

Abstract
Objective:
To evaluate the role of celecoxib on 15-lipoxygenase-1 (15-LOX-1) expression, protein levels, and rates of apoptosis in colorectal cancer cell lines. Also, to evaluate the expression of 15-LOX-1 in human normal mucosa, adenoma, and carcinoma with correlation to overall survival.
Summary Background Data:
The function of 15-LOX-1 is to maintain normal rates of apoptosis (programmed cell death). Decreased apoptosis is one mechanism of cancer growth and dissemination. It is our hypothesis that expression of 15-LOX-1 is reduced in human colorectal cancer (CRC) and the administration of celecoxib can reverse this process and induce apoptosis.
Methods:
Effect of celecoxib in cell culture: The effect of 40 μmol/L celecoxib was compared with untreated controls in tissue culture utilizing HT-29 and DLD-1 CRC cell lines. Expression of 15-LOX-1 protein was measured by immunoblot. Induction of apoptosis was evaluated by annexin V staining. All data are presented as mean ± SEM, with significance defined as P < 0.05. 15-LOX-1 in human CRC: From February 1998 to January 2002, 126 patients underwent surgical resection of either colorectal adenomas (n = 24) or carcinomas (n = 102), or both (n = 25). Tissue was macrodissected, snap frozen, and stored at −80°C. After tissue processing, RNA was extracted and gene expression of 15-LOX-1 was quantified utilizing ABI prism real-time quantitative RT-PCR. Significance evaluated by the Wilcoxon signed rank test.
Results:
Effect of celecoxib in cell culture: After 72 hours of treatment with celecoxib, immunoblot demonstrated a 1.5- to 2-fold increase in15-LOX-1 protein expression in HT-29 and DLD-1 cells, respectively. Celecoxib produced greater than a 2-fold increase in the rate of apoptosis compared with control cells in both cell lines (P < 0.05). 15-LOX-1 in human CRC: The mean age of the patients was 62 ± 1 years; 78% were white and 48% were female. The mean size of the polyps and cancers were 3.0 ± 0.4 and 5.0 ± 0.1 cm, respectively. Expression of 15-LOX-1 relative to S9 was 30 in normal mucosa and significantly down-regulated to 11 in adenomas and 16 in carcinomas (P < 0.05).
Conclusions:
15-LOX-1 gene expression is significantly reduced in both human colorectal adenomas and carcinomas and associated with decreased survival. Administration of celecoxib restores 15-LOX-1 protein expression and induces apoptosis. Down-regulation of 15-LOX-1 is an early event in the adenoma to carcinoma sequence, and reversal with celecoxib may represent one mechanism for chemoprevention of polyps or treatment of carcinomas.
It is our hypothesis that expression of 15-LOX-1 is reduced in human colorectal cancer (CRC), and the administration of celecoxib can reverse this process and induce apoptosis. In cell culture, celecoxib increased 15-LOX-1 expression and induced apoptosis. In human tumors, 15-LOX-1 was significantly down-regulated in adenomas and carcinomas and was associated with a decreased overall survival.
Eicosanoid mediators have been implicated in the development and progression of many cancers including colorectal cancer (CRC). The cyclooxygenase (COX) and lipoxygenase (LOX) pathways are the 2 major enzyme systems involved with the metabolism of polyunsaturated fatty acids.1,2 The most well-known system involves the COX-2 enzyme, which is primarily responsible for the conversion of arachidonic acid to prostaglandin E2 (PGE2)3 which has been implicated in colorectal tumor growth and proliferation.4,5 There are numerous studies which suggest that COX-2 overexpression is associated with increased tumor growth in a number of different histologies.6–8 In vitro data have associated the receipt of nonsteroidal anti-inflammatory drugs (NSAIDs), including selective COX-2 inhibitors, with decreased growth and proliferation.7,9–11 Interestingly, not all CRC cell lines express high levels of COX-2 and yet, they are still shown to have decreased growth after treatment with NSAIDs. This has led to investigations demonstrating COX-2-independent pathways associated with response to NSAIDs. Recent studies in the LOX family of enzymes has identified 15-lipoxygenase-1 (15-LOX-1) as a protein that is associated with cellular differentiation and maintenance of normal apoptotic rates.12 Further work in a relatively small number of patients has suggested that 15-LOX-1 is down-regulated in human CRC, although little data exist relating this to survival.12,13 Finally, cell culture studies have suggested that 15-LOX-1 can be up-regulated in response to NSAIDs with a concomitant increase in its active metabolite 13-hydroxyoctyldecanoic acid (13-S-HODE) leading to increased apoptosis.14 The present study was performed to evaluate the pro-apoptotic effect of celecoxib on 15-LOX-1 protein expression in cell lines that express high and low levels of COX-2. Furthermore, this study plans to document decreased 15-LOX-1 and 13-S-HODE in a large sample of human CRC with correlation to patient survival.
