

Abstract
Objective:
To determine whether insulinlike growth factor-I (IGF-I) and hepatocyte growth factor (HGF) cooperate to induce migration and invasion of human colorectal carcinoma (CRC) cells and whether the effects of IGF-I and/or HGF are mediated through activation of the urokinase plasminogen activator (uPA)/uPA receptor (uPAR) system, a central mediator of tumor-cell migration and invasion.
Summary Background Data:
CRC cells must invade through the basement membrane of the colon and migrate to form metastases. CRC cells are known to overexpress IGF-I receptor (IGF-IR), c-Met, and uPAR, 3 cell-surface receptors known to mediate cell migration and invasion. We hypothesized that IGF-IR and c-Met cooperate to induce migration and invasion in CRC cells and that this signaling is dependent on uPAR.
Methods:
KM12L4 human CRC cells were treated with IGF-I, HGF, or IGF-I + HGF in transwell migration and invasion chambers; cells that had migrated or invaded were counted. To determine the role of c-Met in IGF-I-induced migration and invasion, c-Met was inhibited by infection of cells with an adenovirus containing a c-Met ribozyme; transwell assays were then repeated. To determine the role of the uPA/uPAR system in IGF-I-induced CRC cell migration and invasion, transwell assays were repeated after pretreating cells with the uPA inhibitor amiloride or with neutralizing antibodies to uPA and uPAR.
Results:
IGF-I and HGF, alone or in combination, increased cell migration and invasion. The c-Met ribozyme inhibited IGF-I- and HGF-mediated migration and invasion, indicating that c-Met is essential for these processes. uPA and uPAR inhibition blocked IGF-I- and HGF-mediated migration and invasion, suggesting that uPAR is downstream of IGF/IGF-IR and HGF/c-Met in the signaling pathways that mediate cell migration and invasion.
Conclusions:
IGF-I and HGF cooperate to induce migration and invasion of CRC cells, and c-Met and uPA/uPAR are required for IGF-I-mediated migration and invasion. In our in vitro model of CRC migration and invasion, uPA and uPAR appear to be downstream of IGF-IR and c-Met and are required for migration and invasion. Elucidation of the pathways that contribute to tumor progression and metastasis should provide a foundation for the rational development and use of targeted therapies for CRC.
This study shows that insulinlike growth factor-I (IGF-I) and hepatocyte growth factor (HGF) cooperate to induce migration and invasion of human colorectal carcinoma cells. The HGF receptor, c-Met, is required for IGF-I-mediated migration and invasion, and urokinase plasminogen activator and its receptor are activated by IGF-I and HGF and are necessary for IGF-I- and HGF-induced migration and invasion.
In 2004, there were an estimated 147,000 new cases of colorectal carcinoma (CRC) and 57,000 deaths from this disease, ranking it third among causes of cancer-related death in the United States.1 Significant advances in systemic therapy for metastatic CRC, including targeted therapies, have improved survival, but even with combination therapy the median survival is only about 15 to 21 months.2,3 To continue to improve our therapies for metastatic CRC, we need a better understanding of the factors that lead to tumor progression and metastasis. In particular, the mechanisms regulating CRC cell invasion through the basement membrane of the colon and migration of the cells to form metastases need to be further investigated.
Insulinlike growth factor-I (IGF-I) and its tyrosine kinase receptor (IGF-IR) have been implicated in the development and progression of a variety of human cancers,4–12 including CRC.13–15 IGF-I has been shown to be an important mediator of tumor cell migration and invasion,16–20 but the downstream pathways by which IGF-I induces these processes have not been fully elucidated. Hepatocyte growth factor/scatter factor (HGF) and its tyrosine kinase receptor c-Met have also been implicated in the pathogenesis of a wide variety of human malignancies,21–25 including CRC.26 Similar to IGF-I, HGF/c-Met signaling is known to induce tumor-cell migration and invasion.11,25,27–30
Recently, cooperation between receptors and their signaling pathways has been shown to be important in regulating cellular responses to various ligands. We theorized that IGF-IR and c-Met cooperate in mediating migration and invasion of human CRC cells, given the following findings. First, IGF-I and HGF lead to activation of the urokinase plasminogen activator (uPA)/uPA receptor (uPAR) system in various malignancies.4,11,17,27,31,32 This is central to our hypothesis in that uPA has been shown to cleave pro-HGF to active HGF.33 Second, IGF-I signaling results in induction of hypoxia inducible factor-1α in pancreatic carcinoma cells,10 and hypoxia, likely acting via hypoxia inducible factor-1α, has been shown to increase c-Met levels in human lung, hepatocellular, and other carcinomas.34 Third, growth factor receptor-binding protein 2-associated binder-1 functions as the main substrate and docking protein regulating downstream signaling by c-Met and has been shown to function as a signaling intermediate for IGF-I as well.35 Fourth, IGF-I and HGF have been shown to function as comitogens in a rat hepatoma cell line.36
In the current study, we investigated the hypotheses that IGF-IR and c-Met cooperate to mediate migration and invasion of human CRC cells and that uPA/uPAR activation is required for IGF-I- and HGF-mediated migration and invasion. We used a c-Met ribozyme to inhibit c-Met function in KM12L4 human CRC cells and performed transwell migration and invasion assays. The c-Met ribozyme experiments demonstrated that c-Met function is critical for IGF-I-mediated cell migration and invasion and for constitutive invasion. In experiments inhibiting uPA or uPAR, we demonstrated that migration and invasion mediated by IGF-I and HGF are dependent on uPA/uPAR activation. This suggests that uPA and uPAR are downstream of IGF-I and IGF-IR and of HGF and c-Met. To our knowledge, this study is the first to identify tyrosine kinase receptor cooperation between IGF-IR and c-Met in human CRC and the role of uPA/uPAR in mediating IGF-I and HGF/c-Met effects.
MATERIALS AND METHODS
Cell Lines and Cell Culture Conditions
The human CRC cell line KM12L437 was kindly provided by I. J. Fidler, PhD, DVM (The University of Texas M. D. Anderson Cancer Center, Houston, TX). Cells were cultured and maintained in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 2units/mL of a penicillin-streptomycin mixture (Flow Laboratories, Rockville, MD), vitamins (Life Technologies, Inc., Grand Island, NY), 1 mM sodium pyruvate, 2 mM L-glutamine, and nonessential amino acids and incubated in 5% CO2–95% air at 37°C.
Transwell Migration and Invasion Assays
To assess cell migration in vitro, KM12L4 cells (1.5 × 105 cells in 500 μL MEM supplemented with 1% FBS) were placed in the top chamber of transwell migration chambers (8-μm BioCoat Control Inserts; Becton Dickinson Labware, Bedford, MA). The lower wells were filled with 750 μL MEM supplemented with 10% FBS plus one of the following: (1) phosphate-buffered saline (PBS), (2) 100 ng/mL recombinant human IGF-I (R&D Systems, Minneapolis, MN), (3) 100 ng/mL recombinant human HGF (R&D Systems), or 4) 100 ng/mL IGF-I + 100 ng/mL HGF. After 48hours, unmigrated cells were removed from the upper surface of the transwell membrane with a cotton swab, and migrated cells on the lower membrane surface were fixed, stained, photographed, and counted under high-power magnification.
