


Abstract
The recent availability of bacterial genome sequence information permits the identification of conserved genes that are potential targets for novel antibiotic drug discovery. Using a coupled bioinformatic/experimental approach, a list of candidate conserved genes was generated using a Microbial Concordance bioinformatics tool followed by a targeted disruption campaign. Pneumococcal sequence data allowed for the design of precise PCR primers to clone the desired gene target fragments into the pEVP3 ‘suicide vector’. An insertion–duplication approach was employed that used the pEVP3 constructs and resulted in the introduction of a selectable chloramphenicol resistance marker into the chromosome. In the case of non-essential genes, cells can survive the disruption and form chloramphenicol-resistant colonies. A total of 347 candidate reading frames were subjected to disruption analysis, with 113 presumed to be essential due to lack of recovery of antibiotic-resistant colonies. In addition to essentiality determination, the same high-throughput methodology was used to overexpress gene products and to examine possible polarity effects for all essential genes.
INTRODUCTION
The publication of the genome of the first free-living organism, Haemophilus influenzae, (1) initiated the age of bacterial genomics (see http://www.tigr.org/tdb/mdb/mdbcomplete.html). The dozens of public and proprietary bacterial genomes have made available a wealth of new information and promise to greatly enhance our knowledge of a wide variety of different microbes. Along with this new genomic information, methodologies and tools have either been improved or developed de novo in order to analyze and apply the new data. Bioinformatics, the melding of biological sequence information, specific analysis software and high-speed computer tools, has become a crucial part of the genomic analysis process, both in assembling and interpreting vast amounts of sequence information. An important consequence of the microbial genomics era is that it will provide a number of essential, validated targets as possible candidates for the discovery of new antimicrobials. These validated drug targets will encode proteins either required for cell growth in vitro or for survival and/or virulence in the infected host. Presumably, screening against such novel targets for functional inhibitors will result in the discovery of novel therapeutic compounds active against bacteria, including the increasing number of antibiotic-resistant clinical strains that are the focus of much concern in clinical infectious disease.
An example of a bioinformatic approach to target identification was reported recently in a concordance analysis of microbial genomes (2). This system performs a FASTA comparison of multiple genomes at the amino acid level, building a relational database. The user queries this database and, by a web-based interface, retrieves related sequences with user-specified scores. This particular concordance demonstration compared the Escherichia coli genome against Bacillus subtilis, H.influenzae, Helicobacter pylori and Mycobacterium tuberculosis and subtracted out sequences with similarity greater than a selected exclusion criterion against the eucaryotic yeast Saccharomyces cerevisiae. This step was taken to reduce the potential cross-reaction of inhibitors with eucaryotic proteins. Genes in common with all five bacterial species were identified and a subset of sequences eliminated because of similarity to yeast sequences. The utility of such an approach was demonstrated by examination of the sequences selected, which included known drug targets. For example, the gyrA gene encoding DNA gyrase, the target of the quinolone class of antibiotics, was identified, as was murA, the target for fosfomycin. Several previously reported essential genes were also found, including dnaA, ftsZ and mraY. Different organisms and more or less stringent criteria can be used for such analyses depending on the desired end-point.
However, despite the best of bioinformatic approaches, it is necessary to validate the essentiality of antibacterial targets by experimental means, and several methods to accomplish this have been previously described (3–7). One approach made possible by the availability of genomic information is targeted gene disruption or ‘gene knockouts’. One can attempt to specifically disrupt the desired gene in the chromosome using a selectable marker, such as drug resistance. If one can successfully recover the antibiotic resistance in a disrupted gene, this implies non-essentiality for that target, while lack of recovery of antibiotic-resistant colonies suggests essentiality. In this work, we describe a method to identify conserved bacteria-specific genes followed by high-throughput gene disruptions using the Gram-positive bacterium Streptococcus pneumoniae as a model system. Most of the steps in this process were successfully adapted to a 96-well microtiter plate format. Over 300 conserved candidate genes were disrupted in a relatively short time period, resulting in the identification of 113 conserved essential genes. The functions of the gene products encompassed a broad variety of cellular processes, including cell wall biosynthesis, DNA replication, cell division and fatty acid biosynthesis among others. In addition, a similar high-throughput approach was used for gene product expression and polarity analysis of the identified target genes.
MATERIALS AND METHODS
Bioinformatic analyses
Genome sequence data were obtained from a proprietary database (Pathogenome; Genome Therapeutics Corp., Waltham, MA) or obtained from the public domain (e.g. GenBank and TIGR). Genes were found using programs such as MAGPIE (8), loaded into the Concordance database, and desired genes selected using various parameters (2; see Table 1). Following experimental work, genes of interest were further analyzed with a variety of search methods. For global sequence similarity, conserved essential gene sequences were searched against a non-redundant protein database using the BLAST2 algorithm (9). In addition, similar genes in the Concordance database were identified using the ‘Neighbors’ function (2). To determine if the predicted gene sequences were full length and of proper reading frame, both BLAST searching and CLUSTAL multiple sequence alignments (10) were used along with rules for bacterial start site prediction (11). Putative gene function was assigned based on sequence similarities to genes with established function. Annotations were examined for potential transitive errors with MAGPIE and other informatics tools. For local sequence similarity, sequences were searched for Prosite (12) and Pfam (13) motifs. In addition to sequence similarity searching methods, other analyses were performed in an effort to try to determine the function of the conserved essential genes. Protein threading analyses were performed, using the ProCeryon fold recognition program (M.Sippl, unpublished results). Since genes of related function are often organized into operons in bacteria, operon structure in S.pneumoniae was examined and compared to that found in other bacteria (MAGPIE). Finally, metabolic pathway analysis was performed using Pathway Tools from DoubleTwist, which is based on the EcoCyc system (14).
