


Abstract
The ability of the Cre/lox system to make precise genomic modifications is a tremendous accomplishment. However, recombination between cis-linked heterospecific lox sites limits the use of Cre- mediated exchange of DNA to systems where genetic selection can be applied. To circumvent this problem we carried out a genetic screen designed to identify novel mutant spacer-containing lox sites displaying enhanced incompatibility with the canonical loxP site. One of the mutant sites recovered appears to be completely stable in HEK293 cells constitutively expressing Cre recombinase and supports recombinase-mediated cassette exchange (RMCE) in bacteria and mammalian cell culture. By preventing undesirable recombination, these novel lox sites could improve the efficiency of in vivo gene transfer.
INTRODUCTION
The Cre–lox site-specific recombination system has been applied to a variety of genetic systems. Cre recombinase, a 38.5 kDa protein from bacteriophage P1, catalyzes reciprocal site-specific recombination between DNA elements termed lox sites (1–3). The Cre recombinase alone is sufficient (4) to carry out recombination between lox sites in vitro and in vivo in both bacterial and eukaryotic systems (5–8). The canonical lox site, loxP, is a 34 bp element composed of two 13 bp inverted repeats separated by an asymmetric 8 bp spacer region (Fig. 1). Each 13 bp inverted repeat serves as a binding site for the recombinase, whereas the 8 bp spacer region participates in strand exchange during the recombination reaction. Because a productive recombination event requires synapsis of lox sites between their spacer regions, it is asymmetry of the spacer region that confers directionality to the recombination reaction (Fig. 2A.1).
Figure 1.

Wild-type and mutant lox sites. The wild-type lox site, loxP, is diagrammed along with three mutant lox sites, lox511 (mutant spacer region; 9), lox66 and lox71 (mutant arms; 11). The left side of the panel shows the sequence of each site. The large arrows bracket the 13 bp palindromic arms that serve as binding sites for the Cre recombinase. The mutant arms are indicated by shading. The smaller arrows depict the arbitrary orientation of the 8 bp asymmetric spacer region. Deviations from the wild-type loxP sequence are indicated by lower case letters. The single base substitution in lox511 is denoted by an asterisk. The right side of the panel depicts schematic representations of each lox site. The arrowhead depicts the relative orientation of the spacer region. Each lox site is subdivided into three regions representing the tripartite nature of the lox site. Base substitutions in the spacer region are indicated by an asterisk. Corresponding mutant arms are shaded.
Figure 2.

Cre-mediated recombination in cis and in trans. (A) Basic Cre-mediated recombination reactions. (1) Deletion; recombination between like oriented loxP sites results in efficient deletion of the intervening DNA. The reverse of this reaction, recombination between wild-type lox sites in trans, is highly unfavorable. (2) Insertion; use of lox sites with complementary mutant arms (lox66 and lox71; 11) allows efficient recombination in trans, here depicted as an insertion event. Recombination generates a wild-type loxP site and a defective site with doubly mutant arms. Because the doubly mutant lox site is no longer an efficient substrate for the recombinase, insertion is favored and the reaction is driven in one direction. (3) Incompatibility; non-homology between interacting lox sites dramatically reduces their ability to carry out productive recombination. By definition, two completely incompatible lox sites linked in cis are stable in the presence of the recombinase. Conversely, promiscuous recombination will occur between incompletely incompatible sites when linked in cis. (B) RMCE. A schematic diagram depicting RMCE, or the process by which one lox site-flanked (or ‘floxed’) cassette is exchanged for another by site-directed recombination. In this example, a kanamycin resistance marker (kanr) from one plasmid is exchanged for the LacZ gene on another plasmid. The RMCE reaction proceeds to the right. This diagram also illustrates the consequences of using less than completely incompatible lox sites. Accumulation of products resulting from promiscuous recombination between cis-linked sites is shown at the left.
Cre recombinase can mediate either excisive or inversional intramolecular recombination events between cis-linked loxP sites depending on their relative orientation to one another. DNA flanked by lox sites (‘floxed’) oriented in the same direction is deleted upon recombination (Fig. 2A.1), whereas DNA flanked by lox sites oriented in the opposite direction is inverted upon recombination. Because intramolecular recombination involves the interaction of cis-linked lox sites, the reaction is extremely efficient. In contrast, intermolecular recombination between wild-type lox sites located on different DNA molecules, the reverse of the intramolecular excision reaction, is highly unfavorable (Fig. 2A.2).
Mutational analysis of the loxP site has revealed two classes of mutations that can alter the specificity and directionality of the recombination reaction. The first class consists of mutations within the 8 bp spacer region. Efficient recombination between lox sites requires spacer region homology. lox sites linked in cis whose spacer regions differ in as little as one nucleotide (i.e. lox511; Fig. 1) have dramatically reduced recombination efficiency (Fig. 2A.3), but retain the ability to recombine with homologous lox sites (9). Crystallographic studies of Cre recombinase bound to a lox site suggest that base pairing between exchanging strands within the recombination complex is required for efficient recombination (10). A direct consequence of this requirement is that the 8 bp spacer region not only confers directionality to synapsing lox sites but also specificity to the recombination reaction such that recombination can be precisely directed between specific homologous sites. The ability of a lox site to discriminate against recombination with another lox site with which it does not share spacer region homology is a property we refer to as incompatibility (Fig. 2A.3).
The second class of lox mutations are those within the 13 bp inverted repeats or palindromic arms (lox71 and lox66; Fig. 1) that weaken the affinity of the recombinase for its binding site. Recombination between lox sites containing mutations in complementary palindromic arms generates a lox site with wild-type arms and a lox site with doubly mutant arms (11; Fig. 2A.2). Because the doubly mutant lox site is no longer recognized as an effective substrate for the recombinase, it is not competent to undergo further recombination. Although this type of recombination reaction proceeds at a slightly lower rate, the reaction proceeds almost exclusively in one direction, resulting in the accumulation of recombination products (11). Thus, mutations in the palindromic arms confer unidirectionality to the recombination reaction. lox sites containing arm mutations have been used to stably target site-specific integration of plasmid DNA to both plant and mammalian chromosomes (11,12).
A site-specific recombination reaction that entails the exchange of one DNA cassette for another is termed recombinase-mediated cassette exchange (RMCE) (Fig. 2B). RMCE was originally described in the Cre–lox recombination system for generating combinatorial phage display libraries (13). It has since been adapted for generating isogenic cell lines in both the Cre–lox (14–17) and FLP–frt (18–20) site-specific recombination systems. Generation of an isogenic cell line initially involves the introduction of a floxed cassette at a single chromosomal location by either random integration or homologous recombination. Subsequently the endogenous cassette is replaced by site-directed recombination by co-transfection of a floxed targeting vector and a recombinase expression vector. This approach has typically relied on the use of selectable markers to enrich for clones that have the desired modifications as a result of relatively rare recombination events. The ability to make precise genomic modifications at pre-existing targets integrated into the host chromosome represents a significant accomplishment and demonstrates the tremendous potential of RMCE.