To assess invasion, in vitro invasion assays were performed under the same conditions as the transwell migration assays but using Matrigel-coated transwells (BioCoat Matrigel Invasion Chamber; Becton Dickinson Labware).
c-Met Ribozyme-Expressing Adenovirus
The c-Met ribozyme, designed to inhibit c-Met expression, was constructed by Abounader et al.38 The c-Met ribozyme contains U1snRNA, c-Met antisense sequence, and a hammerhead ribozyme that targets c-Met mRNA at residue 560. The control plasmids lack the ribozyme and the c-Met targeting sequence but contain the U1snRNA and other plasmid sequences. An adenovirus expressing the 560 ribozyme (ad-c-Met ribozyme) and a control adenovirus (ad-pU1) were constructed, and the viruses were harvested and purified as described previously.26
Western Blot Analysis for c-MET
To determine the impact of the c-Met ribozyme, total c-Met protein levels were measured by Western blot analysis. KM12L4 cells at 80% confluence were infected with ad-c-Met ribozyme or ad-pU1 (control) at 50 multiplicities of infection. After 24 hours, cells were lysed with lysis buffer (50 mM HEPES (pH 7.0), 150 mM NaCl, 1.5 mM MgCl2, 1mM EDTA, 10 mM sodium pyrophosphate, 10% glycerol, 1% Triton X-100, 1 mM sodium orthovanadate, and 1 complete Mini Protease Inhibitor Cocktail Tablet [Roche Diagnostics, Basel, Switzerland]). The proteins (50 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane (Immobilon-P Transfer Membrane; Millipore Corp, Bedford, MA). Following blocking with 5% milk in Tris-buffered saline/0.1% Tween 20 (vol/vol), the membrane was probed with anti-c-Met antibody (1:1000 in 5% milk; met (C-28; Santa Cruz Biotechnology, Santa Cruz, CA). The membrane was then washed and treated with horseradish peroxidase–conjugated goat antirabbit immunoglobulin G (IgG; Bio-Rad Laboratories, Hercules, CA) at 1:3000 in 5% milk. Immunoreactive proteins were visualized with an enhanced chemiluminescence detection kit (Amersham Biosciences, Little Chalfont, England). β-actin (Sigma-Aldrich Co, St. Louis, MO) was used for loading control.
c-Met Immune Complex Kinase Assay
A c-Met immune complex kinase assay was performed as described previously.39,40 Briefly, c-Met was immunoprecipitated from 500 μg total cellular protein in 500 μL lysis buffer (detailed above) with 1 μg anti-c-Met antibody and rotated at 4°C for 12 hours. Immune complexes were formed with the addition of 50 μL of 10% (vol/vol) formalin-fixed Staphylococcus aureus (Pansorbin; Calbiochem, San Diego, CA) and rotated at 4°C for 4 hours. Kinase reactions were initiated at room temperature with the addition of kinase reaction buffer (20 mM HEPES, 100 μM sodium orthovanadate, 10 mM MnCl2, 10 mM MgCl2, and 10 μCi [γ-32P]ATP). To analyze phosphorylation of an exogenous substrate, 10 μg acid-denatured rabbit muscle enolase (Sigma-Aldrich Co) was added to the reaction buffer. After 10minutes, reactions were terminated by the addition of SDS sample buffer. Proteins were separated by 8% SDS-PAGE, the gel was dried, and radioactive bands were detected by autoradiography.
uPA and uPAR Inhibition
The uPA inhibitor amiloride41 (Sigma-Aldrich Co) was added to KM12L4 cells in the upper chamber of the transwells at 100 μM to study the effect of uPA inhibition on migration and invasion. To confirm the results obtained with amiloride, migration and invasion assays were performed with specific blocking anti-uPA and anti-uPAR antibodies, using isotype-controlled, nonspecific IgG as a control (all supplied by Andrew Mazar, PhD; Attenuon, LLC, San Diego, CA). The anti-uPA antibody, anti-uPAR antibody, or IgG was added to cells in the upper chamber of the transwells at 10 ng/mL.
Cell Proliferation as Measured by MTT Assay
MTT assays were performed to evaluate the cell proliferative effects of IGF-I, HGF, and IGF-I + HGF on KM12L4 cells and assure that any increase in migration or invasion was not simply due to an increase in cell number. Cells (1.5 × 105) were plated in 96-well plates in 1% FBS-containing MEM (initial conditions in upper chamber of transwells) or 6% FBS-containing MEM (representing equilibrated conditions in transwells), and PBS, IGF-I, HGF, or IGF-I + HGF was added to achieve a final concentration of 100 ng/mL. Assays were performed in quadruplicate. After 48 hours, MTT (Sigma-Aldrich Co) was added at 2 mg/ml in PBS (50 μL/well) then plates were incubated at 37°C for 2hours. Media and MTT were removed, dimethyl sulfoxide was added for 10 minutes, and absorbance was measured at 570 nm. MTT assays were also performed with the ad-c-Met ribozyme-infected cells and the control ad-pU1-infected cells to evaluate the effect of the c-Met ribozyme on cell proliferation and with cells treated with or without the uPA inhibitor amiloride to evaluate its effect on cell proliferation. For each of the MTT assays, the cells were kept under the same culture conditions, plated at the same cell density, and treated for the same time period with the same dose of IGF, HGF, or amiloride to replicate the conditions of the migration and invasion assays.
Statistical Analyses
Statistical analyses were performed using InStat version 3.01 software (GraphPad Software Inc, San Diego, CA). The significance of differences was determined using the Mann-Whitney U test and Student t test, as appropriate. Significance was determined with 95% confidence.
RESULTS
Effect of IGF-I and HGF on Cell Migration and Invasion
To evaluate the effect of IGF-I and HGF on KM12L4 cell migration, a transwell migration assay was performed. IGF-I led to a 17.5- ± 2.3-fold increase in cell migration (P< 0.005 versus PBS), and HGF led to an 11.0- ± 2.2-fold increase in cell migration (P < 0.005 versus PBS; Fig. 1). IGF-I + HGF led to a 29.5- ± 2.4-fold increase in cell migration (P < 0.0001; Fig. 1), which was significantly greater than the increase with IGF-I or HGF alone (P < 0.01).
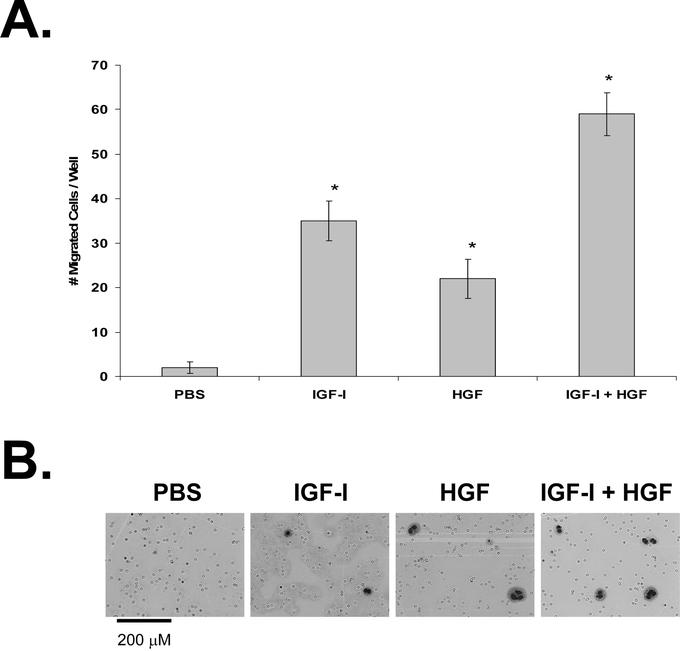
FIGURE 1. Effect of IGF-I and HGF on KM12L4 CRC cell migration. A, A transwell migration assay was performed on KM12L4 cells with PBS, IGF-I, HGF, or IGF-I + HGF. After 48hours, migrated cells were fixed, stained, and counted. IGF-I and HGF increased cell migration, and the effects of IGF-I+ HGF were additive. *P < 0.005 versus PBS (Mann-Whitney Utest); bars, standard error of mean. B, Photographs of migrated cells.
The effect of IGF-I and HGF on KM12L4 cell invasion was evaluated utilizing a Matrigel-coated transwell invasion assay. IGF-I led to a 1.8- ± 0.1-fold increase in cell invasion (P < 0.05 versus PBS), and HGF led to a 2.5- ± 0.2-fold increase in cell invasion (P < 0.005 versus PBS; Fig. 2). IGF-I + HGF led to a 3.0- ± 0.3-fold increase in cell invasion (P < 0.005 versus PBS; Fig. 2), which was significantly greater than the increase with IGF-I alone (P < 0.01).