Table 1. Sample concordance.
| Target organism | Streptococcus pneumoniae |
| Number of sequences | 2345 |
| Compared against | Bacillus subtilis |
| Enterococcus faecalis | |
| Escherichia coli | |
| Staphylococcus aureus | |
| Selection qualifier | Target sequences must match at least two species |
| Selection criterion | Overlap ratio >0.4a |
| Number of sequences which match at least one species | 746 |
| Sequences removed by selection requirement | 265 |
| Excluding organism(s) | Yeast (YPD) |
| Exclusion criterion | Overlap ratio >0.3b |
| Sequences removed by exclusion | 83 |
| Number of sequences in concordance | 398 |
a≥40% amino acid sequence identity.
b≥30% amino acid sequence identity.
Bacterial strains and media
Escherichia coli LE392 (15) was used as the strain for propagation of plasmid pEVP-3 (16). The strain was maintained on Luria (LB) agar (Gibco BRL, Life Technologies, Rockville, MD) and grown in liquid LB, at 37°C with shaking. Streptococcus pneumoniae strain Rx-1 (17) was maintained on blood agar plates at 37°C in 5% CO2. The cells were grown in liquid culture in Todd-Hewitt (Difco, Becton-Dickinson, Sparks, MD) plus 0.5% yeast extract (Difco) at 37°C.
Purification of S.pneumoniae chromosomal DNA
Streptococcus pneumoniae Rx-1 was grown overnight on a blood agar plate at 37°C in 5% CO2. Growth was removed with an inoculating loop and resuspended in 20 ml of Todd-Hewitt, 0.5% yeast extract with 400 µg/ml sterile sodium bicarbonate. The cells were grown at 37°C to an OD600 nm of 0.4–0.6, chilled on ice, harvested by centrifugation at 5000 g for 15 min at 4°C, the pellet resuspended and washed once with 20 ml of ice-cold 10 mM Tris–HCl, pH 8.0, and 1 mM EDTA buffer, centrifuged as above and the resulting pellet quick frozen at –20°C. The cells were thawed and resuspended in 5 ml of the above Tris–HCl, EDTA buffer, and 0.005% sodium deoxycholate and 0.01% SDS added. After incub ation at 37°C for 10 min, during which time the cells lysed, 500 µg/ml proteinase K (Sigma Chemical Co., St Louis, MO) was added and incubated for an additional 10 min. The cell lysate in buffer was gently extracted by inversion with an equal volume of phenol/chloroform/isoamyl alcohol (Gibco BRL). After centrifugation at 8000 g for 10 min, the upper aqueous layer was removed and extracted twice with an equal volume of chloroform/isoamyl alcohol. The final aqueous extract was made up to 0.3 M sodium acetate, and 2.2 vol of ethanol were overlayed. The DNA was spooled onto a glass rod and redissolved in 2 ml of 10 mM Tris–HCl, pH 8.0, 1 mM EDTA overnight at 4°C. This preparation was dialyzed against 400 vol of the same Tris–HCl, EDTA buffer before storage at 4°C. DNA concentration was determined by absorbance at 260 nm and adjusted to 0.5 µg/µl.
Primer construction
Knockout (K/O) primers were designed from sequences of annotated reading frames and were usually ∼25 bp in length, ∼50% GC content and located at least 100 bp internal to the N- and C-termini of the predicted full-length target gene, and generated 300–600 bp fragments. A list of primers used in this work is available as Supplementary Material. Primer design and ordering (Life Technologies) were done electronically and primers were received arrayed in 96-well plate format when appropriate.
Genomic PCR fragment generation
PCRs were run in a 96-well plate format using 50 µl total reaction volumes that contained the following: 36 µl dH2O, 5 µl 10× Vent buffer (New England Biolabs, Beverly, MA), 1 µl K/O forward primer (0.5 µg/µl), 1 µl K/O reverse primer (0.5 µg/µl), 0.5 µl Vent DNA polymerase (2000 U/ml) (New England Biolabs), 1.5 µl each (6.0 µl total) dNTPs (10 mM) (New England Biolabs) and 0.5 µl S.pneumoniae chromosomal DNA (0.5 µg/µl). The PCR program was as follows: 95°C for 5 min, then 30 cycles of 95°C for 1 min, 58°C for 1 min and 72°C for 30 s, followed by 72°C for 10 min and a 4°C hold. PCR Purification Kits, 96-well format and individual columns (Qiagen, Valencia, CA), were used to purify PCR products. Products (5 µl each) were visualized by a 96-well agarose gel electrophoresis system (Life Technologies).
