


Abstract
Previous studies have established a critical role of both TFIIB and RNA polymerase II (RNAPII) in start site selection in the yeast Saccharomyces cerevisiae. However, it remains unclear how the TFIIB–RNAPII interaction impacts on this process since such an interaction can potentially influence both preinitiation complex (PIC) stability and conformation. In this study, we further investigate the role of TFIIB in start site selection by characterizing our newly generated TFIIB mutants, two of which exhibit a novel upstream shift of start sites in vivo. We took advantage of an artificial recruitment system in which an RNAPII holoenzyme component is covalently linked to a DNA-binding domain for more direct and stable recruitment. We show that TFIIB mutations can exert their effects on start site selection in such an artificial recruitment system even though it has a relaxed requirement for TFIIB. We further show that these TFIIB mutants have normal affinity for RNAPII and do not alter the promoter melting/scanning step. Finally, we show that overexpressing the genetically isolated TFIIB mutant E62K, which has a reduced affinity for RNAPII, can correct its start site selection defect. We discuss a model in which the TFIIB–RNAPII interaction controls the start site selection process by influencing the conformation of PIC prior to or during PIC assembly, as opposed to PIC stability.
INTRODUCTION
Accurate initiation of transcription by RNA polymerase II (RNAPII) requires the polymerase itself and a group of general transcription factors (GTFs), including TATA-binding protein (TBP), TFIIB, TFIIE, TFIIF and TFIIH (1–4). The existence of analogous GTFs from different eukaryotic species, including human, rat, fruit fly and yeast, suggests that RNAPII transcription is a highly conserved process. However, one particular feature of transcription appears to be different between the yeast Saccharomyces cerevisiae and other eukaryotes, namely transcription start site determination. In higher eukaryotic cells, transcription generally starts at a fixed distance of ∼30 bp downstream of the TATA element (5), normally utilizing a single initiation site. In S.cerevisiae, transcription occurs within a window located ∼40–120 bp downstream of the TATA box, often with multiple start sites (6,7).
An essential step in transcription initiation is the recruitment of RNAPII to the promoter region. TFIIB plays a critical role at this step by providing a physical link between TBP and RNAPII (8). A large body of studies also suggested that yeast TFIIB is involved in the process of transcription start site selection (9–12). Genetic analysis revealed that mutations in both RNAPII and TFIIB can cause transcription start site alterations in vivo (13). In addition, an in vitro pair-wise replacement experiment using GTFs from S.cerevisiae and Schizosaccharomyces pombe demonstrated that RNAPII and TFIIB are both necessary and sufficient to determine transcription start site profiles characteristic of a given organism (10). Despite different start site usage in S.cerevisiae and other eukaryotes, certain aspects of the start site selection mechanisms may remain conserved. In particular, mutations in human TFIIB corresponding to two genetically isolated mutations in yeast TFIIB also cause detectable, albeit less severe, start site alterations in vitro (14). These findings suggest that understanding how start sites are determined in yeast will also yield information relevant to transcription in mammalian cells.
Despite extensive studies of transcription mechanisms and start site selection in yeast, several questions remain to be answered fully. First, since all the previously described TFIIB mutants only shift start sites further downstream (9,11,12,15–17), it is unclear whether TFIIB is responsible for preventing RNAPII from utilizing DNA sites that are located not only downstream but also upstream of the normal start sites. Second, since the TFIIB–RNAPII interaction can potentially affect both preinitiation complex (PIC) stability and conformation, experiments in vivo that can further differentiate between these two possibilities are of particular importance. Interestingly, two of the genetically isolated TFIIB mutants both have a reduced ability to interact with RNAPII (18). Third, it remains to be determined at what step(s) TFIIB may confer the start site selection properties to RNAPII; in particular, previous studies have shown that mutations in RNAPII can exhibit altered properties in both elongation and start site selection (19,20), suggesting that start site selection defects may be caused by promoter melting/scanning properties.