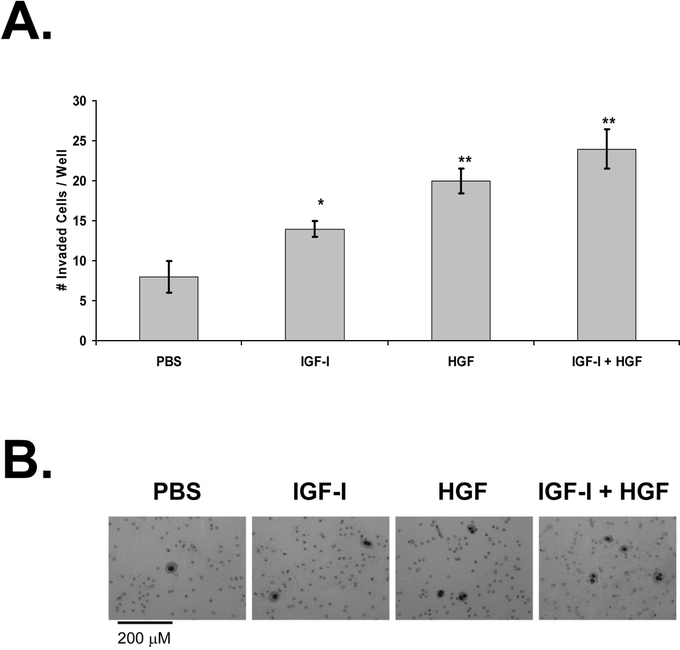
FIGURE 2. Effect of IGF-I and HGF on KM12L4 CRC cell invasion. A, A transwell invasion assay was performed on KM12L4 cells with PBS, IGF-I, HGF, or IGF-I + HGF. After 48hours, migrated cells were fixed, stained, and counted. IGF-I and HGF increased cell invasion, and the effects of IGF-I+ HGF were additive. *P < 0.05, **P < 0.005 versus PBS (Student t test); bars, standard error of mean. B, Photographs of invaded cells.
To demonstrate that the increases in cell migration and invasion were not due to increases in cell proliferation induced by IGF-I and HGF, an MTT assay was performed, with the KM12L4 cells treated with PBS, IGF-I, HGF, or IGF-I + HGF. There was no increase in cell proliferation in any of the groups (data not shown).
Effect of c-Met Inhibition on Cell Migration and Invasion
To test the hypothesis that c-Met function is required for IGF-I-mediated migration and invasion, c-Met was down-regulated with a ribozyme. The ad-c-Met ribozyme resulted in a 15% decrease in total c-Met protein levels compared with the control (ad-pU1; Fig. 3). However, in previous studies, we had observed that a relatively small decrease in protein levels leads to a pronounced effect on kinase activity.26 Therefore, we determined the effect of the ribozyme on intrinsic c-Met kinase activity utilizing an immune complex kinase assay. The ad-c-Met ribozyme decreased c-Met kinase activity by 80% compared with the control (ad-pU1; Fig. 3). These data again demonstrated that a modest reduction in c-Met expression levels leads to substantially reduced c-Met function. An MTT assay demonstrated that the ad-c-Met ribozyme did not change cell proliferation relative to the ad-pU1 control (data not shown).
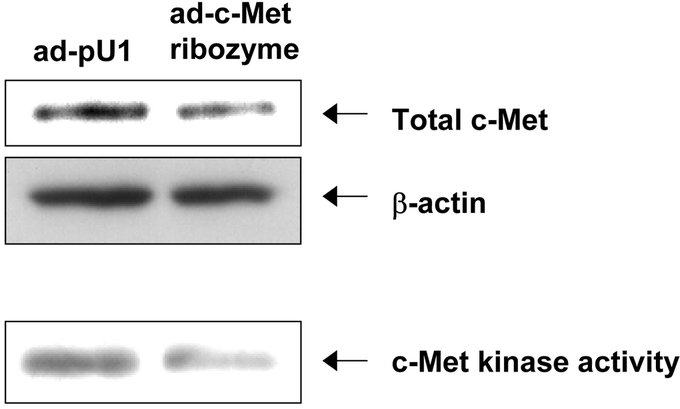
FIGURE 3. Inhibition of c-Met with c-Met ribozyme. KM12L4 CRC cells were infected with an adenoviral vector containing a c-Met ribozyme (ad-c-Met ribozyme) or an empty adenoviral vector (ad-pU1). Western blotting was performed to assess the total c-Met level and an immune complex kinase assay was performed to evaluate c-Met kinase activity. The total c-Met level (upper panel) was reduced 15% by the ad-c-Met ribozyme, whereas c-Met kinase activity (lower panel) was reduced 80%.
Inhibition of c-Met with the ad-c-Met ribozyme led to a 96% ± 4% decrease in HGF-mediated migration (P < 0.005 versus ad-pU1; Fig. 4). This finding confirmed the effect of the ribozyme on c-Met activity because HGF signals through the c-Met receptor and is known to mediate migration and invasion. The ribozyme also inhibited IGF-I-mediated migration (97% ± 3% decrease versus ad-pU1; P < 0.005) and migration mediated by IGF-I + HGF (P < 0.005 versus ad-pU1; Fig. 4). Thus, c-Met activity is required for IGF-I- and HGF-mediated migration and invasion. The ribozyme did not inhibit migration in the PBS-treated cells, suggesting that c-Met activity is not required for constitutive cell migration.
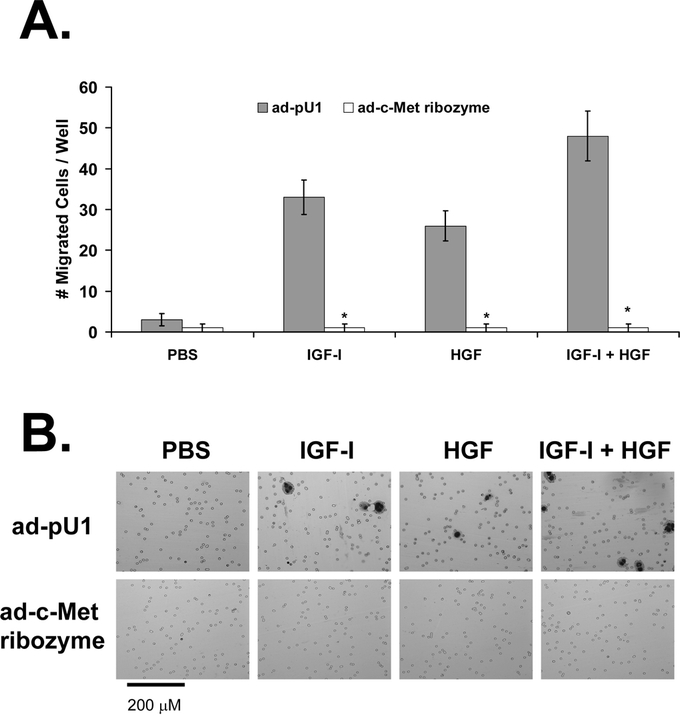
FIGURE 4. Effect of c-Met down-regulation on KM12L4 CRC cell migration. A, KM12L4 cells were infected with an adenoviral vector containing the c-Met ribozyme (ad-c-Met ribozyme) or a control vector (ad-pU1), and then a transwell migration assay was performed. The ad-c-Met ribozyme decreased cell migration in the IGF-I, HGF, and IGF-I + HGF groups by 96% to 98% compared with controls. *P < 0.005 versus ad-pU1 (Mann-Whitney U test); bars, standard error of mean. B, Photographs of migrated cells.
The ad-c-Met ribozyme also led to a 93% ± 7% decrease in HGF-mediated invasion (P< 0.05 versus ad-pU1), a 91% ± 9% decrease in IGF-I-mediated invasion (P< 0.05 versus ad-pU1), and an 89% ± 7% decrease in invasion mediated by IGF-I + HGF (P < 0.05 versus ad-pU1; Fig. 5). Thus, IGF-I and HGF appear to mediate cell invasion via a c-Met-dependent pathway. Furthermore, c-Met appears to be necessary for constitutive cell invasion since the ribozyme led to an 82% ± 12% decrease in invasion in the PBS-treated control group (P < 0.05 versus ad-pU1; Fig. 5).