Ligation reactions
Blunt-end ligation reactions were set up in a 96-well plate format as follows: 10 µl genomic PCR fragment (∼100–500 ng), 1.0 µl pEVP3 SmaI-digested vector (∼10 ng), 1.5 µl 10× ligation buffer (New England Biolabs), 1.0 µl T4 DNA ligase (400 000 U/ml) (New England Biolabs) and 1.5 µl dH2O for 15 µl total reaction volumes. Reactions were incubated at 14°C overnight.
Escherichia coli transformations
Bacterial transformations with the ligated pEVP3 plasmid were set up in a 96-well plate using a 3 µl ligation reaction and 50 µl E.coli LE392 competent cells (E.coli Genetic Stock Center, Yale University, New Haven, CT). Plates were incubated on ice for 30 min, followed by heat shock at 42°C for 90 s, and returned on ice for 2 min. Aliquots of 100 µl SOC medium (Life Technologies) were added to the cells and plates were incubated at 37°C on a platform shaker for 1 h. Each transformation reaction (total volume) was plated on LB/chloramphenicol (15 µg/ml) agar in six-well plate format and grown overnight at 37°C to allow for colony formation.
Colony PCR
Screening for the recombinant clones that contained inserts was done by colony PCR in 96-well plate format. Transformed E.coli colonies were picked by sterile pipet tip and resuspended in 50 µl dH2O. These colony suspensions were stored at 4°C in 96-well plates and subsequent positive clones of interest were plated onto solid medium and saved. PCRs included the following: 36.5 µl dH2O, 0.5 µl pEPV3 forward primer (0.25 µg/µl), 0.5 µl pEPV3 reverse primer (0.25 µg/µl), 1.5 µl each (6.0 µl total) dNTP (10 mM), 0.5 µl Vent DNA polymerase, 5 µl 10× Vent buffer and 1 µl of a 1:50 cell dilution for a 50 µl total reaction volume. The pEPV3 cloning site flanking forward primer was 5′-CAT CAAGCTTATCGATACCGTCG-3′ and the reverse flanking primer 5′-CACAGTAGTTCACCACCTTTTCCC-3′ (Life Technologies). The PCR program was 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 58°C for 1 min and 72°C for 30 s, then 72°C for 10 min with a 4°C hold. PCR products (10 µl each) were visualized by 96-well agarose gel electrophoresis after staining with ethidium bromide. Constructs that were negative for a targeting DNA fragment insert produced a 100 bp control fragment. Those that were positive for an insert produced an ∼400–700 bp fragment, depending on the targeting insert size. Miniprep cultures of positive clones were inoculated directly from cell dilution plates.
Miniprep and clone confirmation
Positive constructs were inoculated into 5 ml LB/chloramphenicol medium and incubated overnight at 37°C with aeration. Miniprep DNA was prepared by standard procedure using the Qiagen 96-well Turbo Prep System or individual columns. PCR with ORF-specific primers was used to confirm the presence of the correct insert. Reactions in 96-well plates included: 35.5 µl dH2O, 5 µl 10× Vent buffer, 1 µl K/O forward primer (0.5 µg/µl), 1 µl K/O reverse primer (0.5 µg/µl), 0.5 µl Vent DNA polymerase (2000 U/ml), 1.5 µl each (6.0 µl total) dNTPs (10 mM) and 1.0 µl miniprep DNA for a 50 µl total reaction volume. The PCR program and agarose gel analysis of products was as described above.
Streptococcus pneumoniae competent cell preparation and gene disruption
One colony, from a fresh blood agar plate, was resuspended in 1.5 ml Todd-Hewitt medium (Difco) plus 0.5% yeast extract (Difco). An aliquot of 100 µl of this dilution was used to inoculate 50 ml of the same medium and grown overnight at 37°C. The next morning, 5 ml of the overnight culture was added to 45 ml fresh medium and grown at 37°C to an OD600 of 0.25 (∼4–5 h). Sterile glycerol to a final concentration of 10% was added, cells were aliquoted in 1 ml volumes, frozen in a dry ice–ethanol bath and stored at –80°C.
Pneumococcal transformations were set up in 96-well plate format and included the following: 1 µg miniprep pEVP3 plus insert DNA (∼10 µl Qiagen miniprep DNA) plus 200 µl S.pneumoniae Rx-1 competent cells diluted 1:10 in competence medium (18,19). Competence medium contained Todd-Hewitt broth plus 0.5% yeast extract, 0.2% BSA, 0.01% CaCl2 and 100 ng/ml peptide pheromone Csp-1 (H-EMRLSK FFRDFILQRKK-OH) (ResGen, Huntsville, AL). A non-essential (pEPV3::lytA) and essential (pEPV3::ftsZ) disruption control construct was run with each experiment. Cells were incubated at 37°C for 2.5–3 h without shaking. All 200 µl were plated on Todd-Hewitt agar with 0.5% yeast extract and 2 µg/ml chloramphenicol and incubated for 24 h at 37°C in a CO2 incubator. Plates were then examined and colony numbers compared to controls. The lytA non-essential controls typically gave 150–200 colonies per transformation and the ftsZ positive controls gave 0 colonies per transformation. Experimentally, most non-essential genes gave 200–300 colonies per transformation (lytA was found to be on the low end of the spectrum) and essential genes gave results similar to ftsZ. An occasional single chloramphenicol-resistant colony was seen for some experiments, presumably due to contamination or non-homologous integration of the construct. For verification, all essential genes were subjected to a second independent gene disruption experiment.