The experiments described in this report further address the questions outlined above. In particular, we report new TFIIB mutants that cause novel transcription start site profiles characteristic of an upstream shift. In addition, we take advantage of an artificial recruitment system in which the RNAPII holoenzyme is directly and stably recruited to DNA through a covalent link to a DNA-binding domain. We show that TFIIB mutations can still exert their effects on the start site selection process even though the requirement for TFIIB becomes relaxed. Using an in vivo footprint assay, we further demonstrate that the promoter melting/scanning step is not altered by TFIIB mutations. Finally, we show that overexpressing E62K, a genetically isolated TFIIB mutant with a reduced affinity for RNAPII, can correct its start site selection defect in vivo. Our results are consistent with a model in which the TFIIB–RNAPII interaction may control the start site selection process by influencing the conformation of RNAPII prior to or during PIC assembly, as opposed to PIC stability.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strain 604 (MATα ura3-52 leu2 his3 trp1 cyh2r sua7) with wild-type TFIIB expressed from a Ura3/ARS-CEN plasmid was used as the parent strain to generate all our mutant TFIIB strains (21). Mutant TFIIB genes were generated using PCR-based mutagenesis methods. The mutant DNA was inserted into a Trp1/2 µm plasmid. A plasmid shuffling procedure (22) was used to generate yeast strains with mutant TFIIB but lacking the endogenous SUA7 gene.
Primer extension
Total RNA was isolated using an acidic phenol method as previously described (21). Primer extension experiments using 50 µg total RNA were carried out as described previously (23). The sequences of the primers were: for the endogenous ADH1 gene, 5′-CGTAGAAGATAACACCTTTTTG-3′; for the lacZ reporter gene, 5′-CACCAGTGAGACGGGCAACAG-3′. DNA samples were subjected to 9% acrylamide sequencing gel electrophoresis in the presence of 7 M urea.
Protein purification and interaction assay
GST fusion proteins were expressed in Escherichia coli strain DH5α and purified as previously described (21). Different amounts of GST–TFIIB protein (as specified in the legend to Fig. 5) were bound to 40 µl of glutathione–Sepharose 4B beads and incubated with 1 µg partially purified RNAPII (kindly provided by Drs Bushnell and Kornberg) at 4°C for 2 h. The beads were washed thoroughly and eluted with 45 µl of SDS–PAGE loading buffer. Aliquots of 15 µl of the supernatant were subjected to SDS–PAGE and assayed by western blot using antibodies against GST (Santa-Cruz) and Rpb1 (Babco).
Figure 5.

Normal interaction between TFIIB mutants and RNAPII in vitro. Partially purified yeast RNAPII was used in a GST pull-down assay with different bacterially expressed GST–TFIIB fusion proteins. The proteins were detected in a western blot using antibodies against the largest subunit of RNAPII and GST. In this experiment, increasing amounts of GST–TFIIB derivatives were used: lanes 3–6, 1 µg; lanes 7–10, 2 µg; lanes 13–16, 4 µg; lanes 17–20, 8 µg. To ensure a comparable loading, decreasing amounts of the pull-down products were loaded onto the gel: lanes 3–6, 1/4 the pull-down product; lanes 7–10 and 13–16, 1/8 the product; lanes 17–20, 1/16 the product. GST alone in lane 12 is a smaller protein compared with GST–TFIIB and thus is not seen in this picture.
Quantitative S1 analysis of mRNA
The S1 analysis of mRNA was performed as described previously (21). The oligonucleotide probe used to detect the lacZ mRNA level has 40 nt matching the sequence of lacZ with six mismatched nucleotides at the 3′ end (underlined). The probe used to detect the endogenous ADH1 gene mRNA level has 30 nt matching the sequence of ADH1 with six mismatched nucleotides at the 3′ end (underlined). The sequences of the probes were: lacZ, 5′-GGAAGGGCGATC GGTGCGGGCCTCTTCGCTATTACGCCAGCAATAG-3′; ADH1, 5′-GGAACTGGAATATCTTTGTATTCCAACTTA CCAAAA-3′
Analysis of promoter melting pattern
In vivo footprinting was carried out according to a method described previously (5) with the following modifications. Yeast cells were grown in selective medium to OD600 = 1.0. An aliquot of 100 OD-ml yeast cells was resuspended in 1 ml of growth medium. Permanganate treatment was carried out by adding 85 µl of freshly made 0.35 M KMnO4 solution to the cells and incubation at 25°C for 2 min. DNA was prepared and treated with piperidine as described (5). Primer extension analysis to detect the in vivo footprint patterns was carried out as described previously (24). The DNA was subjected to 9% acrylamide sequencing gel electrophoresis in the presence of 7 M urea.
