

Abstract
Objectives:
To evaluate whether aggressive, undifferentiated neuroblastomas express tumor suppressor protein PTEN (phosphatase and tensin homolog deleted on chromosome ten) and to examine the effects of gastrin-releasing peptide (GRP) on PTEN gene and protein expression.
Summary Background Data:
We have previously shown that neuroblastomas secrete GRP, which binds to its cell surface receptor (GRP-R) to stimulate cell growth in an autocrine fashion. However, the effects of GRP on expression of the tumor suppressor gene PTEN have not been elucidated in neuroblastomas.
Methods:
Paraffin-embedded sections from human neuroblastomas were analyzed for PTEN and phospho-Akt protein expression by immunohistochemistry. Human neuroblastoma cell lines (SK-N-SH and SH-SY5Y) were stably transfected with the plasmid pEGFP-GRP-R to establish GRP-R overexpression cell lines, and the effects of GRP on PTEN gene and protein expression were determined.
Results:
A decrease in the ratio of PTEN to phospho-Akt protein expression was identified in poorly differentiated neuroblastomas. An increase in GRP binding capacity was confirmed in GRP-R overexpressing cells, which demonstrated an accelerated constitutive cell growth rate. PTEN gene and protein expression was significantly decreased in GRP-R overexpressing cells when compared with controls.
Conclusions:
Our findings demonstrate decreased expression of the tumor suppressor protein PTEN in more aggressive undifferentiated neuroblastomas. An increase in GRP binding capacity, as a result of GRP-R overexpression, down-regulates PTEN expression. These findings suggest that an inhibition of the tumor suppressor gene PTEN may be an important regulatory mechanism involved in GRP-induced cell proliferation in neuroblastomas.
The authors identified presence of decreased expression of tumor suppressor protein PTEN in a higher percentage of undifferentiated neuroblastomas. Human neuroblastoma cells stably transfected to overexpress gastrin-releasing peptide receptor demonstrated increased cell growth rate, decreased expression of PTEN gene, and protein. PTEN may be an important tumor-suppressor gene in neuroblastomas.
Neuroblastoma is the most common extracranial solid tumor in infants and children, and despite recent advances in chemotherapeutic regimen, the overall survival remains dismal.1 Derived from neural crest precursors, the clinical behavior of neuroblastoma is quite variable, depending on histopathological features.2 Well-differentiated tumors with neuroblastic rosettes and formation of ganglion cells lined with mature nerve fibers behave benignly and can even undergo spontaneous regression. In contrast, poorly differentiated neuroblastomas, characterized by small cells lacking special arrangement, are highly aggressive and chemoresistant, frequently resulting in tumor relapse.2 This variable neuroblastoma tumor behavior suggests that malignant transformation of cells may result, in part, from a failure to respond to normal signals regulating morphologic differentiation.
As a neuroendocrine tumor, neuroblastoma is known to produce and secrete various peptides,3 including gastrin-releasing peptide (GRP).4 GRP, the mammalian equivalent of bombesin, is a neuroendocrine peptide shown to have a growth stimulatory effect on various cancer cell types. We and others have shown that bombesin exerts trophic actions on gastric,5 prostate,6 and small-cell lung cancers.7 Recently, our laboratory has further demonstrated that GRP and its cell surface receptor, GRP-R, are abundantly expressed in human neuroblastomas.4 Moreover, we found that more aggressive undifferentiated tumors express an increased level of GRP-R and that GRP acts as an autocrine growth factor to stimulate neuroblastoma cell growth.4 However, the exact cellular mechanisms involved in this process are not clearly elucidated.
Upon activation, phosphatidylinositol 3-kinase (PI3-K) family enzyme phosphorylates lipid second messengers, which recruit serine/threonine kinase Akt (protein kinase B) to the cell membrane through binding to its plexin homology domain.8 This complex activates the phosphoinositide-dependent kinases (PDK)-1 and PDK-2, which then activate Akt through phosphorylation at 2 sites.9 Phosphorylated Akt (phospho-Akt) has been widely implicated as a cell survival factor under multiple conditions, including exposure to cytotoxic agents and hormone manipulations. PI3-K has been shown to be a mitogenic signaling to stimulate tumor cell growth.9–12 In neuroblastoma cells, PI3-K has also been implicated as an important key cell survival pathway.13,14 Tumor suppressor gene PTEN (phosphatase and tensin homolog deleted on chromosome ten), which is known to negatively regulate PI3-K, has been described to be an important gene in various cancers.15–17 However, the exact role of PTEN in neuroblastoma is not clearly defined. Further, the potential regulatory action of GRP on PTEN expression in neuroblastoma has not been previously described.
In this study, we examined paraffin-embedded archival samples of neuroblastomas to compare the expression of tumor suppressor protein PTEN between differentiated and undifferentiated tumors. In addition, we established stably transfected neuroblastoma cell lines overexpressing GRP-R to further elucidate the role of PI3-K/PTEN pathway in GRP-induced neuroblastoma cell growth stimulation.
MATERIALS AND METHODS
Materials
The LY294002 and PD98059 compounds were purchased from Cell Signaling (Beverly, MA). Anti-GFP monoclonal antibody was purchased from BD Biosciences (Clontech, Palo Alto, CA) and polyclonal antibodies to phospho-Akt (ser 473), total Akt and PTEN monoclonal antibody were purchased from Cell Signaling. All secondary antibodies against mouse and rabbit IgG were purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA). Anti-β-actin monoclonal antibody was from Sigma (St. Louis, MO). Restriction enzymes NheI and EcoR were purchased from New England BioLabs Inc (Beverly, MA). Plasmid pEGFP (N3) was purchased from BD Biosciences. PTEN plasmid (pBP-PTEN-HA) was a gift from Dr. Cavernee (University of California, San Diego). All other reagents were molecular biology-grade and obtained from either Sigma or Amresco (Solon, OH).