MATERIALS AND METHODS
Human Tissue Studies
From February 1998 through January 2002, 126 patients with AJCC (American Joint Committee on Cancer) stages I to IV, primary colorectal carcinomas were harvested under an IRB-approved consent process. At the time of surgery, both normal mucosa and carcinoma were macroscopically dissected from the colon or rectum. Dissected specimens were then cut into 5-mm cubic blocks, snap frozen under liquid nitrogen, and stored at −80°C.
Gene Expression
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. RNA samples were dissolved in water, quantitated, and brought to stock concentration of 50 ng/μL. Quantitative real-time reverse transcription PCR for 15-LOX-1 gene expression was performed using ABI Prism 7700 Detection System (Perkin-Elmer Applied Biosystems, Foster, CA). A ribosomal gene, S9, was used as an internal control for gene expression normalization. Previous studies from our laboratory confirmed the utility of this gene in CRC.15
Eicosanoid Measurement by Mass Spectrometry
All tissue samples were removed from −80°C storage and immediately placed on ice prior to extraction. Tissue samples were weighed in the range of 0.07 to 0.10 g wet weight. Water (2 mL) was added to each tissue sample followed by the addition of 50 μL of a 250 μmol/L stock solution of 12(S) hydroxy-16-heptadecynoic acid as the internal standard (final concentration 25 μmol/L). Samples were then homogenized on ice at 30,000 no/min by a tissue tearor (BioSpec Products Inc) hand homogenizer. Homogenized tissues were then extracted 3 times with 3 mL hexane:diethyl ether (60:40 vol/vol). Tissue extracts were vortexed 30 seconds followed by centrifugation at 3200 rpm for 5 minutes. Pooled supernatants were transferred to clean tubes and dried under nitrogen. The dried residues were reconstituted in 500 μL of 100% MeOH and vortexed followed by centrifugation at 3200 rpm for 15 minutes. Supernatants were removed and filtered through a 0.45-μm filter, and placed into HPLC vials for LCMS analysis.
Methanol extracts were separated on a Hewlett Packard 1050 HPLC system using a 10 cm × 4.6-mm-i.d., C8 Aquapore (Varian) reverse-phase column with a gradient elution. The column eluate is split so that 10 μL/min enters the IonSpray interface of a PE Sciex (Concord, Ontario, Canada) API III triple quadrupole mass spectrometer. The orifice potential is set at −60 V and the voltage on the spraying needle at −2700 V. Negative ion spectra are recorded using the multiple reaction ion monitoring (MRM) technique. The following m/z parent/daughter ion combinations are used to selectively and quantitatively measure each component in the standards and samples: internal standard 281.0 to 213.0, 13-S-HODE 295.0 to 277.0.
In Vitro Studies
Cell Lines
HT-29 and DLD-1 CRC cell lines were obtained from the ATCC and cultured in modified RPMI with 10% fetal calf serum, 1 mmol/L pyruvate, 10 mmol/L HEPES, and penicillin/streptomycin. Celecoxib was obtained from XXX in dissolved in DMSO to a stock concentration of 100 mmol/L. Cells were grown to 60% to 70% confluence and were treated with 40 μmol/L celecoxib for 72 hours. The concentration and duration of treatment with celecoxib was chosen based on preliminary data (not shown). We tested the induction of apoptosis at all combinations of 20 and 40 μmol/L and duration of treatment from 24, 48, and 72 hours. Celecoxib treatment at a concentration of 40 μmol/L for 72 hours reliably induced apoptosis. Gene expression profiles were performed that revealed moderate 15-LOX-1 expression in both cell lines. However, HT-29 had relatively high expression of COX-2 compared with almost no detectable expression of COX-2 in DLD-1.
Gene Expression Studies
Quantitative RT-PCR as described above.