We were initially interested in using RMCE as a method for generating recombinant adenoviral vectors using lox sites containing both spacer and palindromic arm mutations (Fig. 2A.2). Our preliminary work with plasmid constructs demonstrated that these mutant lox sites could be used to efficiently drive RMCE in Cre-expressing bacteria, as illustrated in Figure 2B. However, these lox sites undergo promiscuous recombination in mammalian cells, resulting in deletion of the floxed cassette (15,17; our unpublished observations). This undesirable recombination is detectable in bacteria (21; our unpublished observations), but not nearly to the extent that is observed in mammalian cells. The fundamental difference between our approach to using RMCE to generate recombinant adenovirus vectors and that employed to generate isogenic cell lines is the use of genetic selection. While the incorporation of genetic selection allows for the recovery of relatively rare recombination events, the absence of genetic selection reveals that incompatibility between loxP and lox511 is incomplete. This suggested to us that efficient RMCE in mammalian cells in the absence of genetic selection would require mutant lox sites that perform with much higher fidelity than lox511. This prompted us to re-examine lox spacer region mutations and their effects on incompatibility.
We designed a genetic screen for identifying functional lox sites with mutations in their spacer regions that demonstrate greater incompatibility with loxP than lox511. Several mutant spacer-containing lox sites were identified that demonstrate complete incompatibility with loxP in Cre-expressing bacteria. One mutant spacer-containing lox site was further characterized in recombinant adenovirus vector transfections using Cre-expressing HEK293 cells and shown to support stable recombination. These incompatible lox sites should prove useful in improving methods for directing efficient in vivo transfer of genetic material in mammalian cells.
MATERIALS AND METHODS
Generating a library of mutant spacer-containing lox sites
A single-stranded oligonucleotide was synthesized as depicted in Figure 3A. The spacer region was synthesized with degenerate nucleotides at positions –4, –3, –2, +2, +3 and +4, while limiting position –1 to thymine and limiting position +1 to either adenine or thymine (W). Outside the palindromic arms we incorporated an EcoRI site and a BamHI site at the 5′ and 3′ ends of the oligonucleotide, respectively. PCR amplification was used to convert this oligonucleotide into a double-stranded DNA molecule. The primers used for this PCR reaction were 5′-GCGCGAATTCTGCGCATAAC-3′ and 5′-AACTGGATCCGTCCTATACC-3′. The PCR product was subcloned as an EcoRI–BamHI fragment into pPG3-loxP to generate pPG3-loxR/loxP (Fig. 3B) using standard recombinant DNA techniques (22).
Figure 3.

A genetic screen for incompatible lox sites. (A) Generating a degenerate spacer library. A synthetic oligonucleotide based on lox66 (see Fig. 1) but containing degenerate nucleotides in the spacer region (NNNTWNNN, where N is any base and W is either thymidine or adenosine) was used as a template in PCR with corresponding flanking primers. The PCR product, loxR, contains EcoRI and BamHI sites at its termini that facilitated its cloning into the plasmid construct, pPG3loxP, to generate a plasmid library of degenerate spacer-containing lox sites, pPGloxR/loxP. (B) A schematic diagram depicting the approach used to screen for incompatible lox sites. Individual clones from the plasmid library of degenerate spacer-containing lox sites, pPGloxR/loxP, were passaged through 294-CRE bacterial cells (23) by transformation. After recovery, the resulting plasmid DNAs were linearized by restriction digestion and analyzed by agarose gel electrophoresis. Two possible Cre-mediated recombination pathways are depicted: site-directed recombination between promiscuous lox sites or homologous recombination between reiterated portions of the 3′ end of the kanr gene. The extent to which either of these recombination events occurs can be distinguished by restriction analysis with XmnI and EcoRI. (C) Shown is the restriction analysis of a panel of representative clones cut with XmnI. The 4.9 kb fragment corresponds to the size predicted for the parental plasmid. The 3.7 kb fragment corresponds to the size predicted for the parental plasmid after deletion of a unit length of the kanr gene (1.2 kb). Note that the XmnI digest does not distinguish whether the 3.7 kb band results from promiscuous site-directed or homologous recombination. (D) As in (C) except that samples were cut with EcoRI. In addition to the 4.9 (parental) and 3.7 kb (deletion) bands seen with XmnI digestion in (C), a 2.4 kb band is also detected. This band corresponds to supercoiled plasmid DNA that, as a result of homologous recombination, has lost the EcoRI site and is refractory to EcoRI digestion. Clones that lack an apparent 3.7 kb fragment on EcoRI digestion would be considered candidates containing high fidelity incompatible lox sites.
Plasmids
The plasmid construct pPG3-loxR/loxP (Fig. 3B), made to test the compatibility of the mutant lox sites with loxP, was based on the cloning vector pGEM7zf+ (Promega Biotech). The kanr gene (kanr GenBlock; Amersham Pharmacia Biotech) was subcloned into the polylinker of pGEM7zf+ in the same transcriptional orientation as the ampr marker. It was cloned into the NsiI site of pGEM7zf+ as a PstI fragment and the interval between XhoI and HindIII deleted by fill-in with Klenow and ligation with T4 ligase, leaving the 3′ end of the kanr gene intact. A double-stranded oligonucleotide containing the lox71 site (Fig. 1) was inserted at the XbaI site of pGEM7zf+. The kanamycin resistance marker, kanr, was subcloned into the polylinker of pGEM7zf+ in the same transcriptional orientation as the ampr marker. The kanr gene was inserted as a HincII fragment into the pGEM7zf+ polylinker in which XhoI and HindIII were digested and blunt-ended by treatment with Klenow to generate the parental construct pPG3-loxP. pPG3-loxP contains unique EcoRI and BamHI sites that facilitated the directional cloning of the PCR-amplified mutant spacer-containing lox sites described above. The construct pPG3-loxR/loxR was then generated to test whether or not sites that were incompatible with loxP were functional. PCR amplification was used to amplify the 3′ end of the kanr gene along with the mutant lox site (Fig. 4A) and this PCR product was used to replace the HindIII–BglII interval in pPG3-loxR/loxP. The primers used for this PCR reaction were 5′-GATTTTGAGACACAACGTGGCT-3′ and 5′-GCGCGCAAGCTTTTGCCATTCTCACCGGA-3′. pPG3-loxR/loxR (NotI) was constructed to differentiate between homologous recombination and Cre-mediated recombination. It was made by changing the EcoRI site in pPG3-loxR/loxP to NotI after the PCR amplification step (Fig. 4A) and before the subsequent subcloning step to make pPG3-loxR/loxR. pGEM3-loxR/loxP was constructed to examine the incompatibility of the mutant sites with loxP while eliminating homologous recombination as an alternate recombination pathway. The construct pGEM3-loxR/loxP was generated by subcloning the EcoRI–BglII fragment from pPG3-loxR/loxP, which includes only the kanr gene flanked by the mutant lox site and loxP, into a pGEM3 cloning vector (Promega Biotech), thus eliminating the reiterated 3′ end of the kanr gene that was present in pPG3-loxR/loxP (Fig. 5A).