Immunohistochemical Analysis
Protein staining was performed using DAKO EnVision Kit (Dako Corp., Carpinteria, CA). Briefly, paraffin-embedded sections (4 μm) were incubated with monoclonal antibodies for anti-PTEN (1:400 dilutions) from Cell Signaling or antiphospho-Akt (1:400 dilutions) from Santa Cruz for 1 hour at room temperature. After 3 washes with PBS, the sections were incubated for 30 minutes with secondary antibody labeled with peroxidase. Lastly, 1 to 3 drops peroxidase substrate DAB was added for staining. All sections were counterstained with hematoxylin and observed by light microscopy. For negative controls, primary antibody was omitted from the above protocol.
Plasmid Construction
Human GRP-R cDNA was amplified by PCR from plasmid pGEM4-GRP-R, which was a gift from Dr. James Battey (Department of Health and Human Service, National Institute on Deafness and Other Communication Disorders). Plasmid pEGFP-GRP-R was constructed by inserting GRP-R cDNA fragment to plasmid pEGFP (N3) at restriction enzyme sites NheI and EcoR I. Plasmid DNA was confirmed by DNA sequencing.
Cell Culture and Transfection
Neuroblastoma cell lines, SK-N-SH and SH-SY5Y, were purchased from American Type Culture Collection (Manassas, VA). Cells were maintained in RPMI 1640 medium with l-glutamine (Cellgro Mediatech, Inc, Herndon, VA) supplemented with 10% fetal bovine serum (FBS) (Sigma). The cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were transfected with plasmid DNA in 6-well plates using Lipofectin (Invitrogen, Rockville, MD) according to the manufacturer's instructions. Stable transfection cell lines were established by selecting out transfected cells with G418 (Cellgro Mediatech) at 300 μg/mL for 2 weeks. Cells were passed twice weekly using Trypsin 0.25% with EDTA 0.1% (Cellgro Mediatech).
Immunoblot
Briefly, whole cell lysates were collected using a cell lysis buffer [20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% SDS, 1% sodium deoxycholate, 1% Triton X-100, aprotinin, leupeptin, phenylmethylsulfonyl fluoride, and 1 mM sodium orthovanadate]. PMSF (1 mM) was added immediately prior to use. Protein (50 μg) was run on a SDS-PAGE gel and transferred onto PVDF membranes and probed with antibodies. Blots were developed using ECL (Amersham Inc., Piscataway, NJ) and exposed to film (Hyperfilm-ECL; Amersham). Blots shown are representative of 3 independent experiments.
Fluorescence Imaging
To visualize the GRP-R and GFP expression, stably transfected neuroblastoma cells were grown on coverslips in 12-well plate for 24 hours. Cells on coverslips were then rinsed once with PBS, and placed facing onto a glass slide. Fluorescent signals were revealed under the fluorescence microscope (Nikon Eclipse E600), using afluorescein isothiocyanate/GFP filter with maximum excitation at 488 nm and maximum emission at 525 nm.
GRP-R Binding Assay
To extract cell membrane proteins, cells were cultured in 150-mm plates until 90% confluent and then rinsed once with ice-cold PBS, scraped in extraction buffer (100 mM HEPES, pH 7.4, and 1-mM EGTA) with protein cocktail inhibitors (Roche, Indianapolis, IN), and 1-mM PMSF (Sigma). Lysates were homogenized with Dounce homogenizer for 25 strokes and centrifuged at 800× g for 10 minutes at 4°C. The supernatants were centrifuged at 75,000× g for 45 minutes at 4°C. The membrane protein pellets were resuspended in extraction buffer supplemented with 12% sucrose. Protein concentration was measured by Bradford method and aliquots were snapped frozen in liquid nitrogen, kept at −80°C prior to binding assay.
GRP-R ligand binding was quantitated by analysis of binding to the radiolabeled antagonist 125I-GRP (2000 Ci/mmol) from Amersham. Cellular membrane protein (100 μg) was incubated for 60 minutes at 30°C with 50 pM of 125I-GRP in binding solution containing 10-mM HEPES (pH 7.4), 100-mM NaCl, 0.3% BSA, 3-mM MgSO4, 1-mM EGTA in a total volume of 50 μL. Nonspecific binding was determined in the presence of 100-nM unlabeled GRP. The binding reaction was terminated by adding 4 mL of ice-cold solution B (20-mM Tris-HCl, pH 8.0, 100-mM NaCl, 25-mM MgCl2), followed by filtration over GF/F glass-fiber filters (Whatman) and washed 3 times with 4 mL each of ice-cold solution B. 125I-GRP was quantitated on a COBRATM II Auto-Gamma counter.
Cell Proliferation and BrdU Incorporation Assays
Cells were seeded in 96-well plates at a density of 104 cells per well in RPMI culture medium with 10% FBS and grown for 4 days. Cell number was assessed by using Cell-Counting Kit-8 (Dojindo Molecular Technologies, Inc, Gaithersburg, MD) daily. Each assay point was performed in triplicate, and the experiment was repeated 3 times for each cell line. The values, corresponding to the number of viable cells, were read at OD450 with EL808 Ultra Microplate Reader (BioTek Instrument, Inc, Winooski, VT). BrdU incorporation was determined by Cell Proliferation ELISA, BrdU kit (Roche). Briefly, SH-SY5Y cells were transfected with pBP2 (control), and pBP-PTEN-HA vector by electroporation. After 48 hours of transfection, BrdU was added to the cell culture for 6 hours and then BrdU incorporation measured according to the manufacturer’s instruction.
Gene Array Analysis
GEArray Q Series MicroArray (SuperArray, Frederick, MD) was used according to the manufacturer's protocol. Total RNA from stable transfection cells was isolated with RNAqueous (Ambion, Austin, TX). In total, 2 μg of RNA was labeled and hybridized to the membrane. The membranes were exposed to film, and changes in gene expression were determined and normalized to the signal intensity of control, housekeeping gene GAPDH with densitometric analysis using Kodak 1D Image Analysis Software (version 3.6; Rochester, NY).