RESULTS
Novel TFIIB mutants affecting start site selection in vivo
Almost all the previously reported TFIIB mutants defective in start site selection in vivo have individual amino acid substitutions. In addition, these mutants share a common defect in that they all shift start sites downstream. To generate TFIIB derivatives with potentially novel and/or more severe defects in start site selection, we took the drastic approach of inserting five alanines at various locations in a region previously proposed to be critical for start site selection (12,15; Fig. 1B). This region is located immediately downstream of the zinc ribbon structure in the N-terminal domain of TFIIB and is highly conserved in eukaryotes. In particular, two amino acid residues in this region (highlighted in Fig. 1A), Glu62 (E62) and Arg78 (R78), are proposed to form a salt bridge essential for start site selection in yeast (13). For our TFIIB mutants, a string of five consecutive alanines was inserted at locations surrounding these two residues. As shown in Table 1, all these TFIIB mutants were able to support cell growth in the absence of the chromosomal/endogenous SUA7 gene (which encodes TFIIB) (11). However, three of these mutants, 53A5, 63A5 and 79A5, conferred a cold-sensitive phenotype; yeast cells containing these TFIIB derivatives but lacking endogenous SUA7 failed to grow (or grew very poorly) at 18°C. In addition, cells containing two other mutants, 58A5 and 70A5, exhibited a heat/temperature-sensitive phenotype and failed to grow at 37°C (Table 1).
Figure 1.

Schematic diagram of the structural motifs of yeast TFIIB (A) and locations of alanine insertions (B). The C-terminal core domain of TFIIB contains two imperfect repeats folded into two compact sub-domains. The N-terminal domain contains a zinc ribbon structure, followed immediately by a region that is highly conserved among eukaryotes and is important for start site selection in vivo. Highlighted are residues in this region corresponding to E62 and K78 of yeast TFIIB, two residues that are proposed to form a salt bridge.
Table 1. Growth phenotypes of TFIIB derivatives.
| TFIIB derivative |
Temperature |
||
|---|---|---|---|
| 37°C | 30°C | 18°C | |
| Wild-type | ++++ | ++++ | ++++ |
| 53A5 | ++++ | ++++ | – |
| 58A5 | – | +++ | +++ |
| 62A5 | ++++ | ++++ | ++++ |
| 63A5 | +++ | ++++ | +/– |
| 68A5 | ++++ | ++++ | ++ |
| 70A5 | – | +++ | +++ |
| 76A5 | ++++ | ++++ | ++++ |
| 77A5 | ++++ | ++++ | ++++ |
| 78A5 | ++++ | ++++ | ++++ |
| 79A5 | ++++ | ++++ | +/– |
To analyze how the newly generated TFIIB mutants affect start site selection in vivo, we determined the transcription initiation sites of the endogenous ADH1 gene in yeast cells containing these mutants but lacking endogenous SUA7. The start site usage at the ADH1 promoter is sensitive to mutations in RNAPII and TFIIB and, therefore, has been widely used in previous studies (9,11,23,25,26). Another important feature of ADH1 is that, unlike many other yeast promoters which have multiple TATA boxes, the ADH1 promoter contains only one functional TATA element (16). In wild-type yeast cells (Fig. 2A, lane 1), ADH1 transcription initiates at two major positions, –27 and –37, with the A residue of the translation initiation codon ATG designated as +1. To better compare the start site selection profiles, we also generated intensity scanning graphs for ADH1 in both wild-type cells and cells containing selected mutants (Fig. 2B).
Figure 2.