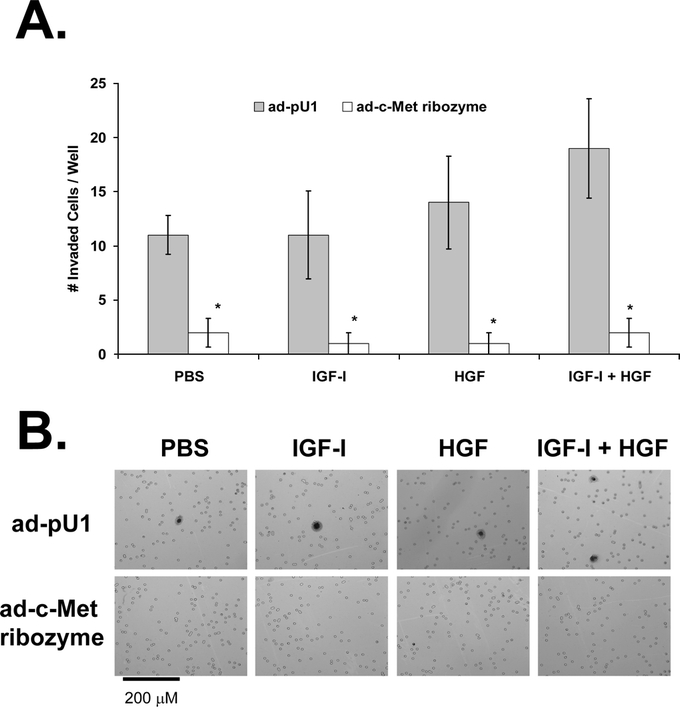
FIGURE 5. Effect of c-Met down-regulation on KM12L4 CRC cell invasion. A, KM12L4 cells were infected with an adenoviral vector containing the c-Met ribozyme (ad-c-Met ribozyme) or a control vector (ad-pU1), and then a transwell invasion assay was performed. The ad-c-Met ribozyme decreased cell invasion by 89% to 93% compared with controls. *P < 0.05 versus ad-pU1 (Mann-Whitney U test); bars, standard error of mean. B, Photographs of invaded cells.
Roles of uPA and uPAR in Cell Migration and Invasion
To determine the role of uPA in cell migration and invasion, we pretreated cells with the uPA inhibitor amiloride and then stimulated them with PBS (control), IGF-I, HGF, or IGF-I + HGF in transwells. Amiloride led to a 47% ± 10% decrease in migration of IGF-I-treated cells (P < 0.05 versus control), a 79% ± 7% decrease in migration of HGF-treated cells (P < 0.0001 versus control), and a 49% ± 7% decrease in the migration of IGF-I + HGF–treated cells (P < 0.0001 versus control; Fig. 6). Thus, IGF-I- and HGF-mediated cell migration are dependent on uPA.
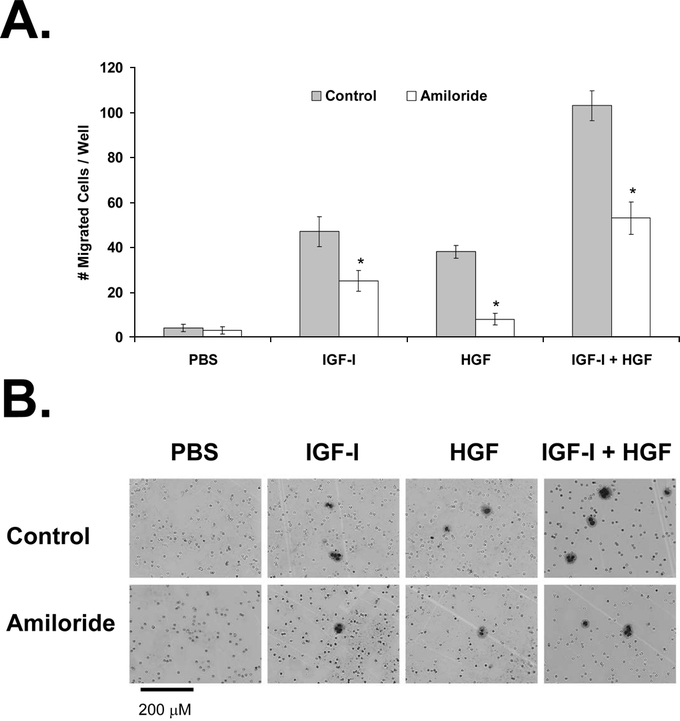
FIGURE 6. Effect of uPA inhibition on KM12L4 CRC cell migration. A, A transwell migration assay was performed on KM12L4 cells with or without the uPA inhibitor amiloride. Amiloride decreased cell migration induced by IGF-I, HGF, and IGF-I + HGF. *P < 0.05 versus control (Student t test); bars, standard error of mean. B, Photographs of migrated cells.
IGF-I-mediated cell invasion was almost completely inhibited by amiloride (94% ± 6% decrease, P < 0.005 versus control; Fig. 7). Amiloride also led to a 70% ± 9% decrease in HGF-mediated invasion (P < 0.005 versus control) and an 80% ± 8% decrease in IGF-I + HGF–mediated invasion (P < 0.001 versus control; Fig. 7). This experiment demonstrated that uPA is required for IGF-I- and HGF-mediated cell invasion.
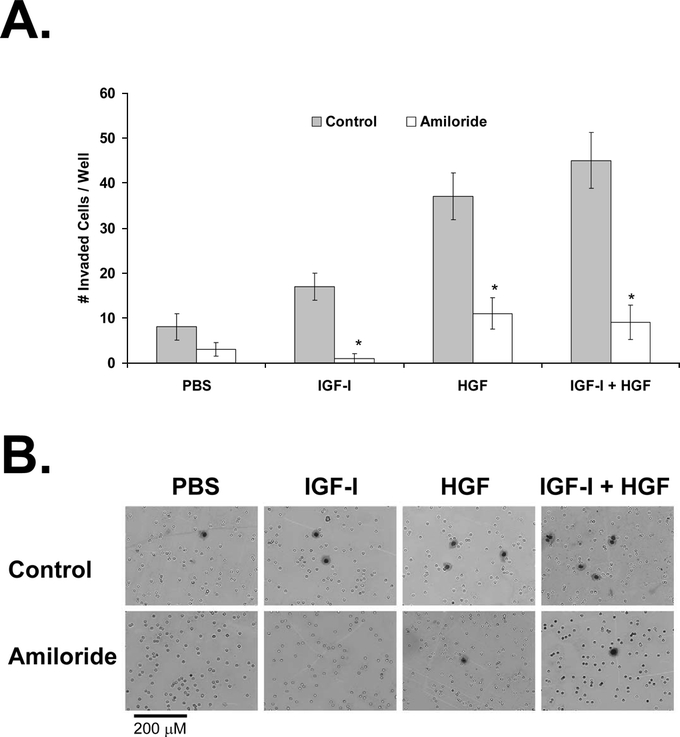
FIGURE 7. Effect of uPA inhibition on KM12L4 CRC cell invasion. A, A transwell invasion assay was performed on KM12L4 cells with or without the uPA inhibitor amiloride. uPA inhibition with amiloride led to a significant decrease in cell invasion mediated by IGF-I, HGF, and IGF-I + HGF. *P < 0.005 versus control (Mann-Whitney U test); bars, standard error of mean. B, Photographs of invaded cells.
To confirm the role of uPA in IGF-I-mediated cell migration and invasion, we pretreated cells in transwells with neutralizing antibodies to uPA or uPAR and then treated the cells with IGF-I (or PBS as a control). Nonspecific isotype-controlled IgG was used as a negative control, and amiloride was used as a positive control. The increase in migration induced by IGF-I, compared with PBS, was unaffected by pretreatment with nonspecific IgG (Fig. 8). The positive control, amiloride, led to a 92% ± 8% decrease in IGF-I-mediated cell migration (P < 0.005 versus IgG; Fig. 8). The anti-uPA antibody inhibited IGF-I-mediated migration by 85% ± 10% (P < 0.005 versus IgG), and the anti-uPAR antibody inhibited IGF-I-mediated migration by 92% ± 8% (P < 0.005 versus IgG; Fig. 8). This experiment demonstrated that IGF-I-mediated cell migration is dependent on uPA/uPAR signaling.