Figure 4.

Secondary genetic screen for mutant lox site function. (A) A schematic diagram depicting the approach used to reiterate the mutant spacer-containing lox sites to determine whether they retain their ability to mediate Cre-mediated recombination. The relative position of PCR primers used to amplify the interval of pPGloxR/loxP containing the mutant lox site are shown as arrows. The amplified product reintroduces a HindIII site present within the intact kanr gene and contains portions of the 3′ and 5′ ends of the kanr gene flanking the mutant spacer-containing lox site. The amplified PCR product was used to replace the lox66 site as a HindIII–BamHI fragment by subcloning between the HindIII and BglII sites of pPGloxR/loxP. The resulting pPGloxR/loxR constructs were passaged through 294-CRE bacterial cells (23) by transformation and the resulting plasmid DNAs analyzed by restriction digestion. (B) Restriction analysis of pPGloxR/loxR constructs before and after passage through 294-CRE bacterial cells. Plasmids were linearized with XmnI and fractionated by agarose electrophoresis. The 4.9 kb bands correspond to the intact parental pPGloxR/loxR. The 3.7 kb bands correspond to the expected size of pPGloxR/loxR after deletion of the kanr marker. Each mutant spacer lox site examined retained recombinatorial capacity that was indistinguishable from that of the wild-type lox site (data not shown).
Figure 5.

Analysis of constructs containing different combinations of mutant spacer-containing lox sites. (A) A schematic diagram depicting the approach used to generate different combinations of the mutant spacer- containing lox sites to test for incompatability. To eliminate the propensity of the reiterated 3′ end of the kanr gene to promote homologous recombination, the EcoRI–BglII fragment of pPGloxR/loxP was subcloned into the pGEM3 cloning vector in which the HindIII site had been converted to BglII. Using the same strategy as in Figure 4A, constructs were generated containing various combinations of mutant spacer-containing lox sites as in Figure 4B. (B) Southern analysis of plasmid constructs after passage through the 294-CRE bacterial strain. XmnI-linearized parental (4.2 kb) and recombined (3.0 kb) plasmid DNA was visualized by hybridization to a 32P-labeled probe specific to the vector backbone. Specific lox site combinations are indicated above each lane. loxP in lane 1 refers to a construct with only a single loxP site and serves as a no recombination control. For the nucleotide sequence of mutant spacers tested see Table 1.
Bacterial recombination assay
lox site-containing constructs were transformed into a bacterial strain (294-CRE) that stably expresses Cre recombinase (23). The isogenic bacterial strain 294-FLP was used as a non-Cre-expressing control (23). Both bacterial strains, 294-CRE [MM294 (F–, λ–, supE44, endAI, thi-1, hsdR17, lacZ::cI857-Cre)] and 294-FLP [MM294 (F–, λ–, supE44, endAI, thi-1, hsdR17, lacZ::cI857-Flp)], were gifts from Francis Stewart and Frank Buchholz (EMBL, Heidelberg, Germany). Transformed bacteria were plated on LB agar plates containing the appropriate antibiotics and grown overnight at 37°C. Single colonies were then picked and allowed to grow in liquid culture overnight at 30°C. Plasmid DNA was then isolated by a standard mini-prep DNA extraction procedure, linearized by digestion with either XmnI or EcoRI and its size determined by agarose gel electrophoresis.
Southern blot analysis
Digested DNAs were size fractionated by agarose gel electrophoresis, blotted to Biotrans nylon membrane (ICN) by capillary transfer and UV-crosslinked. The membranes were incubated overnight at 37°C in a hybridization solution (6× SSC, 10× Denhardt’s solution, 0.1 mg/ml salmon sperm DNA, 0.1% SDS) containing 2 × 106 c.p.m./ml 32P-end-labeled T7 sequencing primer (5′-TAATACGACTCA CTATAGG-3′), which anneals to the pGEM7zf+ vector backbone. The blots were washed in 6× SSC at 25°C and radioactive bands quantified on a phosphorimager (Molecular Dynamics Storm 860).
Dideoxynucleotide sequencing
The sequence for the mutant spacer-containing pPG3-loxR/loxP constructs was determined using a Sequenase Sequencing Kit (US Biochemicals) using the manufacturer’s recommendations. The synthetic oligonucleotide 5′-TAA ATCAGCATCCATGTTGG-3′, which anneals to a unique portion of the kanr gene in pPG3-loxR/loxP, was used as a sequencing primer. Sequencing reactions were fractionated on 8% acrylamide–urea–TBE gels and the sequence was determined after autoradiography.
Recombinant adenoviruses
A left end adenovirus construct containing the adenovirus serotype 5 sequence from bp 1 to 358 (SacII) and from bp 3330 (BglII) to 5700 (XhoI) was used as the base vector for constructing all recombinant adenoviral vectors. A polylinker inserted between the SacII and BglII sites facilitated the construction of pAdCMV-LacZlox66 (Fig. 7B). This vector contains a 1.1 kb immediate early promoter from cytomegalovirus (CMV) driving the expression of the Escherichia coli β-galactosidase gene. The lox66 site was cloned 3′ of the β-galactosidase gene as a BamHI–BstBI fragment derived from annealing two complementary synthetic oligonucleotides. lox71 and the mutant spacer-containing lox sites loxm2/71 and lox511/71 were cloned in a similar fashion between the CMV promoter and the β-galactosidase gene as XbaI– KpnI fragments to generate pAdCMVlox71LacZlox66, pAdCMVloxm2/71LacZlox66 and pAdCMVlox511/71LacZlox66, respectively. Cre-expressing HEK293 cell lines were screened using a recombinant adenoviral construct, AdCUL. AdCUL contains the firefly luciferase gene in the antisense orientation flanked by inverted lox66 and lox71 mutant arm-containing lox sites. The CMV promoter drives expression of the floxed cassette. Cre-mediated recombination between lox66 and lox71 inverts the floxed cassette into the sense orientation, resulting in expression of the luciferase reporter gene. Methods for generating recombinant adenovirus LacZ expression vectors have been described previously (24).