Transcription start sites of the endogenous ADH1 gene. (A) Shown are primer extension results to determine ADH1 transcription start sites in cells (grown at 30°C) containing the indicated TFIIB mutants. As a control, cells that lack the RNAPII subunit RPB9 were also used in the same analysis. Two major start sites of ADH1, –27 and –37, are marked. (B) Shown are intensity scanning graphs for ADH1 start site usage in cells containing selected mutants.
Our newly generated TFIIB mutants can be grouped into three classes according to their effects on start site usage at the ADH1 promoter (Fig. 2). First, three of the mutants, 62A5, 77A5 and 78A5, have relatively little or no effect on start site selection (Fig. 2A, lanes 5, 10 and 11), demonstrating that these locations of TFIIB can tolerate alanine insertions without causing gross start site selection defects. Second, both 53A5 and 79A5 caused a typical downstream shift of start site usage (lanes 3 and 12). In both cases, the intensity of the –27 start site, as well as that of several minor start sites that are further downstream, was significantly increased, with usage of the –37 site dramatically reduced. Three other mutants also had detectable but less severe defects in shifting start sites downstream (63A5, 68A5 and 76A5). Third, mutants 58A5 and 70A5 caused novel start site selection profiles characteristic of an upstream shift similar (but not identical) to that obtained in cells lacking the RPB9 subunit of RNAPII (23). Specifically, the use of start sites further upstream of the –37 major start site was significantly enhanced in both ΔRPB9 yeast cells (Fig. 2A, lane 2) and cells containing the 58A5 or 70A5 mutants (Fig. 2A, lanes 4 and 8; see also Fig. 2B). In addition, the intensity of the start sites between –27 and –37 was also significantly increased in cells containing these two TFIIB mutants.
Start site selection with an artificially recruited RNAPII holoenzyme
Since TFIIB represents a critical link between TBP and RNAPII, the TFIIB–RNAPII interaction can potentially affect the start site selection process by influencing either PIC stability or conformation. To further differentiate between these two possibilities, we took advantage of an artificial recruitment system, in which the RNAPII holoenzyme is directly recruited to the promoter through a covalent link to a DNA-binding domain. Specifically, we used a LexA–GAL11 fusion protein containing the holoenzyme component of GAL11 fused to the DNA-binding domain of LexA (27–29). Previous studies have shown that transcription supported by artificially recruited RNAPII holoenzyme is more sensitive to distance and promoter architecture than activator-stimulated transcription (30), suggesting that these two processes are mechanistically different (see also 31,32). Using temperature-sensitive alleles for TFIIB and other GTFs (Fig. 3B), we further demonstrate that transcription achieved through these two mechanisms exhibits differential requirements for GTFs (Fig. 3C and data not shown). For example, in cells containing the temperature-sensitive allele TFIIB(70A5), the level of lacZ mRNA induced by LexA–VP16 (see Fig. 3A for reporter diagram) decreased significantly after the cells were shifted to the non-permissive temperature (Fig. 3C, lanes 5–8, top panel). In contrast, when the reporter gene was induced by LexA–GAL11, the lacZ mRNA level remained unchanged after the temperature shift (Fig. 3C, lanes 1–4, top panel). As expected, the endogenous ADH1 gene exhibited a reduction in transcription after the temperature shift in cells containing TFIIB(70A5) (Fig. 3C, lanes 1–8, bottom panels). In cells containing wild-type TFIIB, both lacZ and ADH1 mRNA levels remained constant (Fig. 3C, lanes 9–16). These results demonstrate a more relaxed requirement for TFIIB by the artificial recruitment system, presumably due to the more direct and stable recruitment of RNAPII holoenzyme through a covalent link, as opposed to protein–protein interactions.
Figure 3.