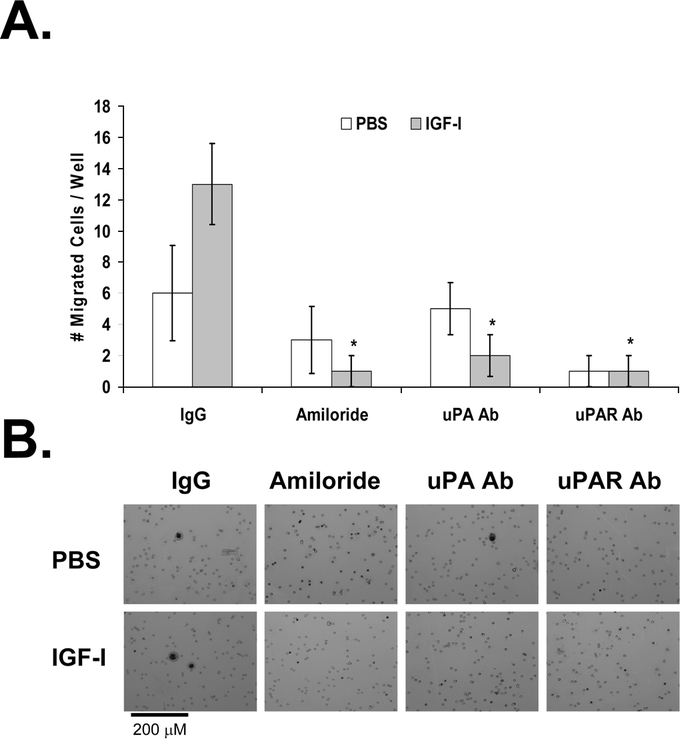
FIGURE 8. Effect of antibodies to uPA and uPAR on IGF-I-induced migration of KM12L4 CRC cells. A, A transwell migration assay was performed on KM12L4 cells with nonspecific IgG, amiloride, anti-uPA antibody (Ab), or anti-uPAR Ab. IGF-I-induced cell migration was blocked by amiloride and by anti-uPA Ab and anti-uPAR Ab. *P < 0.005 versus IgG (Mann-Whitney U test); bars, standard error of mean. B, Photographs of migrated cells.
The anti-uPA and anti-uPAR antibodies had a similar effect on IGF-I-mediated cell invasion. The positive control, amiloride, led to a 75% ± 13% decrease in IGF-I-mediated invasion (P < 0.05 versus IgG), the uPA antibody inhibited IGF-I-mediated invasion by 75% ± 8% (P < 0.05 versus IgG), and the uPAR antibody inhibited IGF-I-mediated invasion by 85% ± 8% (P < 0.01 versus IgG; Fig. 9). This experiment demonstrated that IGF-I-mediated cell invasion is also dependent on uPA/uPAR signaling.
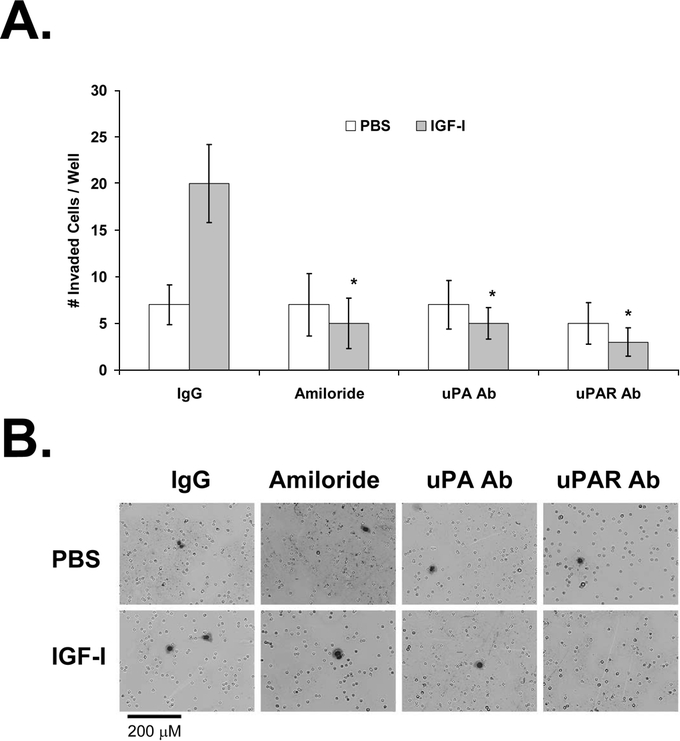
FIGURE 9. Effect of antibodies to uPA and uPAR on IGF-I-induced invasion of KM12L4 CRC cells. A, A transwell invasion assay was performed on KM12L4 cells with nonspecific IgG, amiloride, anti-uPA antibody (Ab), or anti-uPAR Ab. IGF-I-induced cell invasion was blocked by amiloride and by anti-uPA Ab and anti-uPAR Ab. *P < 0.05 versus IgG (Mann-Whitney U test); bars, standard error of mean. B, Photographs of cells.
DISCUSSION
Tumor cell migration and invasion are 2 critical steps in the metastatic cascade. CRCs have been shown to overexpress IGF-IR,42–44 c-Met,45 and uPAR.46–49 These factors are known to mediate cell migration and invasion, but the influence of any one factor on the activity of another is unknown. We hypothesized that IGF-IR and c-Met cooperate to mediate CRC cell migration and invasion and sought to determine the role of uPA/uPAR in this pathway.
In this study, we found that IGF-I and HGF cooperated to induce migration and invasion of human CRC cells. Both IGF-I and HGF, alone and in combination, significantly increased cell migration and invasion. In the ad-c-Met ribozyme experiments, c-Met was required not only for HGF-mediated migration and invasion (as expected) but also for IGF-I-mediated migration and invasion. Inhibition of IGF-I- and HGF-mediated migration and invasion by the uPA inhibitor amiloride or by neutralizing antibodies to uPA and uPAR indicated that uPA and uPAR were essential to the processes. Thus, c-Met appears to be downstream of IGF-IR, and uPAR appears to be downstream of both IGF-IR and c-Met in this signaling pathway (Fig. 10).
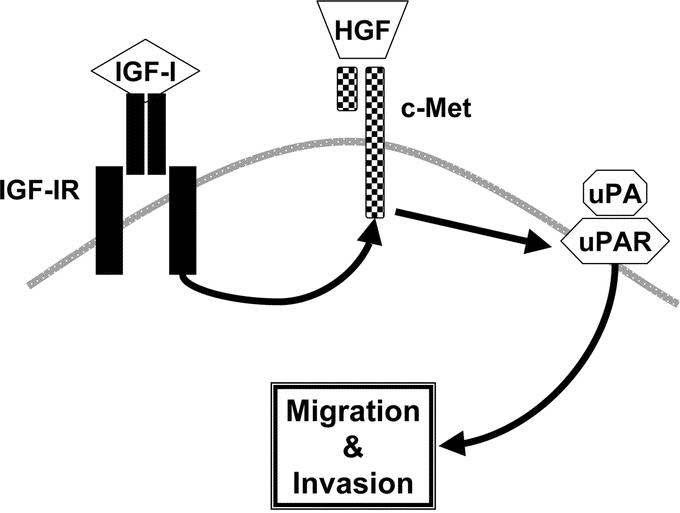
FIGURE 10. Proposed pathway of IGF-IR, c-Met, and uPAR signaling in CRC cell migration and invasion. c-Met appears to be required for IGF-I- and HGF-mediated migration and invasion, and uPAR appears to be downstream of both IGF-IR and c-Met.
Our finding that IGF-I induces migration and invasion of human CRC cells has been reported by others.50–52 However, the link between IGF-I and uPAR in CRC is new. Our finding that HGF increases cell migration and invasion is well documented in the literature.53–55 However, to our knowledge, this study is the first to demonstrate cooperation between IGF-IR and c-Met in any malignancy.
IGF-IR and c-Met may cooperate via interaction of their signaling pathways. Cooperation of cell surface receptors has been shown between the tyrosine kinase receptor RON and epidermal growth factor receptor,56 IGF-IR and epidermal growth factor receptor,57 estrogen receptor and IGF-IR,58 and G protein–coupled receptors and IGF-IR.59 Although cooperation between IGF-IR and c-Met signaling is the most likely explanation for our findings, an alternative explanation could be that HGF/c-Met signaling is a critical downstream regulator of CRC cell migration and invasion. This seems plausible since HGF (initially termed scatter factor)60–62 was originally identified as a factor converting Madin-Darby canine kidney cells into migratory fibroblasts,63,64 a process dependent on epithelial-mesenchymal transition.65 Epithelial-mesenchymal transition is now known to be a key process in cell motility, and its regulation by HGF/c-Met may be a critical downstream step in CRC migration and invasion.