Figure 7.

Stability of floxed cassettes in HEK293Cre cells. (A) A schematic diagram of the strategy used to assess the relative stability of incompatible lox sites in the context of the adenoviral chromosome. Recombinant adenoviral vectors containing floxed LacZ cassettes were transduced into HEK293Cre cells as viral DNA and the emergent virus characterized by plaque assay. X-Gal staining was used to distinguish unrecombined (LacZ+) from recombined (LacZ–) genomes. (B) The top of the panel shows the schematic diagram of the recombinant adenovirus (AdCMV-LacZlox66) used as the basis of three viral chromosomes constructed to test the stability of various lox site combinations on floxed LacZ cassettes. The plaquing data is represented as the total numbers of blue and clear plaques scored. Percent incompatibility is defined as (no. of blue staining plaques/total no. of plaques) × 100. lox71 has the same spacer region sequence as loxP (see Fig. 1) and provides a positive control for Cre-mediated recombination; lox511/71 (identical to lox71 except for a single base substitution; see Fig. 1) was included to show the consequences of using an incompletely incompatible site. loxm2/71 has the same arm mutations as lox71 but differs in spacer region sequence at six of eight positions (see Table 2).
Cre-expressing HEK293 cells
The Cre recombinase gene was subcloned from pMC-1 (25) as a HindIII–BamHI fragment into the β-actin promoter-driven expression vector pβApr-1-neo (26), to generate pβApr-Cre. HEK293 cells were transfected with pβApr-Cre by calcium phosphate precipitation and stably expressing clones selected in 200 µg/ml G418. A total of 53 G418-resistant colonies stably transfected with the pβApr-Cre-neo construct were screened for relative Cre expression levels by infection with the AdCUL recombinant adenovirus reporter vector, where higher relative luciferase values were assumed to be correlated with higher relative levels of recombinase expression. One of the highest expressing clones, HEK293Cre57, was used exclusively in this study based on its superior growth characteristics.
Testing the stability of mutant lox site-containing recombinant adenoviruses
Virus preparations were purified twice by banding on CsCl gradients and the mature viral bands diluted with 2 vol of H2O before ethanol precipitation. After centrifugation the viral pellets were resuspended in 10 mM Tris, pH 8.0, 1 mM EDTA. SDS was added to a final concentration of 0.5% (w/v) and viral capsids were digested with proteinase K overnight at 37°C. After sequential extractions with equal volumes of phenol/chloroform/isoamyl alcohol (25:24:1) and chloroform/isoamyl alcohol (24:1) the viral DNA was recovered from the aqueous phase by ethanol precipitation. Purified viral DNA was resuspended in water and 5 µg was transfected as calcium phosphate precipitates into 60 mm dishes containing HEK293Cre cells seeded at 1 × 106 cells/dish the previous day. Precipitates were removed from cells after 5 h by washing with phosphate-buffered saline, 0.6 mM EDTA, and the medium was replaced. On day 7 post-transfection the plates were buffered by the addition of 1 drop of 2 M Tris, pH 9.0, and freeze-thawed at –20°C to generate crude viral lysates. The viral lysates were plaque titered by plaque overlay and β-galactosidase-expressing plaques detected by addition of X-Gal as previously described (24).
Recombinase-mediated cassette exchange assay
HEK293 and HEK293Cre cells were split and 60 mm dishes seeded at a density of 1 × 106 cells/dish the day before transfection. A total of 10 µg pGEM3loxm2/66kanrlox71 shuttle plasmid DNA and 1 µg AdCMVloxm2/71LacZlox66 viral DNA were added to each plate as calcium phosphate precipitates and processed as described above. After plaque titering, viral plaques were amplified and isolated viral DNA was analyzed by restriction digestion.
RESULTS
Rationale for genetic screen
We have observed that loxP and lox511 undergo promiscuous recombination in a Cre-expressing HEK293 cell line. Promiscuous recombination between the most commonly used incompatible lox sites therefore represents a limitation to RMCE if there is no genetic selection against such events. The basic question we asked is this: do mutant spacer-containing lox sites exist that when linked in cis with loxP are completely stable in the presence of Cre? To answer this question, we developed a genetic screen to detect low level promiscuous recombination in Cre-expressing bacteria and used it to screen a library of spacer regions to find those that did not exhibit detectable recombination.
Generation of a mutant spacer-containing lox site library
To generate a library of mutant spacer-containing lox sites, a synthetic oligonucleotide encompassing the lox66 sequence (see Fig. 1) was used as a template for PCR amplification (Fig. 3A). The nucleotide positions within the template corresponding to the spacer region sequence were synthesized as degenerate nucleotides, with the exception of positions –1 and +1, which were limited to either adenine or thymine. These limitations were based on previous mutational analysis of the spacer region which suggested that A/T nucleotides in the central –1 and +1 positions of the spacer region might play a role in promoting melting of the spacer region during strand exchange (9). PCR amplification provided an efficient method for converting the degenerate template into a large pool of randomized spacer-containing lox sites. The randomized lox sites (loxR) were subcloned into pPG3-loxP, a vector constructed for testing mutant spacer-containing lox sites for incompatibility with loxP and collectively referred to as pPG3-loxR/loxP (Fig. 3B).
Identifying incompatible lox sites
Seventy individual clones of pPG3-loxR/loxP were screened for their incompatibility with the wild-type loxP site. Each construct was transformed into the Cre-expressing bacterial strain 294-CRE (23) and the plasmid DNA isolated from transformants analyzed by restriction digestion to determine the extent to which the interval of DNA flanked by the degenerate lox site and loxP was deleted. Analysis of 13 representative mutant lox site-containing constructs is shown in Figure 3C and D. Linearization by EcoRI digestion revealed varying extents of recombination among the different clones. A 4.9 kb band corresponding to unrecombined parental pPG3-loxR/loxP plasmid was observed for most clones, while a 3.7 kb band corresponding to the recombined pPG3-loxR/loxP (deletion of the 1.2 kb fragment corresponding to the kanr gene; Fig. 3D) (see Fig. 3B for a schematic of pPG3-loxR/loxP) was seen in only three of the clones. These data suggest that while most of the sites exhibit incompatibility with loxP, three of the clones display some level of compatibility (Fig. 3D, m63, m64 and m71). These data are consistent with our expectation that a low degree of sequence identity in the spacer region would result in a high frequency of incompatible sites. Interestingly, an unexpected band of apparent 2.4 kb in size is detectable for all clones and figures predominantly in some lanes (Fig. 3D, m64, m67, m70 and m72). Restriction analysis with a second restriction enzyme was performed to identify the nature of the 2.4 kb band. When the same clones were linearized with XmnI (a unique restriction site located in the ampr gene) the 4.9 and 3.7 kb bands are seen, whereas the 2.4 kb band is noticeably absent (Fig. 3C). Moreover, a 3.7 kb band is detectable for clones with XmnI digestion that is absent with EcoRI. Because the unique XmnI and EcoRI sites are outside the floxed interval, they should linearize both parental and deleted plasmids, and yet they exhibit strikingly different restriction patterns.