Protein Quantification by Western Immunoblot
Anti-15-LOX-1 antibodies were obtained from Cayman chemical (Ann Arbor, MI) and were used at a dilution of 1:1000 in 5% milk. Antibeta-actin antibodies were obtained from Sigma (St. Louis, MO) and were used at a dilution of 1:5000 in 5% milk. Control and treated DLD-1 and HT-29 cells were lysed in RIPA buffer (Sigma) supplemented with 1% Triton-X-100 and protease inhibitor cocktail (Roche, Indianapolis, IN). Proteins separated on SDS-PAGE gels were transferred onto an Immobilon-P polyvinylidene difluoride membrane (Millipore, Bedford, MA). The horseradish peroxidase-conjugated second antibodies were diluted 1:2000 (Amersham Biosciences, Piscataway, NJ). PerkinElmer Life Sciences Renaissance Enhanced Luminol Reagents (Boston, MA) were used as substrates for detection.
Apoptosis Quantification
The percentage of cells undergoing apoptosis was quantified by the annexin V assay. The cells are stained appropriately and then underwent flow cytometry. Cells in the “early” and “late” apoptotic phase were summed and reported as a percentage of the whole. Annexin V was chosen because it was available in our facility and gave reproducible results.
Statistics
Statistical analysis was performed using SPSS Base 10.0 program (SPSS Inc, Chicago, IL). The nonparametric Wilcoxon signed-rank test was used for comparison of mean values of normal mucosa and tumor samples. Survival analysis was performed utilizing the log-rank test and graphical representation was performed by the method of Kaplan and Meier. Groups were defined based on the tumor to normal ratio either above or below the median value. Significance was defined as a P value less than 0.05.
RESULTS
Cell Culture Studies
HT-29 and DLD-1 CRC cells were grown and treated with celecoxib for 72 hours. Figure 1 demonstrates that both cell lines were induced to enter apoptosis after treatment with celecoxib compared with control cells. Baseline apoptotic percentages were approximately 5% and significantly increased to approximately 14% after treatment (P < 0.05). 15-LOX-1 protein expression was measured in treated and untreated HT-29 and DLD-1 cells. Figure 2 demonstrates a significantly increased protein expression in both HT-29 and DLD-1 cells compared with untreated control cells.
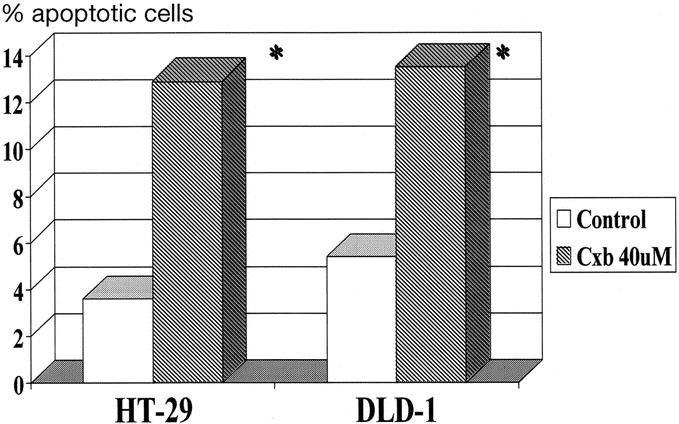
FIGURE 1. Induction of apoptosis by celecoxib in HT-29 and DLD-1 colorectal cancer cells (n = 2 for each cell line). *P < 0.05.
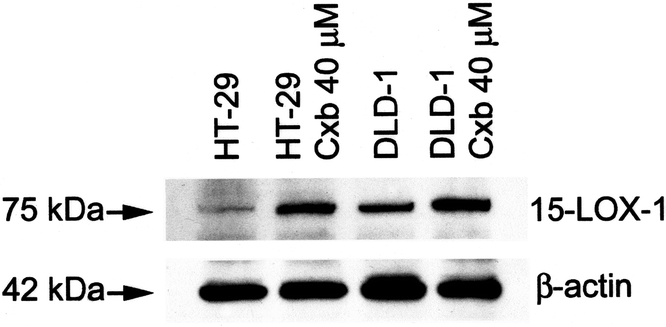
FIGURE 2. A representative Western immunoblot of 15-LOX-1 protein in untreated and treated HT-29 and DLD-1 CRC cell lines.
Human Tumor Studies
From February 1998 through January 2002, 126 patients underwent resection of a primary CRC or adenoma. Table 1 shows clinicopathologic characteristics of the patients. It is important to note that the average size of the adenomas was 3 cm, which implies that these are subject to significant risk of the development of CRC. Figure 3 demonstrates a significantly decreased expression of the 15-LOX-1 gene in colorectal adenomas and carcinomas compared with matched normal mucosa (P < 0.001). There were no significant differences between adenomas and carcinomas. Also, there was no significant differences when analyzed by AJCC stage of the tumors. Figure 4 demonstrates a significantly decreased level of 13-S-HODE in adenomas and carcinomas compared with normal mucosa (P < 0.05). Adenomas and carcinomas were analyzed together due to a lack of significant difference in gene expression studies of 15-LOX-1, and relatively small numbers of each tissue type. Figure 5 demonstrates the overall survival of stage IV patients based on the level of 15-LOX-1 gene expression. This figure demonstrates that patients with 15-LOX-1 greater than the median tumor to normal ratio had significantly increased overall survival.