Polarity analysis of essential ORFs
Primers for the N- and C-termini were designed from curated sequence data of full-length genes. Appropriate restriction enzyme sites were added to primers for cloning and subsequent expression of protein in the E.coli BL21 pET vector system (pET21b and pET21+; Novagen). PCR of full-length genes from genomic DNA was as described above. Full-length genes were cloned as blunt fragments into pEPV3 for polarity testing in S.pneumoniae, similarly to the procedure described above for gene disruptions.
Expression of essential gene products
In a separate reaction, fragments were also digested with appropriate restriction enzymes and ligated with a similarly digested pET expression vector. High-throughput procedures for ligation and transformation were similar to those described previously. Ligations with pET vectors were transformed into E.coli DH5α for the identification of positive clones and then into appropriate strain backgrounds (E.coli BL21 [λDE3]) for protein expression according to the manufacturer’s recommendations. A small-scale (5 ml) induction experiment was run on all new expression constructs and analyzed by SDS–PAGE for protein overexpression and correct size of the expressed protein.
DNA sequence analysis
All expression constructs underwent DNA sequence analysis to confirm insert sequences. Constructs were sequenced using vector-specific primers, followed by primer walking with target gene-specific primers. The sequences were analyzed for proper reading frame and compared to other available sequence data.
RESULTS
In order to select potential target genes for essentiality analyses, the recently described Microbial Concordance tool (2) was employed. Entire genomes can be compared against other genomes of choice to obtain a list of genes common among several pathogens with the desired percent identity. In a similar manner, genes that are more specific to bacteria can be selected for by filtering out eucaryotic genes above a predetermined percent identity. Several different concordance analyses were run and the results were combined in order to obtain a list of candidate bacterial genes with potential broad spectrum. The concordance also screened genes for similarity against possible eucaryotic homologs, with the aim of minimizing potential adverse effects for a novel inhibitor by undesirable cross-reactivity. These genes were then selected as candidates for the high-throughput disruption experiments to determine essentiality. An example of a more recent concordance is illustrated in Table 1. The target genome, S.pneumoniae, was compared against the B.subtilis, Enterococcus faecalis, E.coli and Staphylococcus aureus (20) genomes. The selection criterion was 40% global amino acid sequence identity with matches in at least two bacterial species. Streptococcus pneumoniae sequences that were >30% identical to yeast proteins were excluded to reduce the possibility of target cross-reactivity. The result of this particular concordance analysis was a list of 398 candidate genes. Following several rounds of concordance analyses, a set of 347 candidate open reading frames was selected for subsequent targeted disruptions to establish essentiality.
The target genes were next analyzed to verify probable start and stop codons for the open reading frames. Subsequently, primers were designed from the annotated reading frame sequences and those were selected that were at least 100 bp internal to the start and stop codons of each gene. These 300–600 bp fragments were inserted into the pEVP3 vector (21) and initially transformed into E.coli. Selection for chloramphenicol-resistant transformants and screening in a 96-well format agarose gel allowed ready identification of pEVP3 plasmids with inserts of pneumococcal DNA (Fig. 1). The pEVP3 vectors that had inserts were subsequently transformed into S.pneumoniae. The cloned gene fragment promotes single crossover homologous recombination that, in a manner resembling a Campbell-like insertion (22), introduces via insertion–duplication the pEVP3 vector into the chromosome, effecting the disruption (Fig. 2). A cell in which a non-essential gene has been disrupted will survive the disruption and upon incubation form a macroscopic colony on a chloramphenicol-containing agar plate, by virtue of the chloramphenicol resistance encoded by the pEVP3 recombinant plasmid integrated into the chromosome. A disrupted essential gene, on the other hand, will not permit colony formation and thus no chloramphenicol-resistant colonies will be recovered. Table 2 lists the resulting 113 genes derived from the above essentiality testing. Table 3 lists the 234 genes disrupted that were non-essential. Table 4 catagorizes the essential genes based on available annotations.
Figure 1.
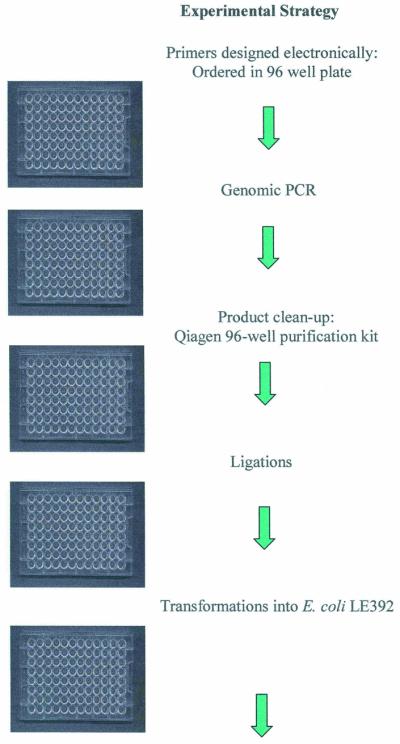

Experimental strategy for high-throughput disruption of pneumococcal target genes. Steps performed in 96-well microtiter plates are indicated. Escherichia coli transformations were plated on six-well plates while S.pneumoniae transformations were plated on standard size 15 × 100 mm plates.
Figure 2.