Recombinase-dependent homologous recombination
One explanation to reconcile the differences between the XmnI and EcoRI restriction patterns is that plasmid constructs are undergoing both homologous and site-specific recombination in the 294-CRE bacterial strain. Homologous recombination between the reiterated 3′ end of the kanr gene and the floxed full-length kanr gene generates the same size plasmid as would site-specific recombination between lox sites. The 2.4 kb band seen in the EcoRI digest is supercoiled plasmid, representing the population of plasmids having undergone homologous recombination and become refractory to EcoRI digestion. In the XmnI digest, the 3.7 kb band represents the sum of plasmid molecules undergoing recombination (homologous and site-specific). Therefore, although all clones exhibit some degree of homologous recombination, this can be distinguished from site-specific recombination by EcoRI digestion.
Although the region of homology in pPG3-loxR/loxP is significant (∼600 bp), the levels of homologous recombination observed are nonetheless unexpectedly high. Some sites (e.g. m67 and m70; Fig. 3D) actually promote homologous recombination, implying that the Cre–lox system in this context can facilitate homologous recombination. To test this possibility we passaged pPG3-loxR/loxP constructs that showed various levels of homologous recombination through the isogenic bacterial strain 294-FLP (23), which expresses the Flp instead of the Cre recombinase. The results showed that no recombination of any kind was detected (data not shown), indicating that in this context homologous recombination is not only mediated by, but is dependent on, the Cre recombinase.
Testing for functional lox site spacer mutations
A non-functional lox site would be expected to exhibit the same phenotype as a genuinely incompatible site in the initial genetic screen. To confirm that a mutant spacer-containing lox site was indeed functional, each mutant lox site that displayed no detectable recombination with loxP was tested for Cre-mediated recombination with itself. The scheme for reiterating and functionally testing each mutant lox site is illustrated in Figure 4A. These reiterated constructs are referred to as pPG3-loxR/loxR. Four of the pPG3-loxR/loxR constructs were passaged through 294-CRE bacteria and the restriction analysis of the resulting plasmid after linearization with XmnI is shown in Figure 4B. This analysis confirmed that all four mutant spacer-containing lox sites examined (m2, m3, m7 and m11) were indeed efficient substrates for Cre-mediated recombination.
In order to distinguish between homologous recombination and site-directed recombination in the four pPG3-loxR/loxR constructs, the mutant spacer-containing lox site PCR products outlined above were subcloned into pPG3-loxR/loxP in which the unique EcoRI site had been changed to NotI. Thus, when the mutant site was reiterated in this new construct, pPG3-loxR/loxR now had a unique EcoRI site within the lox-flanked interval (as a result of the subcloned PCR product) but outside the region of homologous recombination and a unique NotI site outside the region of site-directed recombination and in the region of homologous recombination (Fig. 4A). To demonstrate that the recombination observed was in fact site-directed and not homologous, the passaged DNA was separately digested with EcoRI and NotI. The digests showed that all four constructs had retained the NotI site and lost the EcoRI site (data not shown), indicating that recombination was entirely site-directed.
Direct comparison of mutant spacer-containing lox site incompatibility
We next compared the mutant spacer-containing sites isolated with a previously characterized mutant spacer-containing lox site. lox511, one of the first incompatible spacer mutation-containing lox sites described (9), differs from loxP by a single nucleotide substitution in the spacer region (Fig. 1). It has been used in RMCE reactions in bacteria as a method for generating recombinatorial single chain antibody libraries (13) and in creating isogenic cell lines (14–16). However, our attempts to use lox511 for RMCE in HEK293 cells constitutively expressing the Cre recombinase were not successful, consistent with previous observations (15,17). In our experiments, exposure of HEK293Cre cells to Cre over a period of 7–10 days resulted in a very high frequency of promiscuous recombination between lox511 and loxP (see Fig. 7B). Although the deletion reaction was detected at low levels in 294-CRE bacteria, extended exposure to Cre in HEK293 cells allowed the deletion reaction to predominate. As a result, the low level of compatibility between lox511 and loxP compromised the stability of the floxed DNA.
In order to directly compare the levels of incompatibility between our novel mutant lox sites and lox511, we subcloned each floxed DNA cassette into the cloning vector pGEM3 (Promega Biotech), thereby eliminating the homologous region that was present in the original pPG3-loxR/loxP vector and ensuring that the rearrangements observed were limited to site-directed recombination. We also generated constructs with every possible combination of paired mutant spacer-containing lox sites (m2/m3, m2/m7, m2/m11, m3/m7, m3/m11 and m7/m11) following a procedure similar to that used to construct pPG3-loxR/loxR (Fig. 5A).
We passaged each construct through 294-CRE bacteria and linearized the resultant plasmid DNAs by XmnI digestion. Southern analysis was performed using an oligonucleotide probe complementary to a position outside the floxed cassette, as this is a more sensitive method for detecting low levels of the deletion reaction. Figure 5B shows the results of the Southern analysis. Lane 1 shows a negative control construct which contains only one lox site (loxP) and therefore no recombination would be expected, as is observed. Lane 2 is the previously characterized lox511 spacer mutation with loxP. The faint 3 kb band that is present shows that lox511 is not fully incompatible with loxP. Lanes 3–6 tested the compatibility of each mutant lox site (m2, m3, m7 and m11) with loxP. As no 3 kb band is observed, it is clear that all the new lox sites are incompatible with loxP. The paucity of signal for m3/loxP is due to a loading artifact. Overexposure of this blot and subsequent experiments show that m3/loxP behaves identically to m2/loxP, m7/loxP and m11/loxP (data not shown). Lanes 7–9 and 11–13 tested the incompatibility of the new lox sites with each other. All combinations of sites exhibited promiscuous recombination with one another with the exception of m3/m7 (Fig. 5B, lane 11), where no recombination was evident. A comparison of the nucleotide sequences of all the lox sites is shown in Table 1.