TABLE 1. Clinicopathologic Data

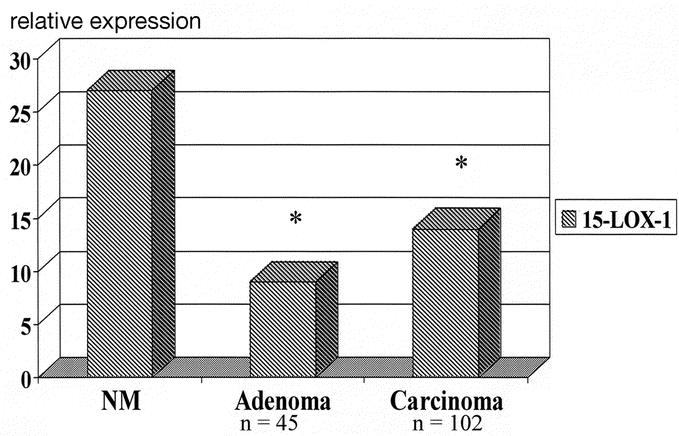
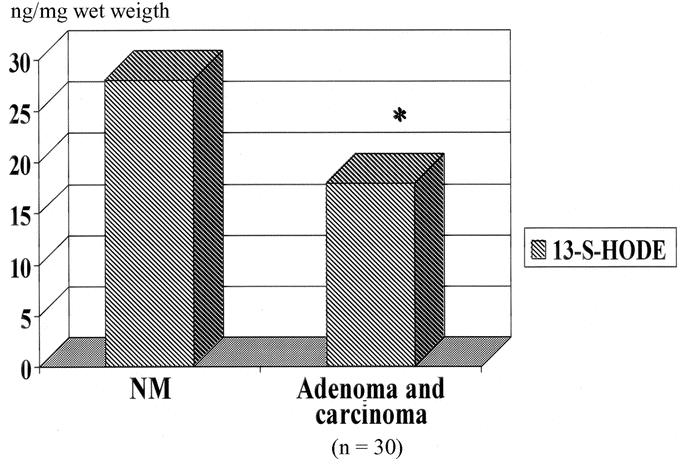
FIGURE 4. 13-S-HODE levels in human colorectal neoplasia (adenoma and carcinoma) compared with normal mucosa. Significant reduction in 13-S-HODE levels (P < 0.05).
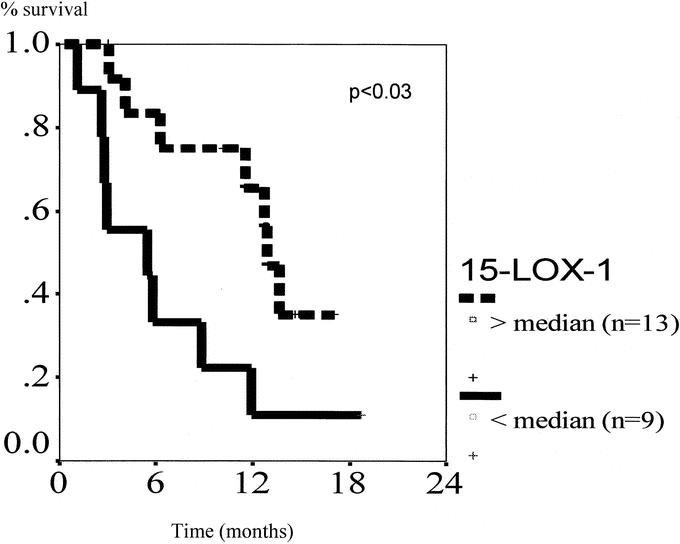
FIGURE 5. Overall survival of AJCC stage IV patients by 15-LOX gene expression. Ratio of tumor/normal mucosa calculated and separated above or below the median value. n = 13 and n = 9 for top and bottom line, respectively (P < 0.02).