Statistical Analysis
In vitro experiments were repeated on at least 2 separate occasions. Scoring index, relative cell growth, and DNA synthesis were expressed as mean ± SEM; statistical analyses were performed using one-way analysis of variance for comparisons between the treatment groups. A P value of <0.05 was considered significant.
RESULTS
PTEN and Phospho-Akt Protein Expression in Human Neuroblastomas
We first wanted to determine whether the PTEN and phospho-Akt proteins are expressed in human neuroblastoma sections. Twenty-four archival paraffin-embedded tumor sections, consisting of differentiated (n = 10) and undifferentiated (n = 14) neuroblastomas from patients of varying ages, were analyzed by immunohistochemical analysis. The histopathologic classification of tumor differentiation was based on well-described features such as nuclear size and shape, cell shape, degree of neuropil formation, and organization.
A decreased level of PTEN protein expression was noted in more undifferentiated tumors when compared with matured, well-differentiated tumors (Fig. 1A; representative sections from 24 samples). When assessed and scored by a blinded pathologist, undifferentiated tumors demonstrated significantly decreased PTEN protein expression (Fig. 1B). In contrast, the level of phospho-Akt protein expression, as an indication of PI3-K pathway activation, was similarly noted in both groups of undifferentiated and differentiated tumors. These findings suggest that down-regulation of tumor suppressor protein PTEN, a negative regulator of PI3-K, may be an important cellular mechanism in more advanced, poorly differentiated tumors.
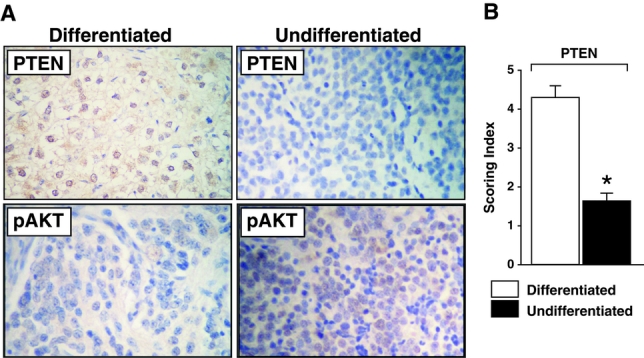
FIGURE 1. PTEN/pAkt protein expression in neuroblastomas. A, A representative histologic section from differentiated (n = 10) versus undifferentiated (n = 14) neuroblastomas demonstrating differential PTEN protein expression by immunohistochemical staining (×100). An increased expression of PTEN protein (shown by the brown staining) is noted in differentiated neuroblastomas when compared with undifferentiated tumors. In contrast, phosphorylated Akt (pAkt) protein expression was not significantly altered. B, Scoring index for PTEN protein expression [scale of 1 (low) to 5 (high) was used by a blinded pathologist] (mean ± SEM; * P < 0.05 versus differentiated tumors).
Overexpression of GRP-R in SK-N-SH and SH-SY5Y Neuroblastoma Cell Lines
GRP is a mitogenic growth factor for various normal, as well as neoplastic cells;18–20 its cell surface receptor, GRP-R, has been found in higher levels in many cancer cell types,21 including neuroblastoma.4 Therefore, to further elucidate a role of endogenously produced GRP in the regulation of PI3-K pathway in neuroblastoma cells, we used 2 well-characterized human neuroblastoma cell lines (SK-N-SH and SH-SY5Y) known to express GRP, and established stably transfected GRP-R overexpressing cell lines. This provided an important tool to further examine the molecular mechanisms of GRP ligand-dependent uncontrolled cell growth in neuroblastoma cells. Under the selection condition, GFP-tagged GRP-R existed in more than 50% cell population by observing with fluorescence microscope. GRP-R proteins were localized to cytoplasm membrane in both SK-N-SH and SH-SY5Y cell lines (Fig. 2A). In contrast, GFP proteins in control cells (pEGFP) were widely distributed in the cytoplasm and nuclei.
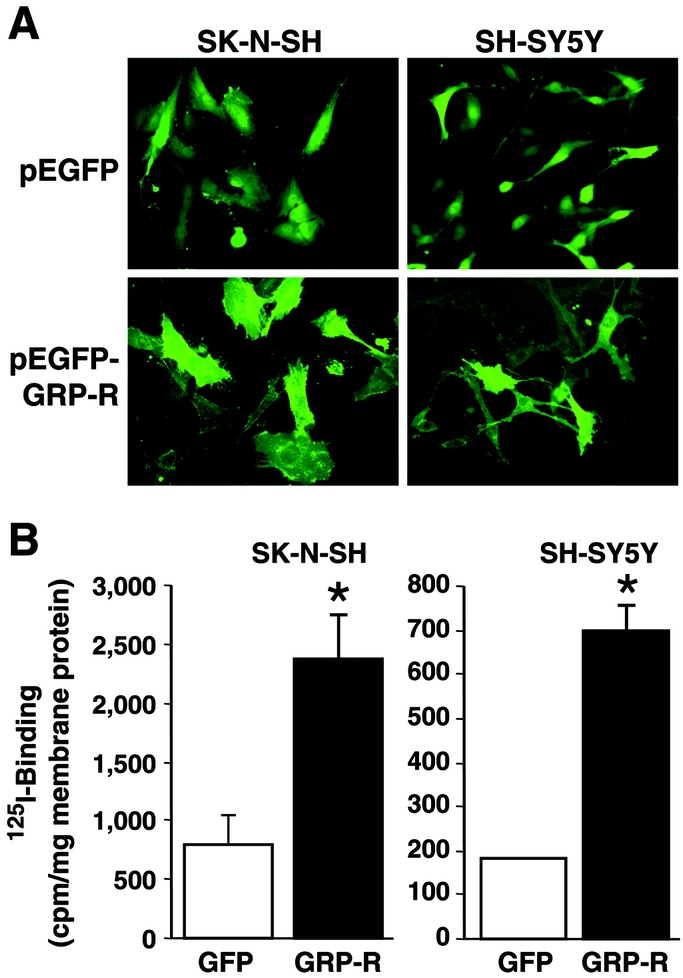
FIGURE 2. GRP-R-overexpressing SK-N-SH and SH-SY5Y neuroblastoma cells. A, A representative image demonstrating increased cell cytoplasmic membrane green fluorescence with GRP-R overexpression (GRP-R) when compared with control cells (GFP alone) in both SK-N-SH and SH-SY5Y cells. B, Competition binding assay for GRP using 125I-labeled GRP demonstrates an increased GRP-R binding capacity in both cell lines (mean ± SEM; *P < 0.05 versus GFP).