Table 1. Comparison between wild-type (loxP) and mutant lox spacer regions.
| lox site | Spacer sequence | Incompatible with |
|---|---|---|
| loxP | GCATACAT | m2, m3, m7, m11 |
| lox511 | GtATACAT | Not tested |
| m2 | tggTttcT | loxP |
| m3 | tggTAtta | loxP, m7 |
| m7 | ttcTAtcT | loxP, m3 |
| m11 | tggTAtcg | loxP |
Lower case letters indicate nucleotides that differ from the loxP spacer sequence.
RMCE in Cre-expressing bacteria
Although these novel mutant spacer-containing lox sites were found to be functional and incompatible with loxP, we wanted to confirm that they could also facilitate RMCE. We selected the m2 mutation for further analysis. The vector pAdCMVloxm2/71LacZlox66, containing m2/lox71 and lox66 flanking the LacZ gene under the control of the CMV promoter, was constructed to serve as the recipient plasmid for the RMCE reaction (Fig. 6A). pGEM3loxm2/66kanrlox71 was constructed as the shuttle vector to deliver the kanr gene cassette (Fig. 6A). The two constructs were transformed into 294-CRE bacteria and co-transformants recovered by selection in both ampicillin and kanamycin. pGEM3loxm2/66kanrlox71, which contains only the kanr gene, should be lost under ampicillin and kanamycin selection following RMCE. pAdCMVloxm2/71LacZlox66, which already contains the ampr gene, should gain the kanr gene after RMCE. Figure 6B shows the results of this experiment. Digestion at a unique ScaI site in each plasmid was used to linearize the constructs. The results show that both the shuttle and recipient vectors are stable in the presence of the Cre recombinase. When both constructs are exposed to Cre together, a prominent 7.2 kb band is observed, corresponding to the predicted size of pAdCMVloxm2/71LacZlox66 after exchange of the LacZ cassette with the kanr cassette (pAdCMVkanr; Fig. 6A). These data demonstrate that m2 can efficiently facilitate RMCE in bacteria.
Figure 6.
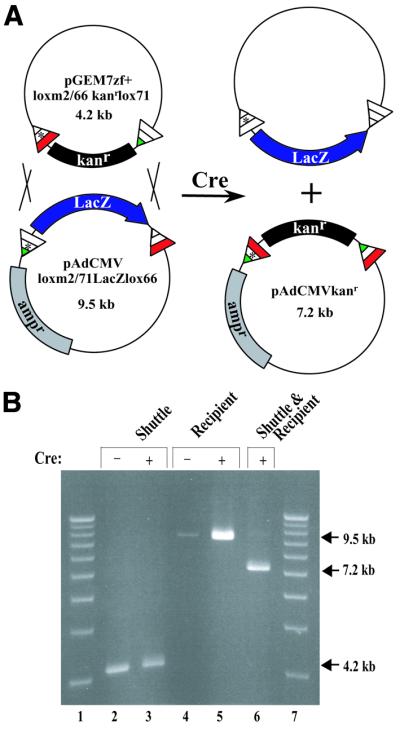
RMCE in Cre-expressing bacteria using incompatible lox sites. (A) A schematic diagram depicting RMCE between a shuttle plasmid containing a floxed kanr marker (pGEM7zf+loxm2/66 kanrlox71) and a recipient adenoviral left end construct containing a floxed LacZ gene (pAdCMVloxm2/71LacZlox66). (B) The parental plasmids diagrammed in (A) were isolated after transformation either separately or together into 294-CRE cells and the resultant plasmid DNA characterized by agarose electrophoresis after linearization with XmnI. Lanes 1 and 7, 1 kb DNA size markers; lanes 2, 3 and 6, the donor plasmid pGEM7zf+loxm2/66kanrlox71; lanes 4–6, the recipient plasmid pAdCMVloxm2/71LacZlox66. The parental plasmids before and after passage through 294-CRE cells are shown in lanes 2 and 4 and lanes 3 and 5, respectively. Lane 6 shows the recombinant product between the parental plasmids after passage through 294-CRE cells.
Stability of cis-linked lox sites in Cre-expressing HEK293 cells
To assess the relative stability of mutant lox sites when linked in cis with loxP, we chose to follow the fate of floxed LacZ-bearing adenoviral chromosomes after passage through Cre-expressing HEK293 cells (Fig. 7A). pAdCMVloxm2/71LacZlox66 (Fig. 6A) was used to generate a replication-defective adenovirus vector (Fig. 7B). Two additional adenovirus vectors based on pAdCMVloxm2/71LacZlox66 were constructed in which loxm2/71 was replaced with either lox71 or lox511/71 (Fig. 7B; see Fig. 1 for sequence). The adenovirus vector containing lox71 was a positive control for Cre recombination. The adenovirus vector containing lox511 was included for comparison. Viral DNA was purified for each construct and transfected into HEK293Cre cells. After 7 days the emergent virus was plaque titered, stained with X-Gal to indicate the presence of the LacZ cassette and scored for the number of blue (unrecombined) or clear (recombined) plaques. Figure 7B shows that the positive control vector containing the lox71LacZlox66 cassette was completely deleted, confirming that Cre-mediated recombination in HEK293Cre cells is efficient. Over 90% of the vectors containing the lox511LacZlox66 cassette are deleted for the floxed cassette. This apparent instability illustrates the problem of using the lox511 spacer region for RMCE in the absence of genetic selection. In contrast, no clear plaques were detected for the vectors containing the loxm2LacZlox66 cassette, demonstrating the complete incompatibility of m2 with loxP.
RMCE in Cre-expressing HEK293 cells
To determine whether m2 could facilitate RMCE in cell culture, AdCMVloxm2/71LacZlox66 viral DNA and the shuttle plasmid pGEM3loxm2/66kanrlox71 were co-transfected into either HEK293 or HEK293Cre cells (Fig. 8A). After 7 days, the emergent virus was plaque titered and β-galactosidase expression detected by staining with X-Gal. Transfections in HEK293 cells resulted in the production of only β-galactosidase-expressing plaques, whereas transfections in HEK293Cre cells displayed both blue and clear plaques (83 and 17%, respectively). Representative blue and clear plaques were picked, expanded, and their viral DNA analyzed by restriction analysis (Fig. 8B). All blue (4/4) and clear (22/22) plaques assayed yielded restriction patterns consistent with having parental and recombinant chromosomes, respectively. This result demonstrates that at least one of the novel incompatible lox sites (m2) isolated from our genetic screen can direct high fidelity RMCE in mammalian cell culture.
Figure 8.