Use of plasmid pEVP3 for disruption of pneumococcal genes. An internal gene fragment was cloned into this vector which is incapable of replication in pneumococci. Selection for the insertion–duplication event was on medium containing chloramphenicol.
Table 2. Conserved essential bacterial genes.


a–, polarity not an issue, see Results; +, polarity is an issue, see Results.
bn.d., not determined.
cGene not present in the TIGR serotype 4 strain.
Table 3. List of S.pneumoniae non-essential genes by gene disruption analysisa.




aNot listed are 60 hypothetical or conserved hypothetical proteins.
Table 4. Functional role of the 113 essential gene disruptionsa.
| Cellular role | Subtotal | Total |
|---|---|---|
| Amino acid biosynthesis | 1 | |
| Aspartate family | 1 | |
| Biosynthesis of cofactors, prosthetic groups and carriers | 4 | |
| Thiamine | 1 | |
| Pantothenate | 3 | |
| Cell envelope | 14 | |
| Biosynthesis of surface poly/liposaccharides | 4 | |
| Biosynthesis of murein sacculus and peptidoglycan | 10 | |
| Cellular processes | 7 | |
| Chaperones | 1 | |
| Cell division | 2 | |
| Protein and peptide secretion | 4 | |
| DNA metabolism | 11 | |
| DNA replication, recombination and repair | 9 | |
| Chromosome-associated proteins | 2 | |
| Energy metabolism | 2 | |
| Pentose phosphate pathway | 1 | |
| Glycolysis/gluconeogenesis | 1 | |
| Fatty acid, lipid and sterol metabolism | 10 | |
| Biosynthesis | 10 | |
| Protein synthesis | 2 | |
| tRNA aminoacylation | 2 | |
| Purines, pyrimidines, nucleosides and nucleotides | 4 | |
| Purine ribonucleotide biosynthesis | 1 | |
| Sugar-nucleotide biosynthesis and conversions | 3 | |
| Regulatory functions | 6 | |
| General | 4 | |
| Protein interactions | 2 | |
| Transcription | 2 | |
| Transcription factors | 2 | |
| Translation | 9 | |
| Translation factors | 5 | |
| Degradation of proteins, peptides and glycopeptides | 1 | |
| Other | 3 | |
| Transport and binding proteins | 4 | |
| Amino acids, peptides and amines | 4 | |
| Unknown | 37 | |
| Unknown | 37 | |
| Total | 113 |
aCategories are based on those proposed by Riley (32).
Similar high-throughput approaches were used to overexpress S.pneumoniae gene products of interest in E.coli and to determine whether there were potential polarity issues for these candidate genes (Fig. 3). Since many bacterial genes are co-transcribed from a common upstream promoter, gene disruptions can cause downstream polar effects that can mislead in the determination of essentiality. By using a full-length target gene sequence cloned into the pEVP3 vector, the insertion–duplication event results in the duplication of the entire target gene (Fig. 4). If chloramphenicol-resistant colonies are now obtained, the assumption is made that the gene product is indeed essential to the bacterium. If no colonies are detected, the assumption is that one or more downstream transciptionally linked genes in an operon are essential for growth (Table 2, +). In the latter case, additional experimental data are required to establish which gene or genes are essential in the putative operon. Genes that display no polarity effects (Table 2, –) can be further advanced as targets for novel antibacterials.
Figure 3.

Scheme for overexpression of target proteins and polarity analysis. A similar high-throughput 96-well microtiter plate approach was used. The regulatable pET vector system was used for protein overexpression.
Figure 4.

Use of plasmid pEVP3 for polarity analysis of pneumococcal genes. The full-length target was cloned into this vector, which is incapable of replication in pneumococci. Selection for the insertion–duplication event was on medium containing chloramphenicol. Arrows indicate direction of transcription. The question mark indicates the possible position of an additional promoter.
For protein overexpression, the full-length target gene sequence was cloned into pET vectors, transformed into E.coli for verification of the construct and transformed into an expression strain of E.coli. Following protein expression and analysis by SDS–PAGE, proteins can be further purified either with Ni–NTA columns for those proteins with a C-terminal 6-histidine tag or via conventional purification strategies for untagged proteins.
DISCUSSION
The recent availability of bacterial genomic sequence information has, among many uses, allowed the identification of potential novel, conserved antibacterial targets (23,24). These novel targets could be the basis for the identification of fundamentally new classes of antibiotics. The need for novel antimicrobials has been emphasized by the continued increase in the number of bacterial pathogens that possess multiple antibiotic resistance (25,26). The combination of bioinformatic and laboratory approaches described here permits the assignment of potential targets conserved among pathogens as either essential or non-essential for in vitro survival on growth media. The previously described Microbial Concordance informatics tool (2) was used to first identify conserved open reading frames based on the assumption that these would have the best chance of yielding a broad spectrum inhibitor of bacterial growth. An entire genome can be compared against multiple genomes of choice to obtain a list of genes common among several pathogens, with the desired percent identity specified by the user. In a similar manner, the concordance permits the identification or subtraction of undesired gene sets, such as bacterial genes with a relatively high similarity to eucaryotic genes. Thus, open reading frames with high sequence identity to eucaryotic gene sequences can be reduced in priority as targets to enhance chances for novel inhibitor selectivity against bacteria. This was done initially with yeast sequence and later with human genomic sequence as it became available. Several concordance iterations and selection criteria were used in this work and results were pooled to yield the final list of 347 genes for target disruption analyses. One advantage of this approach is the flexibility for the user to set desired limits both for sequence identity and sequence exclusion. Since there are no optimal predetermined numbers for both, multiple analyses can be conducted at the bioinformatic level followed by experimental validation. Essential targets can be subsequently rank ordered with regard to their degree of amino acid similarity among pathogens and their distance from eucaryotic sequences.