To determine whether these increased membrane-localized receptors can bind to its ligand, GRP, we next measured the GRP-R binding capacity. Our results showed that there was very low basal GRP binding activity level of 801.1cpm/mg of membrane protein in SK-N-SH control cells (GFP), while GRP-R-overexpressing cells showed much higher binding activity at 2363.5 cpm/mg membrane protein (3-fold increase) (Fig. 2B). Similar results were also noted in SH-SY5Y cells with nearly 4-fold increase in GRP binding activity in GRP-R-overexpressing cells when compared with controls (696 versus 183 cpm/mg membrane protein) (Fig. 2B). These results further demonstrate that human neuroblastoma cells transfected with GRP-R possess increased binding capacity for its ligand, GRP, thus enhancing the cellular response to this endogenously produced trophic peptide. Interestingly, GRP-R overexpressing did not influence the concentration of GRP secretion into the media in either cell lines (data not shown). These stable transfected cell lines were used for subsequent experiments.
GRP-R Overexpression Down-Regulates PTEN Gene and Protein Expression
We next wanted to determine the cellular mechanisms that contribute to GRP-induced neuroblastoma cell growth. This was accomplished by using stable GRP-R overexpression SK-N-SH cells, and performing focused microarray analysis to determine gene-expression change. The array contained 96 genes that play a role in tumor growth regulation. Among them, a tumor suppressor gene PTEN was significantly decreased (9-fold) in GRP-R-overexpressing cells when compared with control cells (Fig. 3A). Using densitometric analysis, nearly 9-fold decrease in PTEN gene expression was noted when compared with housekeeping gene GADPH expression in GRP-R-overexpressing SK-N-SH cells (Fig. 3B).
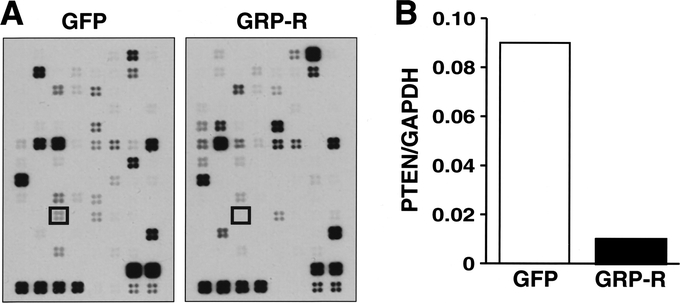
FIGURE 3. GPR-R overexpression decreases in PTEN gene expression. A, Gene array blot demonstrating significantly decreased PTEN gene expression (boxed areas) in GRP-R-overexpressing SK-N-SH cells (GRP-R) when compared with controls (GFP). B, Densitometric analysis of the PTEN gene expression change relative to housekeeping gene GAPDH.
To confirm the findings of gene array data, we next performed Western blot analysis. SK-N-SH cells overexpressing GRP-R demonstrated decreased PTEN protein levels when compared with control cells (GFP vector alone) (Fig. 4). In addition, increasing GRP-R binding capacity for its trophic ligand, GRP, also increased expression of phospho-Akt protein, suggesting that GRP activates PI3-K pathway, as well as down-regulates tumor suppressor gene PTEN in neuroblastoma cells. These findings corroborate immunohistochemical analysis data from human tumor sections, where more aggressive undifferentiated tumors showed decreased level of PTEN protein expression.
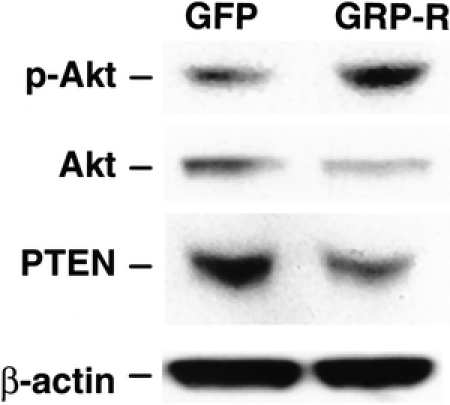
FIGURE 4. GRP-mediated increase in phospho-Akt and decrease in PTEN protein level. GRP-R-overexpressing cells (GRP-R) show an increase phospho-Akt and decrease PTEN protein levels by Western blot analysis, compared with controls (GFP). Total Akt and β-actin protein levels were relatively unchanged.
GRP Stimulates Neuroblastoma Cell Growth via PI3-K Pathway
Finally, it has been shown that mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated (ERK) pathway is typically an important intracellular signaling involved in G-protein-coupled receptor (GPCR)-mediated stimulation of cancer cell growth.22,23 However, we found that GRP-R overexpression activates PI3-K/Akt pathway in neuroblastoma cells, suggesting an important role of this signaling transduction pathway in GRP-induced neuroblastoma cell growth. In addition, we measured cell proliferation of GRP-R-overexpressing cells with or without treatment with LY294002 (10 μM), an inhibitor of PI3-K. GRP-R-overexpressing cells showed an increased basal growth rate, despite the absence of addition of exogenous GRP mitogen when compared with controls (Fig. 5A). More importanty, inhibition of PI3-K pathway with LY294002 compound completely attenuated this growth-stimulatory response in SH-SY5Y cells by day 4 (Fig. 5A). Similar results were also noted in SK-N-SH cells (data not shown). To further confirm a critical role of PI3-K pathway in growth regulation of neuroblastoma cells, we also transiently transfected cells with PTEN plasmid and measured DNA synthesis rate as an index of cell proliferation. Overexpression of PTEN, which negatively regulates PI3-K activity, significantly attenuated DNA synthesis rate by 34% in SH-SY5Y cells, further suggesting an important role of tumor suppressor gene PTEN in neuroblastoma cell growth regulation (Fig. 5B).