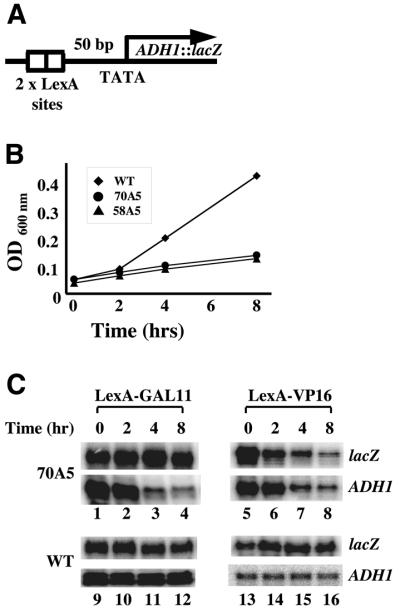
Activator-stimulated transcription and artificial recruitment- supported transcription behave differently. (A) Shown is a schematic diagram depicting the ADH1–lacZ reporter gene. It contains the TATA box, transcription start site and ATG codon from the ADH1 promoter with two LexA-binding sites placed 50 bp upstream of the TATA box. (B) Growth curves of yeast cells containing either wild-type or the indicated TFIIB mutants. Time points at which the optical density (OD600 nm) measurements of the yeast cultures were taken are given; time 0 represents the temperature shift from 30 to 37°C. (C) Yeast cells containing 70A5 but lacking endogenous SUA7 were grown at 30°C and then shifted to 37°C. RNA was isolated at indicated time points to determine the cellular level of lacZ mRNA using an S1 nuclease assay. The reporter gene was activated by either the classical activator LexA–VP16 (lanes 5–8) or the artificial recruiter LexA–GAL11 (lanes 1–4). Cells containing 58A5 exhibited expression profiles of the reporter gene identical to those observed in cells containing 70A5 (not shown). Also shown in this figure are the expression profiles of the reporter gene in cells containing wild-type TFIIB (lanes 9–16). For each time course, the expression level of the endogenous ADH1 gene is shown as a control (bottom panels). These experiments suggest a relaxed requirement for TFIIB by LexA–GAL11-supported transcription. However, LexA–GAL11-supported transcription is not independent of TFIIB because start site alteration defects associated with TFIIB mutations persist even when transcription is activated by LexA–GAL11 under both permissive and non-permissive temperatures (Fig. 4C and data not shown).
We reasoned that if transcription start sites are determined in a manner that is irrespective of PIC stability, start site alterations caused by TFIIB mutations should persist even when the RNAPII holoenzyme is artificially and more stably recruited to DNA. For our experiments, we used an ADH1–lacZ reporter gene (Fig. 3A) which contains the ADH1 core promoter region (including the TATA box, transcription initiation sites and ATG) and two LexA-binding sites located 50 bp upstream of the ADH1 TATA box. As shown in Figure 4, individual TFIIB mutations caused nearly identical start site alterations for both the endogenous ADH1 gene (Fig. 4A) and the ADH1–lacZ reporter gene activated by either LexA–VP16 (Fig. 4B) or LexA–GAL11 (Fig. 4C). These results demonstrate that TFIIB mutations can exert their effects on start site selection irrespective of the RNAPII recruitment mechanism. They are consistent with the hypothesis that the TFIIB–RNAPII interaction controls the start site selection process by influencing the conformation of PIC, as opposed to its stability.
Figure 4.

TFIIB mutations can confer their effects to artificially recruited RNAPII. Shown are primer extension assay results to determine the start sites for the endogenous ADH1 gene (A) and the ADH1–lacZ reporter gene activated by either LexA–VP16 (B) or LexA–GAL11 (C). The yeast cells contained either the wild-type or the indicated TFIIB mutants and were grown at 30°C. The two major start sites, –27 and –37, are marked.
TFIIB mutations do not affect interaction with RNAPII or promoter melting/scanning
To further investigate the role of the TFIIB–RNAPII interaction in start site selection, we determined how our TFIIB mutants interacted with RNAPII in vitro. Recombinant mutant TFIIB proteins were expressed in bacteria as GST fusions and bound to glutathione beads. After incubation with partially purified yeast RNAPII, the bound proteins were analyzed by western blot using antibodies against both GST and RPB1, the largest subunit of RNAPII. An antibody against GST was used in the same western blot analysis because the GST–TFIIB band represented an internal control for the overall pull-down efficiency. In addition, we used different amounts of GST–TFIIB proteins to better compare their strength in interacting with RNAPII. In our experiments, three TFIIB mutants with the most severe start site selection defects were analyzed; two of them have novel start site profiles sharing similarity with that of ΔRPB9 (Fig. 2A). As shown in Figure 5, all these TFIIB mutants interacted with RNAPII in vitro with a similar efficiency compared with wild-type TFIIB. Under the same conditions, a non-functional TFIIB, C48S, which is known to be defective in RNAPII interaction (33), failed to interact with RNAPII (data not shown). These results, together with those published previously (15), further demonstrate that start site selection properties do not correlate with the efficiency of the TFIIB–RNAPII interaction. Although unlikely (10), it is formally possible that an altered interaction with other factors may be responsible for the start site selection defects.