Because of the availability of pneumococcal gene sequence information (27,28), a targeted as opposed to random gene disruption approach (29) was selected. Streptococcus pneumoniae strain Rx-1 (17) was chosen for this work, recognizing that there are DNA sequence polymorphisms among pneumococcal strains and that the use of other strains or essentiality testing methods may yield a few differences in results. Certain key advancements in the biology of S.pneumoniae have made this organism a premier model system for rapid gene knockouts. The identification of a small peptide regulator that induces competence for genetic transformation (18,19) has made manipulation of the system extremely reproducible and convenient. The E.coli plasmid pEVP3, which has a number of key features, including chloramphenicol resistance expression in both Gram-positive and Gram-negative bacterial backgrounds, was designed specifically by Morrison et al. (16) for genetic disruptions in pneumococci. To construct such gene disruptions, the internal gene fragments were cloned into pEVP3 with the recombinant plasmids inserting into the pneumococcal chromosome via homologous recombination into intact genes leading to targeted insertion–partial gene duplication events. The position of the gene disruption event (at least 100 bp from the gene start and stop sites) should be sufficient to ensure that the resulting insertion–duplication event does not restore gene function. It should be noted that our independently derived disruption criteria are similar to those used in another global mutagenesis campaign (30). Clearly an absolute requirement for this process to succeed was the availability of the DNA sequence of the pneumococcal target genes with correctly annotated start and stop signals. This allowed for the synthesis of the gene-specific primers needed for PCR generation of internal gene targeting fragments. We estimate that it would require 6 days for one person to take one plate (96 individual gene knockouts) and complete one cycle, i.e. from genomic PCR to pneumococcal bacterial transformation result. The yield of pEVP3 clones from any given plate on first run was 60–80%. Some genes required more time than others, adding to the complexity of quantitation of the entire effort. Therefore, to do all 347 knockouts again would require six to seven cycles, or 36–42 person-days. We feel that this is an improvement in time and efficiency over other similar target identification and validation attempts.
Using the pneumococcus, both conserved genes of known function as well as conserved genes of unknown function were disrupted in a systematic fashion to establish in vitro essentiality of these individual genes in this organism. In the case of disruptions where the bacteria survived and formed colonies (i.e. non-essential function on an agar plate), the cellular and colony morphologies were examined visually for subtle alterations. Disruption survivors were stocked at –80°C, for potential further examination of the gene disruption by in vivo cell culture or animal model infections. In cases where a disruption in the pneumococci resulted in lethality, a secondary construct using a full-length gene was used in the pneumococcal pEVP3 system to test for possible polar effects caused by plasmid integration. This resulted in a gene duplication of the essential gene versus disruption. If lethality was again the result, the gene was considered to be in an essential operon and required additional experimental analysis.
There are examples where a lethal insertion was ascribed to polarity and the downstream target is known to be essential from our own work. One is a cluster of five genes on the S.pneumoniae genome (SP1588–SP1592), for which we obtained three disruptions (in all genes except 1588 and 1592). An essential disruption within coding region SP1589, a Mur ligase family protein, was an essential event with a polarity effect (+ polarity). An essential disruption occurred in the gene immediately downstream, SP1590, a conserved hypothetical protein which is essential, but with no polarity issues (– polarity), as a disruption of the next gene, SP1591, was determined to be non-essential. From these data, one can infer that the non-polar status of a gene disruption can be confirmed when the next gene (SP1591) is found to be non-essential. However, the final gene going in the same direction in this cluster, SP1592, may still be non-essential too, since it is possible that SP1592 is essential but has an independent promoter driving it that makes it immune to the polarity of upstream insertions. To be conclusive, it is necessary to further examine the polarity issue by experimentation beyond our polarity check method. Such polarity analyses could result in the identification of multiple essential genes within the operon, thus adding genes to the list. Of the 113 genes listed where gene disruptions could not be obtained, 57 were determined to have no issues of polarity (Table 2). These genes are either not in operons, are the last gene in an operon or are in an operon with no downstream essential genes. For these 57 genes lacking polarity effects, as well as the 113 genes found to be non-viable upon disruption, the majority could be assigned a clear or putative role, but roughly one-third were of unknown function (Table 4). Where known, the functions of the 113 conserved essential gene products encompassed a broad variety of cellular processes, including cell wall biosynthesis, DNA replication, cell division and fatty acid biosynthesis, among others (Table 4). Most of these targets have yet to be exploited for drugs in human clinical use.