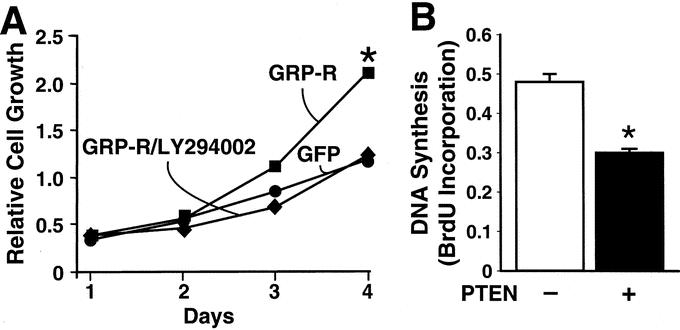
FIGURE 5. Cell proliferation and BrdU incorporation assay. A, SH-SY5Y cells stably transfected to overexpress GRP-R (GRP-R) demonstrate an accelerated basal cell growth rate when compared with control cells (GFP vector alone) by day 4. This stimulated growth rate was attenuated with inhibition of PI3-K (LY294002; 10mM) (GRP-R/LY294002). B, When SH-SY5Y cells were transiently transfected with PTEN plasmid, the DNA synthesis rate was significantly decreased when compared with control at 24-hour time point (mean ± SEM for triplicate determination; *P < 0.05 versus control).
DISCUSSION
In this study, we demonstrate a decreased expression of tumor suppressor protein PTEN in more aggressive undifferentiated neuroblastomas when compared with differentiated tumors. In contrast, the expression of phospho-Akt protein, a downstream effector for PI3-K pathway, did not significantly vary between 2 groups of archival samples examined. We then established GRP-R-overexpressing cell lines to examine the effects of GRP on PI3-K/Akt pathway, as well as its negative regulator, PTEN. We found that increasing GRP-R binding capacity results in activation of PI3-K/Akt pathway along with down-regulation of tumor suppressor gene PTEN, which then ultimately leads to stimulation of neuroblastoma cell growth. This growth-stimulatory effect was attenuated with PI3-K/Akt inhibition. Our findings suggest GRP-induced down-regulation of PTEN may be an important cellular mechanism contributing to cell proliferation in undifferentiated neuroblastomas.
Recent reports suggest that dysregulation of tumor suppressor gene PTEN may be an important cellular mechanism contributing to malignant behavior of human cancers.15,17 In particular, PTEN gene mutation has also been identified in neuroblastoma24,25; however, little is known about the differential expression of PTEN protein in neuroblastomas, correlative to biologic tumor behavior (eg, undifferentiated versus differentiated tumors). In our study, we found a decreased PTEN protein expression in more undifferentiated, clinical aggressive neuroblastomas when compared with their well-differentiated benign counterparts, suggesting that tumor suppressor protein PTEN may be down-regulated in advanced-stage tumors. In contrast, the expression level of phospho-Akt protein, as an indicator of PI3-K pathway activation, was not significantly different among archival tumor samples examined. Specifically, the phospho-Akt protein expression was found in moderate abundance in all tumor sections examined, irrespective of degree of cellular differentiation. These findings suggest that down-regulation of tumor suppressor protein PTEN may be an important mechanism in malignant tumor progression of neuroblastomas.
As a neuroendocrine tumor, neuroblastomas produce and secrete various gut peptides.3 We have previously shown that neuroblastomas express, in particular, GRP and its receptor, GRP-R.4 Additionally, we had also found an increased GRP-R expression in more advanced, undifferentiated tumors, suggesting a role for up-regulation of GRP-R in aggressive tumor behavior in neuroblastomas.4 Accumulated evidence in the literature suggests that the GRP binds to its cell-surface receptor, GRP-R, to exert trophic actions in an autocrine fashion in various cancer cells such as small-cell lung,21 pancreatic,26 breast,27 and colon cancers.20 Moreover, others have also shown that up-regulation of GRP-R may be a critical cellular mechanism contributing to uncontrolled cancer-cell growth.28 In the present study, we also demonstrate that increasing GRP binding activity by up-regulation of cell membrane GRP-R concentration results in stimulation of constitutive neuroblastoma cell growth.
Both SK-N-SH and SH-SY5Y cell lines, which we have previously found to produce and secrete endogenous GRP to act as an autocrine growth factor,4 were stably transfected to overexpress GRP-R. Importantly, these GRP-R-overexpressing cells demonstrated an accelerated basal-cell growth rate even without the addition of exogenous GRP when compared with control cells. Interestingly, endogenous production of GRP in these cells was not affected by overexpression of GRP-R, as measured by GRP secretion into the media (data not shown). These findings further suggest an autocrine loop regulation for GRP as a mitogen in neuroblastomas and that dysregulation of GRP-R may critically contribute to malignant tumor behavior.
The molecular mechanisms involved in GRP-mediated tumor cell growth have not been completely elucidated. GRP-R, a member of GPCR family, is activated upon GRP ligand binding.7 Upon activation, GRP-R activates ERK member of the mitogen-activated protein kinase family by mobilizing intracellular Ca2+ release and Ca2+/calmodulin-independent protein kinases.29 The PI3-K/Akt is an important cell survival pathway, activated in various cancer cell types to affect cell growth, survival, and cell movement.8,13,17 Other trophic growth factors, such as insulin growth factor, have been reported to stimulate neuroblastoma cell growth via activation of PI3-K/Akt pathway;14,30 however, the exact role of GRP on PI3-K/Akt pathway activation in neuroblastoma is not yet fully defined.