An essential step in transcription initiation is the separation of the double-stranded DNA at the promoter region, a process referred to as promoter melting (34). Previous studies have suggested a scanning model in which yeast RNAPII melts the promoter ∼20 bp downstream of the TATA box and scans downstream to search for initiation sites (5,35). To determine whether the promoter melting/scanning step(s) is affected by our TFIIB mutations, we carried out an in vivo permanganate footprint assay. Permanganate preferentially reacts with thymines in single-stranded regions of DNA, a strategy that has been used previously to determine promoter melting profiles in vivo (5). Our initial efforts to determine the promoter melting pattern at the endogenous ADH1 promoter did not reveal useful information because ADH1 is constitutively expressed in yeast cells and, thus, it was not possible to detect transcription-dependent signals. To circumvent this problem, we used our ADH1–lacZ reporter gene (Fig. 3A), which contains the ADH1 core promoter fused to lacZ. Since this reporter is activated by LexA–VP16 but not LexA alone (Fig. 6A), it allowed us to detect transcription-dependent signals in the permanganate footprint assay. As shown in Figure 6B, LexA–VP16 induced permanganate-reactive bands at the ADH1 promoter of the reporter gene (compare lanes 3 and 4). However, despite some relatively minor differences, the profiles induced by LexA–VP16 in cells that contain TFIIB mutants exhibit an overall similarity to that in wild-type cells (lanes 4–6). These results suggest that the mutations in TFIIB do not cause a gross alteration at the promoter melting/scanning step.
Figure 6.
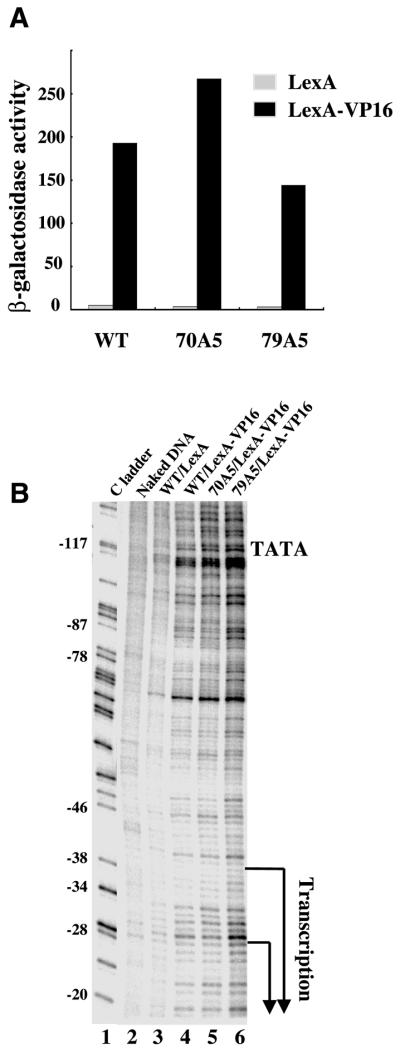
Promoter melting in cells containing various TFIIB mutants. (A) Shown are β-galactosidase activity assay results of the ADH1–lacZ reporter gene in yeast cells (grown at 30°C) containing the indicated TFIIB proteins. The reporter was under the control of either LexA alone or LexA–VP16. (B) Shown are in vivo permanganate footprint results to detect transcription-induced DNA strand separation in the promoter region of the ADH1–lacZ reporter gene. The TATA box and the two major start sites are marked. Yeast cells (grown at 30°C) either contained wild-type TFIIB (WT) or the indicated TFIIB mutants (70A5 and 79A5), with the reporter gene activated by either LexA alone or LexA–VP16. Lane 1 is a DNA sequencing C ladder. Lane 2 shows the results for yeast DNA that was treated with KMnO4 in vitro.