With the development of this high-throughput gene disruption approach, we have rapidly and systematically assessed the essentiality of the computer-generated list of conserved genes of both known and unknown function. An added value of the gene disruption system described here is that a similar high-throughput approach was successfully used to evaluate expression of gene products and to assess polarity issues. Genes of known function that can be assayed and are proved essential have become immediate candidates for further development as high-throughput screens using their protein products. Unknown function genes provide a greater challenge and require multiple strategies aimed at deducing function and generating a screening system. The expectation is that genomic approaches will identify a significant number of new drug targets (31), which will lead to the discovery of several novel agents for the treatment of bacterial infections.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr D. Morrison for providing the pEVP3 plasmid for this work. We acknowledge the technical assistance of K. Kennedy and the support of Dr J. F. Barrett and Dr D. Davison during this work.
REFERENCES
- 1.Fleischmann R.D., Adams,M.D., White,O., Clayton,R.A., Kirkness,E.F., Kerlavage,A.R., Bult,C.J., Tomb,J.F., Dougherty,B.A., Merrick,J.M., McKenney,K., Sutton,G., FitzHugh,W., Fields,C., Gocayne,J.D., Scott,J., Shirley,R., Liu,L., Glodek,A., Kelley,J.M., Weldman,J.F., Phillips,C.A., Spriggs,T., Hedblom,E., Cotton,M.D., Utterback,T.R., Hanna,M.C., Nguyen,D.T., Saudek,D.M., Brandon,R.C., Fine,L.D., Fritchman,J.L., Fuhmann,J.L., Geoghagen,N.S.M., Gnehm,C.L., McDonald,L.A., Small,K.V., Fraser,C.M., Smith,H.O. and Venter,J.C. (1995) Whole genome random sequencing and assembly of Haemophilus influenzae Rd. Science, 269, 496–512. [DOI] [PubMed] [Google Scholar]
- 2.Bruccoleri R.E., Dougherty,T.J. and Davison,D.B. (1998) Concordance analysis of microbial genomes. Nucleic Acids Res., 26, 4482–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akerley B.J., Rubin,E.J., Camilli,A., Lampe,D.J., Robertson,H.M. and Mekalanos,J.J. (1998) Systematic identification of essential genes by in vitro mariner mutagenesis. Proc. Natl Acad. Sci. USA, 95, 8927–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arigoni F., Talabot,F., Peitsch,M., Edgerton,M.D., Meldrum,E., Allet,E., Fish,R., Jamotte,T., Curchod,M.L. and Loferer,H. (1998) A genome-based approach for the identification of essential bacterial genes. Nat. Biotechnol., 16, 851–856. [DOI] [PubMed] [Google Scholar]
- 5.Chalker A.F., Minehart,H.W., Hughes,N.J., Koretke,K.K., Lonetto,M.A., Brinkman,K.K., Warren,P.V., Lupas,A., Stanhope,M.J., Brown,J.R. and Hoffman,P.S. (2001) Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol., 183, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji Y., Zhang,B., Van Horn,S.F., Warren,P., Woodnutt,G., Burnham,M.K. and Rosenberg,M. (2001) Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science, 293, 2266–2269. [DOI] [PubMed] [Google Scholar]
- 7.Judson N., and Mekalanos,J.J. (2000) Transposon-based approaches to identify essential bacterial genes. Trends Microbiol., 8, 521–526. [DOI] [PubMed] [Google Scholar]
- 8.Gaasterland T., and Sensen,C.W. (1996) MAGPIE: automated genome interpretation. Trends Genet., 12, 76–78. [DOI] [PubMed] [Google Scholar]
- 9.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higgins D.G., Thompson,J.D. and Gibson,T.J. (1996) Using CLUSTAL for multiple sequence alignments. Methods Enzymol., 266, 383–402. [DOI] [PubMed] [Google Scholar]
- 11.Hannenhalli S.S., Hayes,W.S., Hatzigeorgiou,A.G. and Fickett,J.W. (1999) Bacterial start site prediction. Nucleic Acids Res., 27, 3577–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofmann K., Bucher,P., Falquet,L. and Bairoch,A. (1999) The PROSITE database, its status in 1999. Nucleic Acids Res., 27, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Howe,K.L. and Sonnhammer,E.L. (1998) The Pfam protein families database. Nucleic Acids Res., 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karp P.D., Riley,M., Paley,S.M., Pellegrini-Toole,A. and Krummenacker,M. (1999) Eco Cyc: encyclopedia of Escherichia coli genes and metabolism. Nucleic Acids Res., 27, 55–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borck K., Beggs,J.D., Brammar,W.J., Hopkins,A.S. and Murray,N.E. (1976) The construction in vitro of transducing derivatives of phage lambda. Mol. Gen. Genet., 146, 199–207. [DOI] [PubMed] [Google Scholar]
- 16.Claverys J.P., Dintilhac,A., Pestova,E.V., Martin,B. and Morrison,D.A. (1995) Construction and evaluation of new drug-resistance cassettes for gene disruption mutagenesis in Streptococcus pneumoniae, using an ami test platform. Gene, 164, 123–128. [DOI] [PubMed] [Google Scholar]