DISCUSSION
This study has demonstrated that 15-LOX-1 expression and its metabolite 13-S-HODE are significantly reduced in a large series of CRCs and adenomas. Furthermore, reduction in 15-LOX-1 expression was significantly associated with reduced overall survival in the patients with stage IV CRC. Celecoxib, a selective COX-2 inhibitor, was found to induce CRC cell lines HT-29 and DLD-1 with relatively high and low COX-2 expression, respectively, to an apoptotic pathway. This was associated with a qualitative increase in 15-LOX-1 protein by immunoblot, suggesting a potential mechanism responsible for regressions seen in polyps. Similarly, this may represent an important tumor suppressor gene that can be targeted for replacement via gene therapy mechanisms or used as a prognostic factor in predicting outcome. Lastly, in vivo testing of the utility of celecoxib to increase 15-LOX-1 expression with restoration of 13-S-HODE levels would be important to evaluate in a prospective randomized trial.
The mechanism by which NSAIDs exert their pro-apoptotic effect remains controversial. Aspirin, which is the prototypical nonselective COX inhibitor, has long been proposed to decrease the risk of polyps and potentially prevent colon cancer.16 Indeed, more recent data confirm this effect in 2 large randomized trials.17,18 Many have suggested that the effect is due to inhibition of COX with resultant decreases in PGE2 production and therefore reduced tumorigenesis. After the realization that 2 isoenzymes of COX existed, investigators found that COX-2 was the inducible form and was increased in both inflammation and cancer. It was also found that inhibition of COX-1 was related to the increased risk of gastritis and gastric ulceration and thus the need for selectivity.19 The genesis of selective COX-2 inhibitors was designed to inhibit overexpressed COX-2 without the deleterious effects of COX-1 inhibition. This was accomplished by the pharmaceutical industry, and studies demonstrated a decreased risk of gastritis while selectively inhibiting COX-2. Prospective trials evaluating the effect of celecoxib on regression of polyps in patients with familial adenomatous polyposis demonstrated a significant reduction in the number of polyps.11 This further supported a role for selective COX-2 inhibitors in the treatment of colorectal neoplasia, but a clear mechanism for polyp regression was not demonstrated in patients with sporadic colorectal neoplasia. While intense work has been performed to delineate the role of cyclo-oxygenase in colorectal neoplasia, a smaller body of evidence has been growing to support the notion that there may be other pathways inhibited by NSAIDs.20 Sulindac sulfone was found to induce apoptosis and inhibit growth of colorectal cell lines without reduction in PGE2 levels.21 The mechanism for this reduction remains elusive; however, most data support that the answer is related to induction of apoptosis, and this is driven by a balance between pro- and anticarcinogenic eicosanoids.2
The relationship of pro- and anticarcinogenic eicosanoids is complex. Multiple studies in cell lines of different histologies have implicated PGE2 and other pro-inflammatory eicosanoids. The main anticarcinogenic enzyme is 15-LOX-1, which catalyzes the conversion of linoleic acid to 13-S-HODE. This pathway was found to be involved with intestinal cell differentiation and maintenance of normal rates of apoptosis.12,22 Further study in a series of experiments utilizing a variety of CRC cell lines, 15-LOX-1 was up-regulated by NSAIDs including celecoxib.23 These studies also documented the specificity of 13-S-HODE for inducing apoptosis and the likely involvement of peroxisome proliferator-activating receptor-delta (PPAR-delta). Our data are supported by this work where we document that the administration of celecoxib is associated with the increase in 15-LOX-1 protein expression in cell lines with and without COX-2 expression. Additionally, we have shown that both cell lines are induced to undergo apoptosis regardless of the COX-2 status. This basic work supports the further evaluation of this pathway in human colorectal adenomas and carcinomas.
In a small series of CRC patients, 15-LOX-1 and 13-S-HODE have been shown to be reduced, theoretically allowing the cancer cell to have a proliferative advantage due to the potential for decreased apoptosis.13 Our data have shown, in a large series of colorectal adenomas and carcinomas, that 15-LOX-1 and 13-S-HODE are significantly reduced, corroborating previous findings. It is important to note that reduction in 15-LOX-1 and 13-S-HODE in adenomas suggests that this alteration is likely an early event in the carcinogenic pathway. Therefore, this pathway holds promise for further investigation as a potential chemopreventive for colorectal polyps. More importantly is that this alteration in the cellular apoptotic control mechanism is related to overall survival in stage IV patients. Patients with tumor to normal ratios that were less that the median value had significantly reduced survival. We are unaware that this finding has been reported in the literature previously and lends credibility to the importance of the pathway. Obviously, the number of patients that were included in the final analysis was small, but stage IV patients represented an “event dense” population and therefore were able to be statistically analyzed. Unfortunately, there have been only 9 events in the stage III patients, making any type of meaningful analysis difficult.