The biological defect of E62K can be corrected by overexpressing the mutant protein
Although our experiments described thus far suggest that the TFIIB–RNAPII interaction does not control the start site selection process by influencing PIC stability, some TFIIB mutants (such as E62K) that are defective in start site selection do have a weakened RNAPII interaction (12,13,18). To further determine whether its reduced ability to interact with RNAPII contributes to the defect of E62K in start site selection, we expressed this TFIIB mutant at different levels in yeast cells (either from a single copy plasmid or a high copy plasmid). We reasoned that if the defect of E62K in start site selection was caused by its reduced affinity for RNAPII, overexpression of this protein in yeast cells may correct, or partially correct, such a defect. Our experiments show that in yeast cells overexpressing E62K from a high copy plasmid, the transcription start site pattern of the endogenous ADH1 gene is corrected to a nearly wild-type profile (Fig. 7A, lanes 1 and 2). In addition, E62K expressed from the high copy plasmid also corrected the temperature-sensitive phenotype conferred by this protein expressed from a single copy plasmid (data not shown). However, the start site selection defect of E62K persists even when the RNAPII holoenzyme is artificially recruited to DNA (Fig. 7B). We will discuss the implications of these findings further in the Discussion.
Figure 7.
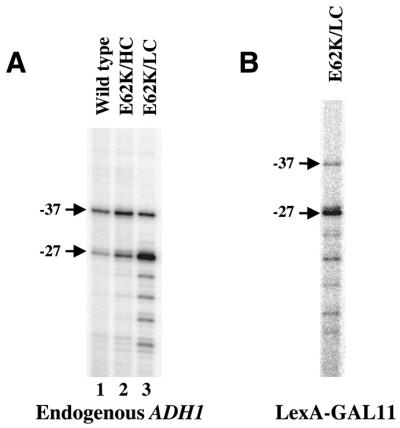
High levels of E62K can correct its start site selection defect. (A) Shown are primer extension assay results to determine the start sites of the endogenous ADH1 gene when the mutant TFIIB E62K was expressed from either a single copy (LC) or a high copy (HC) plasmid. The result for cells containing wild-type TFIIB expressed from a single copy plasmid (lane 1) is also shown. The two major start sites are marked. (B) Shown is the primer extension assay result of the ADH1–lacZ reporter gene activated by LexA–GAL11 in cells containing E62K expressed from a single copy plasmid. Yeast cells were grown at 30°C for experiments in both (A) and (B).
Given the different behaviors of E62K when expressed at different levels in cells, it is important to note that our newly generated TFIIB mutants were all expressed from high copy plasmids. Several of these mutants tested could not support cell growth when expressed from single copy plasmids, thus making it impossible to determine their behavior under these conditions. Regardless, the fact that these mutants exhibited start site selection defects even when overexpressed indicates that their defects are distinct from that of E62K. This suggestion is consistent with the finding that these newly generated TFIIB mutants, unlike E62K, can interact with RNAPII normally in vitro (Fig. 5).
DISCUSSION
The experiments described here argue against the possibility that the TFIIB–RNAPII interaction controls the start site selection process by influencing the stability of the PIC because: (i) TFIIB mutations can cause start site alterations without affecting its interaction with RNAPII (Fig. 5); (ii) the start site selection defects persist even when the RNAPII holoenzyme is more stably recruited to DNA through a covalent link to a DNA-binding domain (Fig. 4). Instead, our data are consistent with the idea that the TFIIB–RNAPII interaction controls the start site selection process by affecting the conformation of the PIC, most likely RNAPII itself. Biochemical studies have demonstrated that TFIIB, at least in a mammalian transcription system, remains associated with RNAPII until transcription initiates (36). If yeast TFIIB is also associated with RNAPII when transcription start sites are selected, TFIIB mutations in the evolutionarily conserved region that interfaces with RNAPII could readily alter the conformation and biochemical properties of RNAPII.