RMCE in Cre-expressing HEK293 cells using incompatible lox sites. (A) A schematic diagram depicting RMCE between a shuttle plasmid containing a floxed kanr marker (pGEM7zf+loxm2/66 kanrlox71) and a parental adenoviral chromosome containing a floxed LacZ gene (AdCMVloxm2/71LacZlox66). A unique BstBI restriction site in the parental chromosome allows sizing of the left terminal restriction fragment. (B) Restriction digestion of viral DNA isolated from plaque-purified virus resulting from co-transfection of HEK293Cre cells with shuttle plasmid and viral DNA depicted in (A). X-Gal staining for the production of β-galactosidase distinguishes parental viral plaques (blue) from putative recombinant plaques (clear). Restriction analysis of representative blue and clear plaques confirms the predicted size for parental (5.1 kb) and recombinant viruses (2.8 kb), respectively.
DISCUSSION
The ability to manipulate DNA via site-specific recombination has become an increasingly powerful tool in molecular biology. Site-specific recombination has the ability to precisely target exogenous DNA to a specific locus, allowing modifications for the functional analysis of a particular gene or control region. The use of heterospecific lox sites has led to the development of RMCE, a method for selectively replacing one cassette for another (13–19). With RMCE, genomic targeting no longer suffers from problems associated with the integration of the entire targeting plasmid. Early versions of RMCE to generate isogenic cell lines used positive selectable markers to select for targeted events that were either left behind (16,18,19) or removed by the action of a second site-specific recombinase system (14).
Recently, a marker-free method has been developed for the FLP-frt recombination system for the purpose of generating stable cell lines using the dual positive and negative selectable marker hygtk as the outgoing cassette in RMCE (20). The successful application of negative selection to enrich for targeted events is dependent on the absolute stability of the heterospecific sites. Attempts to use a similar approach for Cre RMCE has revealed that the extensively used heterospecific sites loxP and lox511 undergo a high rate of promiscuous recombination (15,17). In this instance, the use of a heterospecific site with greater incompatibility than lox511 is desirable. Such a site, lox2722, was initially described in an in vitro recombination system and was predicted to exhibit greater incompatibility than lox511 (27). This has been subsequently confirmed in an in vivo phagemid system used to test the incompatibility of previously characterized heterospecific lox sites with one another (21). Recently, lox2722 has been used successfully in targeting experiments in ES cells using negative selection against the outgoing floxed cassette (17). While the incompatibility between loxP and lox2722 in those experiments was not absolute (90%), they represent a marked improvement over lox511, illustrating the value of using heterospecific lox sites of greater incompatibility.
The use of RMCE to generate recombinant adenoviral vectors in the absence of genetic selection underscores the necessity of using high fidelity heterospecific lox sites. To further maximize the fidelity of heterospecific lox sites, we have developed a genetic screen to identify novel lox sites that are functional and have greater incompatibility with the wild-type lox site, loxP. In our screen, we identified four mutant spacer-containing lox sites that retain function and yet are incompatible with loxP, with two of the four sites being incompatible with each other as well. We demonstrated that the new sites were more incompatible with loxP than a site (lox511) previously reported to be fully incompatible with loxP (9). We subsequently showed that all four sites were functionally indistinguishable from loxP as substrates for Cre recombinase. Using a combination of spacer region and palindromic arm mutations, we demonstrated that for at least one novel lox site, the Cre–lox recombination system could facilitate high fidelity RMCE in both bacteria and cell culture.
Interestingly, in bacteria the Cre–lox system could facilitate homologous recombination in a recombinase-dependent manner when given a significant region of homology (600 bp). The Cre–lox system probably assists in mediating homologous recombination by stabilizing a tertiary complex between two cis-linked lox sites. During site-specific recombination, two Cre-bound lox sites must be brought together in a tertiary complex prior to strand exchange between interacting spacer regions (28). Presumably, in our constructs the regions of DNA adjacent to the lox sites come into close proximity as a consequence of Cre binding, and by virtue of their shared homology an elevated frequency of homologous recombination was observed. This homologous recombination appears to occur whether or not the two synapsing lox sites are capable of undergoing productive site-specific recombination. Some mutated lox sites facilitated homologous recombination to a greater degree than other sites. The increased frequency of homologous recombination may reflect the impaired kinetics of strand exchange between particular spacer regions. Kinetic differences of in vitro reactions between loxP and lox sites containing various mutant spacer regions have been described recently (27). The homologous recombination that we have observed is likely an in vivo manifestation of strand exchange kinetics.
Some generalizations concerning the nature of incompatibility can be made by directly comparing the spacer regions between combinations of lox sites that display incompatibility with those that do not. Table 2 shows a direct sequence comparison between all of the lox site combinations that were tested and whether or not they exhibited incompatibility. In general, incompatible lox sites have limited sequence identity. In our survey, lox sites that shared identity at four or less positions exhibited incompatibility while those lox sites that shared identity at five or more positions were capable of productive recombination at some detectable frequency. However, this interpretation is probably an oversimplification in explaining the basis of incompatibility. loxP and lox511, which only differ at one position, exhibit the least recombination observed between compatible sites we tested, while all other combinations of compatible lox sites tested exhibited far more recombination while sharing less sequence identity. It is perhaps noteworthy that none of the incompatible lox site combinations share sequence identity at positions –3 and +3. Because the nucleotides at these positions are located at the terminal 5′ ends of exchanging strands within the recombination complex, mismatches here may not be as easily resolved by the recombinase, resulting in a kinetic block to productive recombination. This explanation is consistent with the work of Lee and Saito (27), who examined the effects of substituting all possible nucleotides at each position within the spacer region on recombination kinetics using an in vitro recombination system. They reported that a double mutant-containing spacer region, lox2722, with substitutions at –3 and +3 displayed the greatest ‘exclusivity’ with regard to loxP. Although these authors did not present evidence that this was the case in vivo, our experiments suggest that non-identity at these positions may indeed be the most important factor in determining incompatibility.
Table 2. Comparison of incompatible versus compatible lox sites.

Incompatible sites are distinguished from compatible sites as combinations where no detectable recombination was observed when assayed in Cre-expressing bacteria. Vertical lines indicate positions in which spacer regions share sequence identity. The frequency of matches at each position within the spacer region is shown at the right.
Although our in vivo experiments using viral chromosomes demonstrate the increased fidelity of loxm2 as compared with lox511, we did not directly use loxm2 to target a singly integrated chromosome, as would be the case for generating isogenic cell lines. However, because the Cre recombinase binding site does not include nucleotides within the 8 bp spacer region, it would not be expected to influence the reactivity of the lox site in single chromosomes, packaged as chromatin. In contrast, the lox arm mutations we used in our constructs, lox66 and lox71, have already been successfully used to target sites in the chromosomes of both plants (11) and mammalian cell cultures (12). Whereas the frequency of integration between plasmid DNA and a chromosomal target may be quite different than what we observed between plasmid and viral DNA, we expect that the fidelity of incompatible lox sites will not be affected by chromatin.