Future direction of investigations related to 15-LOX-1 will include the significance of the mechanism by which 15-LOX-1 is down-regulated. One recent study suggests that the 15-LOX-1 promoter is hypermethylated, and this leads to decreased transcription.24 Preliminary data from our laboratory evaluated demethylated DNA from cell lines, and there was a significant rise in RNA expression of 15-LOX-1 (data not shown). The prognostic significance of hypermethylation of 15-LOX-1 will be important to determine since we can more easily extract DNA from paraffin blocks and create a larger database of patients with longer follow-up. Second, further testing in a clinical scenario to evaluate the in vivo biologic consequences of NSAIDs will be important. This will be able to test whether or not restoration of apoptosis can actually be achieved with agents such as celecoxib in a human experiment. Evaluation of the complex eicosanoid interactions requires human modeling with biologic endpoints to define the true effects. Lastly, the role of NSAIDs either alone or in combination with cytotoxic agents may play a role in cancer treatment. Modern cancer therapy hopefully will continue to evolve where patients will have their tumor genetically tested for a number of important factors and then the most appropriate combination of agents will be given.
Discussions
Dr. Nipun Merchant (Nashville, Tennessee): I would like to congratulate Dr. Heslin and his colleagues for shedding some insight into the other pathway of arachadonic acid metabolism in colorectal cancer. To my knowledge, this is the first paper that has shown decreased expression of 15 LOX-1 in colonic adenomas as well as implicate its role as a prognostic factor for survival in colon cancer.
While there has been focus on the carcinogenic effects of the COX-2 enzyme and prostaglandin synthesis, evidence clearly is emerging to suggest that the lipoxygenase enzymes and their byproducts also play major roles in colon carcinogenesis. Furthermore, the induction of apoptosis by NSAIDs has focused on the inhibition of cyclooxygenase and prostaglandin synthesis. However, evidence clearly suggests that NSAIDs have other possible molecular targets.
The data presented here highlight the importance of 15 LOX-1 down-regulation in both human adenomas and carcinomas compared with its expression in mucosa. In vitro studies using colon cancer cell lines, DLD-1, which does not express COX-1 or COX-2, and HT-29 cells, which do, treated with the selective COX-2 inhibitor Celebrex, demonstrated an increase in 15 LOX-1 protein expression and a significant increase in apoptosis, thereby implying a role of NSAID-induced apoptosis that is independent of the COX-2 pathway. To that end, I have the following comments and questions.
To support 15 LOX-1 as a specific target of NSAID-induced apoptosis, have you seen increased 15 LOX-1 expression after treatment with nonselective NSAIDs that inhibit both COX-1 and COX-2 as well as sulindac sulfone, which does have COX-1 or COX-2 inhibitory activity? Have you, in your in vitro studies, tried to block 15 LOX-1 activity and re-add 13-S-HODE as well as linoleic acid to see if apoptosis is restored with 13-S-HODE?
I wanted to ask about the dose of Celebrex used in your in vitro studies: 40 micromolar for 72 hours may be toxic to your cells, and you may be seeing some component of necrosis. 15 LOX-1 and 13-S-HODE expression can be seen within 24 to 48 hours after NSAID treatment. Have you tried to see if this effect with Celebrex or other NSAIDs might be dose- or time-dependent at other lower doses and time points?
Increasing evidence suggests that early in the neoplastic process, COX-2 is expressed primarily in the stroma, and only later in the malignant process is it expressed in the epithelial tumors. Have you had the opportunity to do immunohistochemical stains on your polyp and tumor samples to see where 15 LOX-1 is expressed early in late in colon cancer?
Lastly, can you speculate as to the mechanism behind which 15 LOX and 13-S-HODE induce apoptosis with NSAIDs?
There is some evidence that suggests that 13-S-HODE binds to PPAR-delta and down-regulates its activity. Increased levels of PPAR-delta have been shown to promote colon tumorigenesis. We and others have shown that prostaglandins, particularly PGI-2, produced from pericryptal fibroblasts bind to PPAR-delta, increase its expression, and confer an antiapoptotic effect to colon cancer cells.
Once again, I would like to congratulate Dr. Heslin and his colleagues for enlightening us with a potentially novel mechanism in colon tumorigenesis.