In addition to demonstrating cooperation between IGF-IR, c-Met, and uPAR, we have validated these 3 factors as targets for CRC therapy. IGF-IR-directed therapy is currently being investigated extensively in clinical trials. In preclinical studies by our laboratory (unpublished data) and in studies from other laboratories, IGF-IR inhibition has led to decreased growth of human CRCs.13,14 c-Met inhibitors have not been investigated in clinical trials at the time of the writing of this manuscript. Thus, c-Met-directed therapy represents an underdeveloped area and needs to be further explored. Similarly, uPAR inhibition has not been studied in clinical trials and warrants additional investigation.
In conclusion, our study better defines the roles of IGF-IR, c-Met, and uPAR in CRC cell migration and invasion. Elucidation of the pathways that contribute to tumor progression and metastasis should provide a foundation for the rational development and use of targeted therapies for CRC.
ACKNOWLEDGMENTS
The authors thank Rita Hernandez for administrative assistance and Melissa G. Burkett of the Department of Scientific Publications for editorial assistance.
Discussions
Dr. William G. Cance (Gainesville, Florida): Dr. Ellis's group has performed pioneering work in the areas of insulinlike growth factor receptors and their translational aspects in cancer. This is a very significant study because it provides clues to the methods by which tumors invade and metastasize, and this ultimately will lead to the development of novel therapeutics.
As Dr. Ellis has shown, it is naive to think that one signaling pathway in a tumor is going to be able to be inhibited and kill the tumor itself. There will be a convergence of multiple different pathways and probably selection of different pathways within resistant tumor cells that we need to dissect in order to develop drugs for the next generation of therapeutics.
To take our surgical craft to the next level, we must define these translational aspects of human tumors. As we already have heard at the meeting, we can resect most any tumor, but it is the recurrence that causes the problem that ultimately kills the patient. In gastrointestinal tumors, our drugs simply don't work very well. So I have several questions and comments.
You have described 2 novel tyrosine kinase pathways in these cells. What other signaling pathways are operative? Src has been described by your group in colon cancer. Is the IGF-I receptor/Met axis more important than Src? This leads to the question of molecular profiling. Are we getting to a point where you can dissect and profile individual tumors to design a therapy that targets the most significant pathways in individual tumors?
What are your plans on combining different therapeutics? You mentioned downstream pathways of uPAR, which is an outstanding target. What about c-Met and IGF-I receptor? Are there inhibitors for c-Met other than your hammerhead ribozyme? Should you target the ligand or should you target the receptor?
Finally, you touched on the issue of tumor survival as separate from tumor invasion and metastasis. The tumor cells can be resistant to apoptosis via pathways that affect survival, whereas other pathways in the same tumor may affect invasion metastasis, as you have shown. Do you have any evidence for cross-talk of these 2 pathways for invasion and metastasis and for resistance to apoptosis and survival?
Dr. Craig L. Slingluff, Jr. (Charlottesville, Virginia): We are in the early days of a revolution in cancer therapy, with the advent of new targeted molecular therapies for cancer. There are exciting opportunities to develop targeted therapies against common solid tumors based on understanding the molecular pathways for cell signaling that are critical for the lethal events of cell migration and metastasis.
In the present paper, Dr. Bauer, Dr. Ellis, and colleagues have elucidated new aspects of the pathways critical for migration and metastasis in colorectal cancer cells. They are to be congratulated for a clear and definitive manuscript that will contribute to advancement in this field. I thank them for providing me with a full manuscript in advance of the meeting for my review. I do have 3 questions.
The first is a technical question. I noticed in the manuscript that the media that were used for these assays include fetal bovine serum, and I wondered whether there is a possibility of IGF-I or hepatocyte growth factor being in that medium. Is there a way to control for that?
Second, the studies presented here were performed in a single human cell line of human colorectal cancer. They stated in the introduction and in some of the early slides here that IGF-I and HGF have been implicated in the path in colorectal cancer. Pathogenesis is unclear how generalizable these findings are. Malignant cells are notorious for their heterogeneity both within individuals and between individuals in the suppression of cellular receptors and in their dependence on certain signaling pathways. Can the authors define the proportion of colorectal cancers that express IGF-I receptor and c-Met and whether anything is known about the cooperation of these molecules in either other cancer cell lines or in fresh tumor tissue?
The last question is that the prospect of inhibiting these pathways for therapeutic potential is appealing; however, there are concerns about how to apply agents for these pathways. Since we generally believe that micrometastases have established themselves years before they are clinically evident and may well be in place at the time of original diagnosis, what is the prospect for interfering with the processes of invasion of metastasis in patients with colorectal cancer in a timely fashion before they occur?
Dr. W. Roy Smythe (Temple, Texas): I would like to congratulate Drs. Ellis and Bauer on a fine presentation and continued excellent work in translational biology as it pertains to surgical oncology. I would like to ask 1 question that Dr. Cance touched on but in a bit more discreet fashion.
As you know, the problem for adult solid tumors is that 40% to 70% of patients that present, present with metastatic disease. Therapies that are directed at metastasis only may not be as effective as those that target both survival and metastasis.
Receptor tyrosine kinases activate a number of transcription factors, which then obviously activate a number of other processes in the cell, including those that involve cell survival. Are the common transcription factors that IGF and HGF activate that may then in turn activate uPAR and would provide a better target for therapies designed to address this work?
Dr. Courtney M. Townsend, Jr. (Galveston, Texas): I would like to ask 2 questions. One, how do you know that the ribozyme directed against c-MET had no effect on the IGF-I receptor? Two, what is the relevance of this in vitro assay to in vivo invasion and metastasis?
Dr. Todd W. Bauer (Houston, Texas): I would like to thank the discussants for their insightful and interesting comments and questions. I will try to address these in order.
Dr. Cance asked whether additional pathways might be involved by downstream signaling from these receptors; in particular, he questioned whether the Src pathway might be involved. In fact, the IGF-I receptor, c-Met, and uPAR have all been shown to increase Src signaling, which is known to be a promigratory factor. The question then was raised, could we then better inhibit cell migration knowing this? It is our belief that by targeting multiple pathways rather than a single pathway, one can achieve better inhibition of tumor growth and metastases.
Dr. Cance also asked if there were inhibitors for c-Met and whether it made more sense to target the ligand, HGF, or target the receptor. Historically, c-Met has been difficult to selectively inhibit. However, in the past year there has been a report in Clinical Cancer Research (Hov et al. 2004;10:6686–6694) and another in Oncogene (Berthov et al. 2004;23:5387–5393) demonstrating that small-molecule tyrosine kinase inhibitors can effectively inhibit the c-Met receptor. This, I think, makes more sense and is probably going to be more effective than inhibiting the ligand because we have demonstrated that the c-Met receptor itself is likely functioning independent of HGF as a promigratory factor.
Dr. Cance also questioned whether cell survival pathways might be involved with these factors in addition to promigratory or proinvasive pathways. We do believe that these factors all contribute to cell survival.
Indirect evidence of a role of the uPAR receptor in initiating cell survival is very well supported by some of Dr. Cance's work. He has nicely characterized the role of focal adhesion kinase, or FAK, as an anti-apoptotic factor in cells. In addition, it has been demonstrated that uPAR activates FAK signaling, and this would provide indirect evidence that uPAR is a pro-survival factor.
In our laboratory we have also investigated these signaling pathways in a human pancreatic carcinoma cell line. In an orthotopic model of human pancreatic carcinoma, we treated mice with a uPAR inhibitory antibody and demonstrated that the antibody inhibited primary tumor growth by inhibiting tumor cell proliferation, inhibited tumor invasion into the retroperitoneum, and inhibited metastasis to the liver.