In the present study, we attempted to elucidate the molecular mechanism underlying GRP-induced stimulatory growth effects on neuroblastoma cells. Our results show that GRP-R overexpression, resulting in increased GRP binding activity, stimulates constitutive neuroblastoma cell growth rate via activation of PI3-K/Akt pathway. The regulation of GPCR is a complex cellular process. Altering cell-surface GPCR for a particular ligand, such GRP, can potentially influence binding activity for other growth factor (eg, epidermal growth factor) by mechanism of receptor transactivation.31 Therefore, overexpression of GRP-R in our cell model may also, in fact, activate cellular signaling other than PI3-K/Akt pathway in response to GRP. However, we found that GRP-R-overexpression-induced stimulation of SH-SY5Y cell growth is completely attenuated with PI3-K inhibitor, LY294002 compound. In contrast, inhibition of MEK/ERK pathway using PD98059 compound (50 μM) did not attenuate GRP-induced increase in SH-SY5Y cell growth (data not shown), thus further suggesting an important role of PI3-K/Akt in the GRP-mediated neuroblastoma cell growth.
The PTEN tumor suppressor gene encodes a dual-specificity phosphatase with lipid and protein phosphatase activity. PTEN play an important role in carcinogenesis of multiple human cancers.15,17 PTEN modulates cell growth and survival by negatively regulating the key cell survival pathway, PI3-K/Akt signaling pathway; decreased PTEN expression has been shown to promote cancer progression. However, little is known about an exact role of PTEN in neuroblastomas. Our findings in this study suggest that GRP-R overexpression significantly down-regulates PTEN gene and protein expression. Moreover, increasing GRP binding activity with GRP-R overexpression also increases phospho-Akt protein expression, thus further contributing to activation of PI3-K pathway to stimulate neuroblastoma cell proliferation. Taken together, our in vitro results are consistent with the immunohistochemical analysis of human neuroblastoma sections where undifferentiated tumors, previously reported to express high levels of GRP-R, demonstrate a decreased PTEN protein expression. This suggests that GRP-induced activation of PI3-K, as well as PTEN inhibition, may both contribute to tumor-cell progression; therefore, potential treatment strategies targeted to inhibit trophic actions of GRP may prove to be ideal therapy options to restore balance between PI3-K and PTEN pathways in neuroblastomas.
In addition to negative regulatory function on PI3-K/Akt pathway, PTEN also possesses the ability to regulate cancer cell proliferation by directly controlling cell apoptotic pathways. PTEN regulates translocation of Mdm2 into the nucleus.32 PTEN inhibits the nuclear translocation of Mdm2, which then rapidly degrades in the cytoplasm, resulting in activation of p53 tumor suppressor protein.33 Conversely, inhibition of PTEN results in the up-regulation of Mdm2, subsequently leading to p53 degradation and inhibition of cell apoptosis. Thus, PTEN plays an important role as a tumor-suppressor gene in various cancer-cell types. Others have shown that p53 inactivation via mutation or Mdm2 amplification is responsible for relapse in patients with aggressive recurrent neuroblastomas.34 Although it is the beyond the scope of this study, GRP-mediated down-regulation of PTEN may, in fact, also directly inhibit cell apoptosis via up-regulation of Mdm2 contributing to uncontrolled neuroblastoma cell growth.
In conclusion, we have demonstrated relative uniform expression of phospho-Akt protein in neuroblastomas examined. In contrast, expression of tumor suppressor protein PTEN was significantly decreased in more undifferentiated tumors. Furthermore, we established GRP-R-overexpressing cell lines to demonstrate that increasing GRP binding activity results in activation of PI3-K pathway and down-regulation of PTEN in neuroblastoma cells. Moreover, inhibition of PI3-K completely attenuated GRP-mediated neuroblastoma cell growth rate, demonstrating that PI3-K is an important signaling pathway in mitogen-induced stimulation of neuroblastoma cell growth. Collectively, our findings suggest an important role of GRP as an autocrine growth factor for neuroblastoma cells and that treatment strategies targeted to prevent GRP-R activation, therefore affecting tumor suppressor gene PTEN and PI3-K/Akt pathway, may provide novel adjuvant therapy options for infants and children with neuroblastomas.
ACKNOWLEDGMENTS
The authors thank Tatsuo Uchida for statistical analysis and Karen Martin for manuscript preparation.
Discussions
Dr. Kevin P. Lally (Houston, Texas): As mentioned, neuroblastoma does remain a challenging problem for both surgeons and oncologists. Advanced-stage neuroblastoma has survival rates that are well below 50%. There are a number of prognostic factors that have been associated with neuroblastoma, including patient age, histology, N-MYC amplification, and the SHIMADA classification. The authors have performed a very elegant series of experiments examining the mechanisms of tumor growth in neuroblastoma. I do have a few questions.
The first is, they suggest a scoring system in their evaluation of the PTEN expression but these tend to be somewhat arbitrary. Was this validated with a blinded observer or was it only 1 person's viewpoint? They also performed statistics on this evaluation, and was this a linear curve for their grading 1 through 5?
Second, since N-MYC oncogene is a known prognostic factor in neuroblastoma and undoubtedly has an important role in tumor biology, have they looked at the correlation between PTEN expression and amplification of N-MYC?
Third, can they speculate on the role of the PI3-K pathway in tumor differentiation? Neuroblastoma is a very unusual tumor in that a small subset can undergo spontaneous maturation. Do they think that this has a role in these tumors undergoing spontaneous maturation?
Last, in the manuscript they suggest that restoring the PI3-K/PTEN pathway would provide a novel treatment option. How would they suggest we go about that?
Dr. Max R. Langham, Jr. (Gainesville, Florida): Dr. Lally has talked about the clinical problems of neuroblastoma, and it provides a very interesting tumor model for investigation of basic mechanisms of cancer because of the difference in the behavior of a 4-S neuroblastoma in a baby where widely metastatic and very malignant appearing cancer spontaneously regresses, where a patient who is 2 or 3 years of age with a very similar tumor dies quickly despite all therapy.