Dr. Dai H. Chung (Galveston, Texas): I would like to congratulate Dr. Heslin and his colleagues for an excellent study. The authors should be commended for conducting bench research to explore potential molecular mechanisms responsible for colorectal cancer progression and correlating to human tumor sample data for a translational approach.
The authors show that 15-lipoxygenase-1 (15-LOX-1), which degrades linoleic acid to 10-S-hydroxyoctadecadienoic acid (13-S-HODE), induces colon cancer cell apoptosis and that selective cyclooxygenase-2 (COX-2) inhibitor, known toincrease 15-LOX-1 activity, enhances cell apoptotic response. Furthermore, they analyzed human colon cancer samples from 126 patients to show that expression of 15-LOX-1 gene and 13-S-HODE protein levels are significantly decreased in adenomas and carcinomas. In addition, a decreased survival was found in patients with stage IV disease with decreased 15-LOX-1 gene expression.
I have several questions for Dr. Heslin. Have you correlated the Annexin V staining with other methods such as caspase cleavage or DNA fragmentation to confirm the effects of COX-2 inhibitor on colon cancer cell apoptosis? Have you also determined the effects of COX-2 inhibitor on downstream products for 13-S-HODE? I am also curious to know the dose-response results for in vitro experiments using COX-2 inhibitor. Your patient data samples represent tumors from varying clinical stages of colorectal cancer. Since you are suggesting that 15-LOX-1 plays an important role in colorectal cancer progression, have you analyzed and stratified expression changes relative to adenoma versus carcinoma as well as to clinical cancer stage? Have you also examined where exactly 15-LOX-1 expression is found in tumor samples? Lastly, what is the relative significance of 15-LOX-1 with respect to COX-2 in colorectal cancer progression in general?
Dr. Courtney M. Townsend, Jr. (Galveston, Texas): Was histologic verification used to determine that you had the equivalent amounts of viable tissue from each specimen? And how far from either cancer or adenoma were the specimens for normal tissue taken to avoid any kind of field change?
Dr. Martin J. Heslin (Birmingham, Alabama): Thank you for those comments. We appreciate you asking those insightful questions. I will block together some of the questions because they overlap.
We did not look at other nonselective COX-inhibitors; we did not block 15-LOX-1 or add 13-S-HODE. We are in the process of completing those studies.
I think that the effective dose and duration of celecoxib were evaluated and that data weren't shown. We looked at both 24 and 48 hours as well as 20 and 40 micromolar. We were able to induce apoptosis with 40 micromolar at 72 hours. It is possible that the up-regulation of 15-LOX-1 occurs at 24 hours, which would be prior to apoptosis induction.
We did not measure other methods of apoptosis. The measurement of apoptosis can be a difficult process in cell culture. We found that this method provided a reproducible way to put a number on apoptosis in our cell culture.
We have not done further immunohistochemical studies either to define rates of apoptosis in human tissue, as well as the localization of COX-2 and 15-S-HODE. I think that defining the location of these enzymes would be important to do, and we are in the process of getting those slides cut now.
The method of induction, we believe, is through a PPAR-delta mechanism, which has been shown in cell culture. PPAR-delta is elevated in colorectal polyp cancer. It appears that 13-S-HODE decreases PPAR-delta, which slows growth. We will be analyzing the expression of PPAR-delta in our human colorectal cancer specimens in an effort to define its prognostic significance.
You asked about the importance of a COX-2 pathway. We specifically selected DLD-1 and HT-29 cells to try to differentiate a COX effect or not. And as Dr. Merchant elucidated, DLD-1 is a low or no expression of COX-2, and HT-29 is a moderate, high level of COX-2 expression. The goal of this was to emphasize a COX-2-independent effect. The role of COX-2 in our tissue specimens is not shown here, but it did demonstrate that COX-2 was significantly elevated, and most prognostic, in stage 4 patients.
You asked about the role of 15-LOX-1 in stage 1 or 2, 3 or 4 colorectal cancers. I think the best way to answer that question is that 15-LOX-1 is down-regulated in adenomas, which is stage zero. We didn't really have the numbers to split out the different stages of cancer. Since 15-LOX-1 is down-regulated in adenomas or stage zero patients, more than likely it is going to be down-regulated in stage 1, 2, or 3. A correlation with survival couldn't be done in the earlier stages because of the low numbers of patients. There were only 9 patients in the stage 3 group that had died, which would make any kind of survival analysis not meaningful.