Dr. Slingluff noted that the media we used contained fetal bovine serum and he questioned whether this contained small amounts of IGF or HGF. Yes, FBS does contain both of these. We felt this was probably a good replication of the tumor microenvironment. We were studying the effect of addition off a growth factor at much higher concentration, relative to the levels of these factors in FBS.
Dr. Slingluff asked how generalizable our findings were, ie, “Is this only specific to the one tumor cell line we examined? Is this signaling pathway occurring in other cell lines?” Every single colon carcinoma cell line we have tested in our laboratory expresses both the IGF-I receptor and c-Met. We investigated this in a human pancreatic carcinoma cell line, and we have obtained similar results.
Dr. Slingluff asked, “knowing what we know about these signaling pathways, how might we apply this clinically? When in the tumor growth cascade would we apply this therapy?” I think our in vitro data need to be supported by further in vivo studies. Some of our preliminary in vivo work suggests that targeting the uPAR is likely more effective than targeting the c-Met pathway or the IGF-I receptor pathway. Using this as an adjunctive therapy to inhibit tumor recurrence and the outgrowth of micrometastases is an area that warrants further study.
Dr. Smythe then asked whether or not there were likely common downstream pathways which could be targeted to more effectively inhibit cell growth and tumor metastasis. I believe that we have demonstrated that the uPAR pathway is downstream of both the IGF-I receptor and c-Met. FAK and Src are important signaling pathways downstream of uPAR and are good candidates, to address Dr. Smythe's inquiry.
Last, Dr. Townsend asked how we knew that the ribozyme was specifically inhibiting the c-Met receptor and not the IGF-I receptor. That work has been borne out in the preliminary studies that characterized this ribozyme, where the ribozyme was shown to be specific for the c-Met receptor (Abounader et al. JNCI. 1999;91:1548–1556). The ribozyme RNA consensus sequence does not share homology with the IGF-I receptor. Thank you.
Footnotes
Sources of support: National Institutes of Health T32 grant CA-09599 (to TWB) and National Institutes of Health Cancer Center Support grant CA-16672.
Reprints: Lee M. Ellis, MD, Department of Surgical Oncology, Unit 444, The University of Texas M. D. Anderson Cancer Center, P.O. Box 301402, Houston, TX 77230-1402. E-mail: lellis@mdanderson.org.
REFERENCES
- 1.Jemal A, Tiwari RC, Murray T, et al. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. [DOI] [PubMed] [Google Scholar]
- 2.Tournigand C, Andre T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22:229–237. [DOI] [PubMed] [Google Scholar]
- 3.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. [DOI] [PubMed] [Google Scholar]
- 4.Nielsen TO, Andrews HN, Cheang M, et al. Expression of the insulin-like growth factor I receptor and urokinase plasminogen activator in breast cancer is associated with poor survival: potential for intervention with 17-allylamino geldanamycin. Cancer Res. 2004;64:286–291. [DOI] [PubMed] [Google Scholar]
- 5.Min Y, Adachi Y, Yamamoto H, et al. Genetic blockade of the insulin-like growth factor-I receptor: a promising strategy for human pancreatic cancer. Cancer Res. 2003;63:6432–6441. [PubMed] [Google Scholar]
- 6.Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell. 2004;5:221–230. [DOI] [PubMed] [Google Scholar]
- 7.Burroughs KD, Oh J, Barrett JC, et al. Phosphatidylinositol 3-kinase and mek1/2 are necessary for insulin-like growth factor-I-induced vascular endothelial growth factor synthesis in prostate epithelial cells: a role for hypoxia-inducible factor-1? Mol Cancer Res. 2003;1:312–322. [PubMed] [Google Scholar]
- 8.LeRoith D, Roberts CT Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–137. [DOI] [PubMed] [Google Scholar]
- 9.Moschos SJ, Mantzoros CS. The role of the IGF system in cancer: from basic to clinical studies and clinical applications. Oncology. 2002;63:317–332. [DOI] [PubMed] [Google Scholar]
- 10.Stoeltzing O, Liu W, Reinmuth N, et al. Regulation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and angiogenesis by an insulin-like growth factor-I receptor autocrine loop in human pancreatic cancer. Am J Pathol. 2003;163:1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paciucci R, Vila MR, Adell T, et al. Activation of the urokinase plasminogen activator/urokinase plasminogen activator receptor system and redistribution of E-cadherin are associated with hepatocyte growth factor-induced motility of pancreas tumor cells overexpressing Met. Am J Pathol. 1998;153:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karna E, Surazynski A, Orlowski K, et al. Serum and tissue level of insulin-like growth factor-I (IGF-I) and IGF-I binding proteins as an index of pancreatitis and pancreatic cancer. Int J Exp Pathol. 2002;83:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reinmuth N, Fan F, Liu W, et al. Impact of insulin-like growth factor receptor-I function on angiogenesis, growth, and metastasis of colon cancer. Lab Invest. 2002;82:1377–1389. [DOI] [PubMed] [Google Scholar]
- 14.Reinmuth N, Liu W, Fan F, et al. Blockade of insulin-like growth factor I receptor function inhibits growth and angiogenesis of colon cancer. Clin Cancer Res. 2002;8:3259–3269. [PubMed] [Google Scholar]
- 15.Maloney EK, McLaughlin JL, Dagdigian NE, et al. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res. 2003;63:5073–5083. [PubMed] [Google Scholar]
- 16.Puglianiello A, Germani D, Rossi P, et al. IGF-I stimulates chemotaxis of human neuroblasts: involvement of type 1 IGF receptor, IGF binding proteins, phosphatidylinositol-3 kinase pathway and plasmin system. J Endocrinol. 2000;165:123–131. [DOI] [PubMed] [Google Scholar]
- 17.Dunn SE, Torres JV, Oh JS, et al. Up-regulation of urokinase-type plasminogen activator by insulin-like growth factor-I depends upon phosphatidylinositol-3 kinase and mitogen-activated protein kinase kinase. Cancer Res. 2001;61:1367–1374. [PubMed] [Google Scholar]
- 18.Ligensa T, Krauss S, Demuth D, et al. A PDZ domain protein interacts with the C-terminal tail of the insulin-like growth factor-1 receptor but not with the insulin receptor. J Biol Chem. 2001;276:33419–33427. [DOI] [PubMed] [Google Scholar]
- 19.Neudauer CL, McCarthy JB. Insulin-like growth factor I-stimulated melanoma cell migration requires phosphoinositide 3-kinase but not extracellular-regulated kinase activation. Exp Cell Res. 2003;286:128–137. [DOI] [PubMed] [Google Scholar]
- 20.Stracke ML, Engel JD, Wilson LW, et al. The type I insulin-like growth factor receptor is a motility receptor in human melanoma cells. J Biol Chem. 1989;264:21544–21549. [PubMed] [Google Scholar]
- 21.Alami J, Williams BR, Yeger H. Expression and localization of HGF and met in Wilms’ tumours. J Pathol. 2002;196:76–84. [DOI] [PubMed] [Google Scholar]
- 22.Tam NN, Chung SS, Lee DT, et al. Aberrant expression of hepatocyte growth factor and its receptor, c-Met, during sex hormone-induced prostatic carcinogenesis in the Noble rat. Carcinogenesis. 2000;21:2183–2191. [DOI] [PubMed] [Google Scholar]
- 23.Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 2002;109:863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maulik G, Kijima T, Ma PC, et al. Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer. Clin Cancer Res. 2002;8:620–627. [PubMed] [Google Scholar]
- 25.Qian LW, Mizumoto K, Maehara N, et al. Co-cultivation of pancreatic cancer cells with orthotopic tumor-derived fibroblasts: fibroblasts stimulate tumor cell invasion via HGF secretion whereas cancer cells exert a minor regulative effect on fibroblasts HGF production. Cancer Lett. 2003;190:105–112. [DOI] [PubMed] [Google Scholar]
