significant increase in mutant frequency and a spectrum of mutation consistent with the mechanism of known ENU adducts); and others.26 In addition, pilot data on homozygous rats revealed that the mutation frequency of duodenal tissue was similar to background esophageal frequency (approximately 5 × 10−5) and that there was no difference between animals with and without duodenogastroesophageal reflux. Thus, our observations likely reflect the effects of duodenoesophageal reflux rather than any peculiarity of the assay system. Other systems, including those using normally transcribed mammalian “housekeeping” genes such as aprt or hprt, are less desirable because they are not equally expressed in all tissues, are very labor-intensive, costly, and require tissue culture assay techniques.
In summary, the results of this study provide molecular biologic evidence that the reflux of mixed reflux of gastric and duodenal juice into the esophagus is involved in the genesis of esophageal adenocarcinoma. Further characterization of the time course and prevalence of mutational events, mutation patterns of other genes, and studies dissecting the components of the refluxate causing the mutations will further enhance our understanding of the pathogenesis esophageal adenocarcinoma.
Footnotes
Reprints: Jeffrey H. Peters, MD, University of Rochester, 601 Elmwood Ave., Rochester, NY 14642. E-mail: jeffrey_peters@urmc.rochester.edu.
REFERENCES
- 1.Moffat RC, Berkas EM. Bile esophagitis. Arch Surg. 1965;91:963–965. [DOI] [PubMed] [Google Scholar]
- 2.Lillimoe KD, Johnson LF, Harmon JW. Role of the components of the gastroduodenal contents in experimental acid esophagitis. Surgery. 1982;92:276–284. [PubMed] [Google Scholar]
- 3.Pellegrini CA, DeMeester TR, Wernly JA, et al. Alkaline gastroesophageal reflux. Am J Surg. 1978;135:177–184. [DOI] [PubMed] [Google Scholar]
- 4.Kauer WKH, Peters JH, DeMeester TR, et al. Mixed reflux of gastric juice is more harmful to the esophagus than gastric juice alone. The need for surgical therapy reemphasized. Ann Surg. 1995;222:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein HJ, Feussner H, Kauer W, et al. Alkaline gastroesophageal reflux: assessment by ambulatory esophageal aspiration and pH monitoring. Am J Surg. 1994;167:163–168. [DOI] [PubMed] [Google Scholar]
- 6.Beuers U, Throckmorton DC, Anderson MS, et al. Tauroursodeoxycholic acid activates protein kinase C in isolated rat hepatocytes. Gastroenterology. 1996;110:1553–1563. [DOI] [PubMed] [Google Scholar]
- 7.Hirano F, Tanaka H, Makino Y, et al. Induction of the transcription factor AP-1 in cultured human colon adenocarcinoma cells following exposure to bile salts. Carcinogenesis. 1996;17:427–433. [DOI] [PubMed] [Google Scholar]
- 8.Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids; natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. [DOI] [PubMed] [Google Scholar]
- 9.Lagergren J, Bergstrom R, Lindgren A, et al. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med. 1999;340:825–831. [DOI] [PubMed] [Google Scholar]
- 10.Kohler SW, Provost GS, Kretz PL, et al. An in vivo assay using transgenic mice to analyze spontaneous and induced mutations at the nucleic acid level. Strategies in Mol Biol. 1990;3:19–21. [Google Scholar]
- 11.Mirsalis JC, Monforte JA, Winegar RA. Transgenic animal models for measuring mutations in vivo. Crit Rev Toxicol. 1994;24:255–280. [DOI] [PubMed] [Google Scholar]
- 12.Ireland A, Peters JH, Smyrk TC, et al. Gastric juice protects against the development of esophageal adenocarcinoma in the rat. Ann Surg. 1996;224:358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Provost GS, Kretz PL, Hamner RT, et al. Transgenic systems for in vivo mutation analysis. Mutat Res. 1993;288:133–149. [DOI] [PubMed] [Google Scholar]
- 14.Piegorsch WW, Lockhart AC, Margolin BH, et al. Sources of variability in data from a lacI transgenic mouse mutation assay. Environ Mol Metagen. 1994;23:17–31. [DOI] [PubMed] [Google Scholar]
- 15.Callahan JD, Short JM. Transgenic λ/lacI mutagenicity assay: statistical determination of sample size. Mutat Res. 1995;327:201–208. [DOI] [PubMed] [Google Scholar]
- 16.Jones PA, Buckley JD, Henderson BE, et al. From gene to carcinogen: a rapidly evolving field in molecular epidemiology. Cancer Res. 1991;51:3617–3620. [PubMed] [Google Scholar]
- 17.Vogelstein B, Kinzler KW. Carcinogens leave fingerprints. Nature. 1992;355:209–210. [DOI] [PubMed] [Google Scholar]
- 18.Mazur M, Glickman BW. Sequence specificity of mutations induced by benzo(a)pyrene-7,8-diol-9,10-epoxide at endogenous aprt gene in Cho cells. Somat Cell Molec Genet. 1988;14:393–400. [DOI] [PubMed] [Google Scholar]
- 19.Wang P, Bennett RAO, Povirk LF. Melphalan-induced mutagenisis in an SV-40 based shuttle vector. Predominance of A:T-T:A transversions. Cancer Res. 1990;50:7257–7531. [PubMed] [Google Scholar]
- 20.Hsu IC, Metcalf RA, Sun T, et al. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350:427–428. [DOI] [PubMed] [Google Scholar]
- 21.Otzurk M, et al. P53 mutation in hepatocellular carcinoma after aflatoxin exposure. Lancet. 1991;338:1356–1359. [DOI] [PubMed] [Google Scholar]
- 22.Greenblat MS, Bennett WP, Hollstein M, et al. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 23.Hollstein MC, Peri L, Mandard AM, et al. Genetic analysis of human esophageal tumors from two high incidence geographic areas: frequent p53 base substitutions and absence of ras mutations. Cancer Res. 1991;51:4102–4106. [PubMed] [Google Scholar]
- 24.Silverman SJ, Andrews AW. Bile acids: co-mutagenic activity in the Salmonella–mammalian–microsome mutagenicity test. J Natl Cancer Inst. 1977;59:1557–1559. [DOI] [PubMed] [Google Scholar]
- 25.Wantanabe M, Takagi S, Magara J, et al. Cytogenetic analysis of human bile for mutagenicity and co-mutagenicity. Tohoku J Exp Med. 1991;163:255–261. [DOI] [PubMed] [Google Scholar]
- 26.Stratagene Inc. Big Blue Transgenic Rodent Mutagenesis Assay systems. Standardization and Validation Summary, May 1, 1994.