- 17.Pozzi G., Masala,L., Iannelli,F., Manganelli,R., Havarstein,L.S., Piccoli,L., Simon,D. and Morrison,D.A. (1996) Competence for genetic transformation in encapsulated strains of Streptococcus pneumoniae: two allelic variants of the peptide pheromone. J. Bacteriol., 178, 6087–6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Havarstein L.S., Coomaraswamy,G. and Morrison,D.A. (1995) An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl Acad. Sci. USA, 92, 11140–11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrison D.A., (1997) Streptococcal competence for genetic transformation: regulation by peptide pheromones. Microb. Drug Resist., 3, 27–37. [DOI] [PubMed] [Google Scholar]
- 20.Kuroda M., Ohta,T., Uchiyama,I., Baba,T., Yuzawa,H., Kobayashi,I., Cui,L., Oguchi,A., Aoki,K., Nagai,Y., Lian,J., Ito,T., Kanamori,M., Matsumaru,H., Maruyama,A., Murakami,H., Hosoyama,A., Mizutani-Ui,Y., Takahashi,N.K., Sawano,T., Inoue,R., Kaito,C., Sekimizu,K., Hirakawa,H., Kuhara,S., Goto,S., Yabuzaki,J., Kanehisa,M., Yamashita,A., Oshima,K., Furuya,K., Yoshino,C., Shiba,T., Hattori,M., Ogasawara,N., Hayashi,H. and Hiramatsu,K. (2001) Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet, 357, 1225–1240. [DOI] [PubMed] [Google Scholar]
- 21.Lee M.S., Seok,C. and Morrison,D.A. (1998) Insertion-duplication mutagenesis in Streptococcus pneumoniae: targeting fragment length is a critical parameter in use as a random insertion tool. Appl. Environ. Microbiol., 64, 4796–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mortier-Barriere I., Humbert,O., Martin,B., Prudhomme,M. and Claverys,J.-P. (1997) Control of recombination rate during transformation of Streptococcus pneumoniae: an overview. Microb. Drug Resist., 3, 233–242. [DOI] [PubMed] [Google Scholar]
- 23.Galperin M.Y., and Koonin,E.V. (1999) Searching for drug targets in microbial genomes. Curr. Opin. Biotechnol., 10, 571–578. [DOI] [PubMed] [Google Scholar]
- 24.McDevitt D., and Rosenberg,M. (2001) Exploiting genomics to discover new antibiotics. Trends Microbiol., 9, 611–617. [DOI] [PubMed] [Google Scholar]
- 25.Levy S.B., (2002) Factors impacting the problem of antibiotic resistance. J. Antimicrob. Chemother., 49, 25–30. [DOI] [PubMed] [Google Scholar]
- 26.Mazel D., and Davies,J. (1999) Antibiotic resistance in microbes. Cell Mol. Life Sci., 56, 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoskins J., Alborn,W.E.Jr, Arnold,J., Blaszczak,L.C., Burgett,S., DeHoff,B.S., Estrem,S.T., Fritz,L., Fu,D.J., Fuller,W., Geringer,C., Gilmour,R., Glass,J.S., Khoja,H., Kraft,A.R., Lagace,R.E., LeBlanc,D.J., Lee,L.N., Lefkowitz,E.J., Lu,J., Matsushima,P., McAhren,S.M., McHenney,M., McLeaster,K., Mundy,C.W., Nicas,T.I., Norris,F.H., O’Gara,M., Peery,R.B., Robertson,G.T., Rockey,P., Sun,P.M., Winkler,M.E., Yang,Y., Young-Bellido,M., Zhao,G., Zook,C.A., Baltz,R.H., Jaskunas,S.R., Rosteck,P.R.Jr, Skatrud,P.L. and Glass,J.I. (2001) Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol., 183, 5709–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tettelin H., Nelson,K.E., Paulsen,I.T., Eisen,J.A., Read,T.D., Peterson,S., Heidelberg,J., DeBoy,R.T., Haft,D.H., Dodson,R.J., Durkin,A.S., Gwinn,M., Kolonay,J.F., Nelson,W.C., Peterson,J.D., Umayam,L.A., White,O., Salzberg,S.L., Lewis,M.R., Radune,D., Holtzapple,E., Khouri,H., Wolf,A.M., Utterback,T.R., Hansen,C.L., McDonald,L.A., Feldblyum,T.V., Angiuoli,S., Dickinson,T., Hickey,E.K., Holt,I.E., Loftus,B.J., Yang,F., Smith,H.O., Venter,J.C., Dougherty,B.A., Morrison,D.A., Hollingshead,S.K. and Fraser,C.M. (2001) Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science, 293, 498–506. [DOI] [PubMed] [Google Scholar]
- 29.Lee M.S., Dougherty,B.A., Madeo,A.C. and Morrison,D.A. (1998) Construction and analysis of a library for random insertional mutagenesis in Streptococcus pneumoniae: use for recovery of mutants defective in genetic transformation and for identification of essential genes. Appl. Environ. Microbiol., 65, 1883–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutchison C.A., Peterson,S.N., Gill,S.R., Cline,R.T., White,O., Fraser,C.M., Smith,H.O. and Venter,J.C. (1999) Global transposon mutagenesis and a minimal Mycoplasma genome. Science, 286, 2165–2169. [DOI] [PubMed] [Google Scholar]
- 31.Read T.D., Gill,S.R., Tettelin,H. and Dougherty,B.A. (2001) Finding drug targets in microbial genomes. Drug Discov. Today, 6, 887–892. [DOI] [PubMed] [Google Scholar]
- 32.Riley M., (1993) Functions of the gene products of Escherichia coli. Microbiol. Rev., 57, 862–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.