I can't help but emphasize that Dr. Lally talked about one of the major risk factors that clearly stratifies these groups is N-MYC amplification. That gene product was not mentioned in this paper, which describes a hormone or a growth factor, its receptor, second message protein kinases, and their effect on a tumor suppressor gene, none of which have been shown clinically to differentiate populations of neuroblastoma.
Demonstrating that PTEN does differentiate the populations could be a very big step, particularly if it was independent of N-MYC. The authors looked at 24 patients, and it would be extremely helpful to know what the N-MYC status of those patients are and whether or not the PTEN expression was at all related to N-MYC.
In the abstract that was presented but not in the presentation or the manuscript, the authors used a third cell line, BE(2)-C, to overexpress GRP. Of the cell lines that they have chosen, the SK-N-SH cell line is nonamplified from N-MYC status, the SY-S-SH cell line has 1 copy of the N-MYC, and the BE(2)-C is the only cell line they had where the N-MYC was amplified. I would appreciate it if the authors could give us a little bit of at least preliminary data about what the expression of PTEN looked like in BE(2)-C.
A couple of mechanistic questions. If PTEN down-regulates the PI3-kinase, why is the phosphate Akt expression not changed? That seemed a little funny to me. And GRP overexpression did not increase secretion of GRP into the media. Do the authors have any speculation about why overexpression would not cause an increased secretion of the protein?
I enjoyed this paper. I think that the incredible detailed work that is required for this type of investigation will be extremely important in elucidating future therapies for a problem that kills a lot of children. And I would congratulate the authors on their paper.
Dr. Joseph J. Tepas, III (Jacksonville, Florida): I want to congratulate Dr. Chung for walking us through an extraordinarily complex presentation absolutely elegantly.
I have a question which is a little more simplistic and gets to the extrapolation of bench to bedside. How ubiquitous is PTEN? Is this seen in every cell line in the body? Is this something that falls into the category of cellular protection? Or is this unique to the particular tumor line?
Dr. Dai H. Chung (Galveston, Texas): I would like to thank all the discussants for their insightful comments and questions.
Dr. Lally asked about the validity of the quantitative analysis for PTEN and phospho-Akt protein expression by immunohistochemistry. Using a simple scoring index, utilized by a pathologist in a blinded fashion, the relative PTEN and phospho-Akt protein expression was assessed. This allowed us to quantitatively demonstrate that the PTEN, but not phospho-Akt, expression was significantly decreased in undifferentiated neuroblastomas. We have not specifically correlated the PTEN expression to N-myc copies; however, we are currently performing this study to determine whether PTEN expression inversely correlated to N-myc amplification.
In response to whether neuroblastoma differentiation affects PTEN expression, we are conducting this experiment using retinoic acid, a differentiation inducer, to determine whether PTEN expression is regulated by induction of differentiation in neuroblastoma cells. We focused our current study on determining whether gastrin-releasing peptide (GRP) down-regulates PTEN, a tumor suppressor protein, to ultimately stimulate neuroblastoma cell growth.
Dr. Langham also asked whether we examined other neuroblastoma cell lines with varying N-myc expression. We also performed experiments using another neuroblastoma cell line, BE(2)-C, known to express high N-myc copies, and found that GRP treatment down-regulates PTEN expression. So far, we have not found any significant correlation of GRP-induced PTEN down-regulation in respect to N-myc status. GRP antagonists or GRP antibody may be an important novel therapeutic agent in the treatment of neuroblastomas by a mechanism of regulating PTEN/P13-K pathway.
We do not know why there was a more significant downregulation of PTEN by GRP-R overexpression than an activation of P13-K/Akt. Clearly, GRP-induced neuroblastoma cell growth may involve other signaling pathways. The most important finding from our study was that the ratio of PTEN to phospho-Akt was significantly decreased with increasing GRP binding capacity. Therefore, GRP stimulates cell growth by down-regulating a crucial tumor suppressor protein, PTEN, in neuroblastomas.
Dr. Tepas asked whether this finding is unique to neuroblastoma. PTEN dysregulation contributing to tumor growth has been extensively studied in other cancers such as colon, pancreatic, and prostate. However, there are only a few reports examining the role of PTEN in neuroblastomas, and further studies are required to discern the exact role of PTEN as a tumor suppressor protein in neuroblastomas.
Footnotes
Supported by grants RO1 DK61470, PO1 DK35608, RO1 DK48498 from the National Institutes of Health.
Reprints: Dai H. Chung, MD, Department of Surgery, The University of Texas Medical Branch, 301 University Blvd, Galveston, TX 77555-0353. E-mail: dhchung@utmb.edu.