You asked about our tissue acquisition procedures. We do not split the actual tissue specimen in the OR: one half for paraffin section and one half for frozen section. We macrodissect it and take the most visually viable tumor and normal mucosa. Ideally, splitting the tissue would be a good way to do it, but our facilities are not able to accommodate that process. The normal mucosa is taken from the colon adjacent to the tumor. Again, the tissue is macrodissected in the OR. We don't believe there is any level of contamination. But how close is far enough? I don't know the answer to that question.
Footnotes
Reprints: Martin J. Heslin, MD, 1922 Seventh Ave South, KB 321, Birmingham, AL 35294. E-mail: martyh@uab.edu.
REFERENCES
- 1.Kujubu DA, Fletcher BS, Varnum BC, et al. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 2.Shureiqi I, Lippman SM. Lipoxygenase modulation to reverse carcinogenesis. Cancer Res. 2001;61:6307–6312. [PubMed] [Google Scholar]
- 3.Eberhart CE, Coffey RJ, Radhika A, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. [DOI] [PubMed] [Google Scholar]
- 4.Dubois RN, Shao J, Tsujii M, et al. G1 delay in cells overexpressing prostaglandin endoperoxide synthase-2. Cancer Res. 1996;56:733–737. [PubMed] [Google Scholar]
- 5.Pugh S, Thomas GA. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut. 1994;35:675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawaoka H, Kawano S, Tsuji S, et al. Cyclooxygenase-2 inhibitors suppress the growth of gastric cancer xenografts via induction of apoptosis in nude mice. Am J Phys. 1998;274(Pt 1):G1061–G1067. [DOI] [PubMed] [Google Scholar]
- 7.Sheng H, Shao J, Kirkland SC, et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest. 1997;99:2254–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsujii M, Kawano S, Dubois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA. 1997;94:3336–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boolbol SK, Dannenberg AJ, Chadburn A, et al. Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res. 1996;56:2556–2560. [PubMed] [Google Scholar]
- 10.Kawamori T, Rao CV, Seibert K, et al. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res. 1998;58:409–412. [PubMed] [Google Scholar]
- 11.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. [DOI] [PubMed] [Google Scholar]
- 12.Bronstein JC, Bull AW. The correlation between 13-hydroxyoctadecadienoate dehydrogenase (13-HODE dehydrogenase) and intestinal cell differentiation. Prostaglandins. 1993;46:387–395. [DOI] [PubMed] [Google Scholar]
- 13.Shureiqi I, Wojno KJ, Poore JA, et al. Decreased 13-S-hydroxyoctadecadienoic acid levels and 15-lipoxygenase-1 expression in human colon cancers. Carcinogenesis. 1999;20:1985–1995. [DOI] [PubMed] [Google Scholar]
- 14.Shureiqi I, Chen D, Lotan R, et al. 15-Lipoxygenase-1 mediates nonsteroidal anti-inflammatory drug-induced apoptosis independently of cyclooxygenase-2 in colon cancer cells. Cancer Res. 2000;60:6846–6850. [PubMed] [Google Scholar]
- 15.Blanquicett C, Johnson MR, Heslin M, et al. Housekeeping gene variability in normal and carcinomatous colorectal and liver tissues: applications in pharmacogenomic gene expression studies. Anal Biochem. 2002;303:209–214. [DOI] [PubMed] [Google Scholar]
- 16.Baron JA. Aspirin and cancer. Prev Med. 1995;24:121–124. [DOI] [PubMed] [Google Scholar]
- 17.Baron JA, Cole BF, Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–899. [DOI] [PubMed] [Google Scholar]
- 18.Sandler RS, Halabi S, Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348:883–890. [DOI] [PubMed] [Google Scholar]
- 19.Masferrer JL, Zweifel BS, Manning PT, et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piazza GA, Rahm AL, Krutzsch M, et al. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995;55:3110–3116. [PubMed] [Google Scholar]
- 21.Piazza GA, Alberts DS, Hixson LJ, et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997;57:2909–2915. [PubMed] [Google Scholar]
- 22.Bull AW, Branting C, Bronstein JC, et al. Increases in 13-hydroxyoctadecadienoic acid dehydrogenase activity during differentiation of cultured cells. Carcinogenesis. 1993;14:2239–2243. [DOI] [PubMed] [Google Scholar]
- 23.Shureiqi I, Jiang W, Zuo X, et al. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells. Proc Natl Acad Sci USA. 2003;100:9968–9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C, Xu D, Sjoberg J, et al. Transcriptional regulation of 15-lipoxygenase expression by promoter methylation. Exp Cell Res. 2004;297:61–67. [DOI] [PubMed] [Google Scholar]