- 26.Herynk MH, Stoeltzing O, Reinmuth N, et al. Down-regulation of c-Met inhibits growth in the liver of human colorectal carcinoma cells. Cancer Res. 2003;63:2990–2996. [PubMed] [Google Scholar]
- 27.Monvoisin A, Neaud V, De Ledinghen V, et al. Direct evidence that hepatocyte growth factor-induced invasion of hepatocellular carcinoma cells is mediated by urokinase. J Hepatol. 1999;30:511–518. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T, Kanda S, Yamamoto K, et al. Increase in hepatocyte growth factor receptor tyrosine kinase activity in renal carcinoma cells isassociated with increased motility partly through phosphoinositide 3-kinase activation. Oncogene. 2001;20:7610–7623. [DOI] [PubMed] [Google Scholar]
- 29.Saeki H, Oda S, Kawaguchi H, et al. Concurrent overexpression of Ets-1 and c-Met correlates with a phenotype of high cellular motility in human esophageal cancer. Int J Cancer. 2002;98:8–13. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YW, Vande Woude GF. HGF/SF-met signaling in the control of branching morphogenesis and invasion. J Cell Biochem. 2003;88:408–417. [DOI] [PubMed] [Google Scholar]
- 31.Tacchini L, Matteucci E, De Ponti C, et al. Hepatocyte growth factor signaling regulates transactivation of genes belonging to the plasminogen activation system via hypoxia inducible factor-1. Exp Cell Res. 2003;290:391–401. [DOI] [PubMed] [Google Scholar]
- 32.Ried S, Jager C, Jeffers M, et al. Activation mechanisms of the urokinase-type plasminogen activator promoter by hepatocyte growth factor/scatter factor. J Biol Chem. 1999;274:16377–16386. [DOI] [PubMed] [Google Scholar]
- 33.Naldini L, Tamagnone L, Vigna E, et al. Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J. 1992;11:4825–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pennacchietti S, Michieli P, Galluzzo M, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. [DOI] [PubMed] [Google Scholar]
- 35.Winnay JN, Bruning JC, Burks DJ, et al. Gab-1-mediated IGF-1 signaling in IRS-1-deficient 3T3 fibroblasts. J Biol Chem. 2000;275:10545–10550. [DOI] [PubMed] [Google Scholar]
- 36.Price JA, Kovach SJ, Johnson T, et al. Insulin-like growth factor I is a comitogen for hepatocyte growth factor in a rat model of hepatocellular carcinoma. Hepatology. 2002;36:1089–1097. [DOI] [PubMed] [Google Scholar]
- 37.Morikawa K, Walker SM, Nakajima M, et al. Influence of organ environment on the growth, selection, and metastasis of human colon carcinoma cells in nude mice. Cancer Res. 1988;48:6863–6871. [PubMed] [Google Scholar]
- 38.Abounader R, Ranganathan S, Lal B, et al. Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J Natl Cancer Inst. 1999;91:1548–1556. [DOI] [PubMed] [Google Scholar]
- 39.Talamonti MS, Roh MS, Curley SA, et al. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest. 1993;91:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herynk MH, Tsan R, Radinsky R, et al. Activation of c-Met in colorectal carcinoma cells leads to constitutive association of tyrosine-phosphorylated beta-catenin. Clin Exp Metastasis. 2003;20:291–300. [DOI] [PubMed] [Google Scholar]
- 41.Vassalli JD, Belin D. Amiloride selectively inhibits the urokinase-type plasminogen activator. FEBS Lett. 1987;214:187–191. [DOI] [PubMed] [Google Scholar]
- 42.Freier S, Weiss O, Eran M, et al. Expression of the insulin-like growth factors and their receptors in adenocarcinoma of the colon. Gut. 1999;44:704–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hakam A, Yeatman TJ, Lu L, et al. Expression of insulin-like growth factor-1 receptor in human colorectal cancer. Hum Pathol. 1999;30:1128–1133. [DOI] [PubMed] [Google Scholar]
- 44.Michell NP, Langman MJ, Eggo MC. Insulin-like growth factors and their binding proteins in human colonocytes: preferential degradation of insulin-like growth factor binding protein 2 in colonic cancers. Br J Cancer. 1997;76:60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Renzo MF, Olivero M, Giacomini A, et al. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res. 1995;1:147–154. [PubMed] [Google Scholar]
- 46.Pyke C, Ralfkiaer E, Ronne E, et al. Immunohistochemical detection of the receptor for urokinase plasminogen activator in human colon cancer. Histopathology. 1994;24:131–138. [DOI] [PubMed] [Google Scholar]
- 47.Ganesh S, Sier CF, Heerding MM, et al. Urokinase receptor and colorectal cancer survival. Lancet. 1994;344:401–402. [DOI] [PubMed] [Google Scholar]
- 48.Verspaget HW, Sier CF, Ganesh S, et al. Prognostic value of plasminogen activators and their inhibitors in colorectal cancer. Eur J Cancer. 1995;31A:1105–1109. [DOI] [PubMed] [Google Scholar]
- 49.Ganesh S, Sier CF, Heerding MM, et al. Contribution of plasminogen activators and their inhibitors to the survival prognosis of patients with Dukes’ stage B and C colorectal cancer. Br J Cancer. 1997;75:1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andre F, Rigot V, Remacle-Bonnet M, et al. Protein kinases C-gamma and -delta are involved in insulin-like growth factor I-induced migration of colonic epithelial cells. Gastroenterology. 1999;116:64–77. [DOI] [PubMed] [Google Scholar]
- 51.Andre F, Rigot V, Thimonier J, et al. Integrins and E-cadherin cooperate with IGF-I to induce migration of epithelial colonic cells. Int J Cancer. 1999;83:497–505. [DOI] [PubMed] [Google Scholar]
- 52.Andre F, Janssens B, Bruyneel E, et al. Alpha-catenin is required for IGF-I-induced cellular migration but not invasion in human colonic cancer cells. Oncogene. 2004;23:1177–1186. [DOI] [PubMed] [Google Scholar]
- 53.Parr C, Hiscox S, Nakamura T, et al. Nk4, a new HGF/SF variant, is an antagonist to the influence of HGF/SF on the motility and invasion of colon cancer cells. Int J Cancer. 2000;85:563–570. [PubMed] [Google Scholar]
- 54.Hiscox S, Hallett MB, Puntis MC, et al. Inhibition of cancer cell motility and invasion by interleukin-12. Clin Exp Metastasis. 1995;13:396–404. [DOI] [PubMed] [Google Scholar]
- 55.Uchiyama A, Essner R, Doi F, et al. Interleukin 4 inhibits hepatocyte growth factor-induced invasion and migration of colon carcinomas. J Cell Biochem. 1996;62:443–453. [DOI] [PubMed] [Google Scholar]
- 56.Peace BE, Hill KJ, Degen SJ, et al. Cross-talk between the receptor tyrosine kinases Ron and epidermal growth factor receptor. Exp Cell Res. 2003;289:317–325. [DOI] [PubMed] [Google Scholar]
- 57.Gilmore AP, Valentijn AJ, Wang P, et al. Activation of BAD by therapeutic inhibition of epidermal growth factor receptor and transactivation by insulin-like growth factor receptor. J Biol Chem. 2002;277:27643–27650. [DOI] [PubMed] [Google Scholar]
- 58.Dupont J, Karas M, LeRoith D. The potentiation of estrogen on insulin-like growth factor I action in MCF-7 human breast cancer cells includes cell cycle components. J Biol Chem. 2000;275:35893–35901. [DOI] [PubMed] [Google Scholar]
- 59.Dalle S, Ricketts W, Imamura T, et al. Insulin and insulin-like growth factor I receptors utilize different G protein signaling components. J Biol Chem. 2001;276:15688–15695. [DOI] [PubMed] [Google Scholar]
- 60.Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443. [DOI] [PubMed] [Google Scholar]
- 61.Naldini L, Weidner KM, Vigna E, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. EMBO J. 1991;10:2867–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weidner KM, Hartmann G, Naldini L, et al. Molecular characteristics of HGF-SF and its role in cell motility and invasion. EXS. 1993;65:311–328. [PubMed] [Google Scholar]
- 63.Stoker M, Perryman M. An epithelial scatter factor released by embryo fibroblasts. J Cell Sci. 1985;77:209–223. [DOI] [PubMed] [Google Scholar]
- 64.Stoker M, Gherardi E, Perryman M, et al. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327:239–242. [DOI] [PubMed] [Google Scholar]
- 65.Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982;95:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]