REFERENCES
- 1.Haase GM, Perez C, Atkinson JB. Current aspects of biology, risk assessment, and treatment of neuroblastoma. Semin Surg Oncol. 1999;16:91–104. [DOI] [PubMed] [Google Scholar]
- 2.Joshi VV, Silverman JF. Pathology of neuroblastic tumors. Semin Diagn Pathol. 1994;11:107–117. [PubMed] [Google Scholar]
- 3.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264–2279. [DOI] [PubMed] [Google Scholar]
- 4.Kim S, Hu W, Kelly DR, et al. Gastrin-releasing peptide is a growth factor for human neuroblastomas. Ann Surg. 2002;235:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HJ, Evers BM, Guo Y, et al. Bombesin-mediated AP-1 activation in a human gastric cancer (SIIA). Surgery. 1996;120:130–136. [DOI] [PubMed] [Google Scholar]
- 6.Bologna M, Festuccia C, Muzi P, et al. Bombesin stimulates growth of human prostatic cancer cells in vitro. Cancer. 1989;63:1714–1720. [DOI] [PubMed] [Google Scholar]
- 7.Cuttitta F, Carney DN, Mulshine J, et al. Bombesin-like peptides can function as autocrine growth factors in human small-cell lung cancer. Nature. 1985;316:823–826. [DOI] [PubMed] [Google Scholar]
- 8.Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335(pt 1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–267. [DOI] [PubMed] [Google Scholar]
- 10.Roche S, Downward J, Raynal P, et al. A function for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in fibroblasts during mitogenesis: requirement for insulin- and lysophosphatidic acid-mediated signal transduction. Mol Cell Biol. 1998;18:7119–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arase Y, Hiwasa T, Hasegawa R, et al. Prevention of v-Ha-Ras-dependent apoptosis by PDGF coordinates in phosphorylation of ERK and Akt. Biochem Biophys Res Commun. 2000;267:33–39. [DOI] [PubMed] [Google Scholar]
- 12.Gupta AK, McKenna WG, Weber CN, et al. Local recurrence in head and neck cancer: relationship to radiation resistance and signal transduction. Clin Cancer Res. 2002;8:885–892. [PubMed] [Google Scholar]
- 13.Kim S, Kang J, Qiao J, et al. Phosphatidylinositol 3-kinase inhibition down-regulates survivin and facilitates TRAIL-mediated apoptosis in neuroblastomas. J Pediatr Surg. 2004;39:516–521. [DOI] [PubMed] [Google Scholar]
- 14.Kim B, van Golen CM, Feldman EL. Insulin-like growth factor-I signaling in human neuroblastoma cells. Oncogene. 2004;23:130–141. [DOI] [PubMed] [Google Scholar]
- 15.Pandolfi PP. Breast cancer: loss of PTEN predicts resistance to treatment. N Engl J Med. 2004;351:2337–2338. [DOI] [PubMed] [Google Scholar]
- 16.Lee JT Jr, Steelman LS, McCubrey JA. Phosphatidylinositol 3′-kinase activation leads to multidrug resistance protein-1 expression and subsequent chemoresistance in advanced prostate cancer cells. Cancer Res. 2004;64:8397–8404. [DOI] [PubMed] [Google Scholar]
- 17.Fraser MM, Zhu X, Kwon CH, et al. PTEN loss causes hypertrophy and increased proliferation of astrocytes in vivo. Cancer Res. 2004;64:7773–7779. [DOI] [PubMed] [Google Scholar]
- 18.Chu KU, Evers BM, Ishizuka J, et al. Role of bombesin on gut mucosal growth. Ann Surg. 1995;222:94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bold RJ, Kim HJ, Ishizuka J, et al. A human gastric cancer cell line possesses a functional receptor for gastrin-releasing peptide. Cancer Invest. 1998;16:12–17. [DOI] [PubMed] [Google Scholar]
- 20.Narayan S, Guo YS, Townsend CM Jr, et al. Specific binding and growth effects of bombesin-related peptides on mouse colon cancer cells in vitro. Cancer Res. 1990;50:6772–6778. [PubMed] [Google Scholar]
- 21.Zhou J, Chen J, Mokotoff M, et al. Targeting gastrin-releasing peptide receptors for cancer treatment. Anticancer Drugs. 2004;15:921–927. [DOI] [PubMed] [Google Scholar]
- 22.Rice PL, Beard KS, Driggers LJ, et al. Inhibition of extracellular-signal regulated kinases 1/2 is required for apoptosis of human colon cancer cells in vitro by sulindac metabolites. Cancer Res. 2004;64:8148–8151. [DOI] [PubMed] [Google Scholar]
- 23.Sebolt-Leopold JS. MEK inhibitors: a therapeutic approach to targeting the Ras-MAP kinase pathway in tumors. Curr Pharm Des. 2004;10:1907–1914. [DOI] [PubMed] [Google Scholar]
- 24.Munoz J, Lazcoz P, Inda MM, et al. Homozygous deletion and expression of PTEN and DMBT1 in human primary neuroblastoma and cell lines. Int J Cancer. 2004;109:673–679. [DOI] [PubMed] [Google Scholar]
- 25.Wang L, Ignat A, Axiotis CA. Differential expression of the PTEN tumor suppressor protein in fetal and adult neuroendocrine tissues and tumors: progressive loss of PTEN expression in poorly differentiated neuroendocrine neoplasms. Appl Immunohistochem Mol Morphol. 2002;10:139–146. [DOI] [PubMed] [Google Scholar]
- 26.Parekh D, Ishizuka J, Townsend CM Jr, et al. Characterization of a human pancreatic carcinoid in vitro: morphology, amine and peptide storage, and secretion. Pancreas. 1994;9:83–90. [DOI] [PubMed] [Google Scholar]
- 27.Bajo AM, Schally AV, Groot K, et al. Bombesin antagonists inhibit proangiogenic factors in human experimental breast cancers. Br J Cancer. 2004;90:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markwalder R, Reubi JC. Gastrin-releasing peptide receptors in the human prostate: relation to neoplastic transformation. Cancer Res. 1999;59:1152–1159. [PubMed] [Google Scholar]
- 29.Zhang Q, Thomas SM, Xi S, et al. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004;64:6166–6173. [DOI] [PubMed] [Google Scholar]
- 30.Meyer GE, Shelden E, Kim B, et al. Insulin-like growth factor I stimulates motility in human neuroblastoma cells. Oncogene. 2001;20:7542–7550. [DOI] [PubMed] [Google Scholar]
- 31.Lui VW, Thomas SM, Zhang Q, et al. Mitogenic effects of gastrin-releasing peptide in head and neck squamous cancer cells are mediated by activation of the epidermal growth factor receptor. Oncogene. 2003;22:6183–6193. [DOI] [PubMed] [Google Scholar]
- 32.Chang CJ, Freeman DJ, Wu H. PTEN regulates Mdm2 expression through the P1 promoter. J Biol Chem. 2004;279:29841–29848. [DOI] [PubMed] [Google Scholar]
- 33.Zhou M, Gu L, Findley HW, et al. PTEN reverses MDM2-mediated chemotherapy resistance by interacting with p53 in acute lymphoblastic leukemia cells. Cancer Res. 2003;63:6357–6362. [PubMed] [Google Scholar]
- 34.Tweddle DA, Pearson AD, Haber M, et al. The p53 pathway and its inactivation in neuroblastoma. Cancer Lett. 2003;197:93–98. [DOI] [PubMed] [Google Scholar]