The mutant lox sites we have isolated and characterized for RMCE underscore the importance of the lox site spacer region in specifying directed recombination. Infidelity in the spacer region presents a major limitation to many of the potential applications of the Cre–lox system, particularly for RMCE in the absence of genetic selection. Using the previously described mutational analyses of the palindromic arms in combination with the novel spacer mutations described herein should allow the Cre–lox system to carry out efficient high fidelity, site-specific recombination reactions. In the future, this technology should prove useful for the precise manipulation of eukaryotic genomes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Frank Buchholz and Francis Stewart for generously providing the 294-CRE and 294-FLP bacterial strains, and Jason Carnes, Bob Thompson, David Allen and Massimo Buvoli for their critical review of the manuscript. This work was supported by National Heart, Lung and Blood Institute grant HL50560.
REFERENCES
- 1.Austin S., Ziese,M. and Sternberg,N. (1981) A novel role for site-specific recombination in maintenance of bacterial replicons. Cell, 25, 729–736. [DOI] [PubMed] [Google Scholar]
- 2.Hoess R.H., Ziese,M. and Sternberg,N. (1982) P1 site-specific recombination: nucleotide sequence of the recombining sites. Proc. Natl Acad. Sci. USA, 79, 3398–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Craig N.L., (1988) The mechanism of conservative site-specific recombination. Annu. Rev. Genet., 22, 77–105. [DOI] [PubMed] [Google Scholar]
- 4.Hoess R.H., and Abremski,K. (1990) The Cre-lox recombination system. In Eckstein,F. and Lilley,D.M.J. (eds), Nucleic Acids and Molecular Biology. Springer-Verlag, Berlin, Germany, Vol. 4, pp. 99–109.
- 5.Sauer B., (1987) Functional expression of the cre-lox site-specific recombination system in the yeast Saccharomyces cerevisiae. Mol. Cell Biol., 7, 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sauer B., and Henderson,N. (1988) Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl Acad. Sci. USA, 85, 5166–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sauer B., and Henderson,N. (1989) Cre-stimulated recombination at loxP-containing DNA sequences placed into the mammalian genome. Nucleic Acids Res., 17, 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauer B., and Henderson,N. (1990) Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol., 2, 441–449. [PubMed] [Google Scholar]
- 9.Hoess R.H., Wierzbicki,A. and Abremski,K. (1986) The role of the loxP spacer region in P1 site-specific recombination. Nucleic Acids Res., 14, 2287–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo F., Gopaul,D.N. and Van Duyne,G.D. (1997) Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature, 389, 40–46. [DOI] [PubMed] [Google Scholar]
- 11.Albert H., Dale,E.C., Lee,E. and Ow,D.W. (1995) Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J., 7, 649–659. [DOI] [PubMed] [Google Scholar]
- 12.Araki K., Araki,M. and Yamamura,K. (1997) Targeted integration of DNA using mutant lox sites in embryonic stem cells. Nucleic Acids Res., 25, 868–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waterhouse P., Griffiths,A.D., Johnson,K.S. and Winter,G. (1993) Combinatorial infection and in vivo recombination: a strategy for making large phage antibody repertoires. Nucleic Acids Res., 21, 2265–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouhassira E.E., Westerman,K. and Leboulch,P. (1997) Transcriptional behavior of LCR enhancer elements integrated at the same chromosomal locus by recombinase-mediated cassette exchange. Blood, 90, 3332–3344. [PubMed] [Google Scholar]
- 15.Feng Y.-Q., Seibler,J., Alami,R., Eisen,A., Westerman,K.A., Leboulch,P., Fiering,S. and Bouhassira,E.E. (1999) Site-specific chromosomal integration in mammalian cells: highly efficient CRE recombinase-mediated cassette exchange. J. Mol. Biol., 293, 779–785. [DOI] [PubMed] [Google Scholar]
- 16.Bethke B., and Sauer,B. (1997) Segmental genomic replacement by Cre-mediated recombination: genotoxic stress activation of the p53 promoter in single-copy transformants. Nucleic Acids Res., 25, 2828–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolb A.F., (2001) Selection-marker-free modification of the murine β-casein gene using a lox2722 site. Anal. Biochem., 290, 260–271. [DOI] [PubMed] [Google Scholar]
- 18.Schlake T., and Bode,J. (1994) Use of mutated FLP recognition target (FRT) sites for the exchange of expression cassettes at defined chromosomal loci. Biochemistry, 33, 12746–12751. [DOI] [PubMed] [Google Scholar]
- 19.Seibler J., and Bode,J. (1997) Double-reciprocal crossover mediated by FLP-recombinase: a concept and an assay. Biochemistry, 36, 1740–1747. [DOI] [PubMed] [Google Scholar]
- 20.Seibler J., Schubeler,D., Fiering,S., Groudine,M. and Bode,J. (1998) DNA cassette exchange in ES cells mediated by Flp recombinase: an efficient strategy for repeated modification of tagged loci by marker-free constructs. Biochemistry, 37, 6229–6234. [DOI] [PubMed] [Google Scholar]
- 21.Siegel R.W., Jain,R. and Bradbury,A. (2001) Using an in vivo phagemid system to identify non-compatible loxP sequences. FEBS Lett., 499, 147–153. [DOI] [PubMed] [Google Scholar]
- 22.Maniatis T., Fritsch,E.F. and Sambrook,J. (1982) Molecular Cloning: A Laboratory Manual, 1st Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 23.Buchholz F., Angrand,P.O. and Stewart,A.F. (1996) A simple assay to determine the functionality of Cre or FLP recombination targets in genomic manipulation constructs. Nucleic Acids Res., 24, 3118–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaack J., Langer,S. and Guo,X. (1995) Efficient selection of recombinant adenoviruses by vectors that express beta-galactosidase. J. Virol., 69, 3920–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu H., Zou,Y.R. and Rajewsky,K. (1993) Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell, 73, 1155–1164. [DOI] [PubMed] [Google Scholar]
- 26.Gunning P., Leavitt,J., Muscat,G., Ng,S.Y. and Kedes,L. (1987) A human beta-actin expression vector system directs high-level accumulation of antisense transcripts. Proc. Natl Acad. Sci. USA, 84, 4831–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee G., and Saito,I. (1998) Role of nucleotide sequences of loxP spacer region in Cre-mediated recombination. Gene, 216, 55–65. [DOI] [PubMed] [Google Scholar]
- 28.Mack A., Sauer,B., Abremski,K. and Hoess,R. (1992) Stoichiometry of the Cre recombinase bound to the lox recombining site. Nucleic Acids Res., 20, 4451–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]