Our experiments with the mutant E62K suggest that TFIIB may be able to confer its start site selection properties to RNAPII prior to or during PIC formation. Unlike the newly generated TFIIB mutants described in this report (Fig. 5), E62K is impaired in RNAPII interaction (18). Although a post-assembly defect has been detected for E62K in vitro (37) (see also a similar study using E62G; 38), such a defect cannot be the underlying cause for its start site selection defect; if this were the case, increasing the amount of E62K could not have restored the defective PIC once it was already fully assembled. Our finding that the start site selection defect of E62K persists even when the RNAPII holoenzyme is more stably recruited (Fig. 7B) further argues against a PIC stability defect being responsible for the alteration of start sites. Previous studies suggest that TFIIB is the least tightly associated component of the RNAPII holoenzyme: depending on the purification procedures, TFIIB is either present or absent in the holoenzyme complex (39–43). It is possible that RNAPII can adopt different conformations depending on whether the holoenzyme recruited to the promoter contains or lacks E62K, i.e. whether E62K and RNAPII join the PIC together or separately.
An important finding of the current work is that TFIIB mutations can cause an upstream shift of start sites in vivo, suggesting that TFIIB is responsible for preventing RNAPII from utilizing DNA sites that are located both upstream and downstream of the normal start sites. It is currently unknown why some mutations cause an upstream shift of start sites while others cause a downstream shift. Start site selection defects could potentially arise from both relaxation of specificity and an altered DNA preference of RNAPII. In cells lacking RPB9 or containing our new TFIIB mutants 58A5 and 70A5, there is an increased usage of sites that are both upstream and downstream of the normal start sites of ADH1 (Fig. 2A, lanes 2, 4 and 8), suggesting a relaxed specificity. In this context, it is interesting to note that yeast RNAPII lacking the RPB9 subunit has a reduced ability to recognize and stall at potential arrest sites in vitro (19). In addition, structural studies reveal that residues or domains of RNAPII subunits important for start site selection, including RPB9 (20,23,44), are located along the projected DNA path (45,46), suggesting that their alterations can affect protein–DNA contacts.
The distance between the TATA box and the active center of yeast RNAPII is ∼30 bp in the crystal structure (35), but a transcription initiation site placed ∼30 bp downstream of the TATA box is not efficiently utilized in yeast cells (47–49). This suggests that yeast RNAPII cannot efficiently recognize or initiate transcription until its active center is translocated further downstream. A scanning model has been proposed for yeast transcription initiation where yeast RNAPII melts promoter DNA at ∼20 bp downstream of TATA and scans further downstream to initiate transcription (5). It has been shown that an intramolecular interaction exists in both human and yeast TFIIB (21,50–53). It is possible that an open conformation of yeast TFIIB is required to ‘activate’ RNAPII. Perhaps the transition from a closed to an open conformation of TFIIB would allow its N-terminal domain, which is critical for RNAPII interaction (54), to extend downstream of the promoter and, consequently, cause RNAPII to scan. Although our experiments show that TFIIB mutations that cause start site selection defects do not grossly alter the promoter melting/scanning step, the question of why yeast RNAPII needs to undergo a scanning step remains extremely important and requires further biochemical studies.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs D. Bushnell, R. Kornberg, L. Gaudreau, N. Woychick and M. Hampsey for providing us with valuable materials, and M. Ptashne and members of this laboratory for discussions and critical comments on the manuscript. This publication was made possible by NIH grant GM56215 to J.M. as well as NIH grant P30 ES06096 to the University of Cincinnati.
REFERENCES
- 1.Hampsey M., (1998) Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol. Mol. Biol. Rev., 62, 465–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tjian R., and Maniatis,T. (1994) Transcriptional activation: a complex puzzle with few easy pieces. Cell, 77, 5–8. [DOI] [PubMed] [Google Scholar]
- 3.Ptashne M., and Gann,A. (1997) Transcriptional activation by recruitment. Nature, 386, 569–577. [DOI] [PubMed] [Google Scholar]
- 4.Orphanides G., Lagrange,T. and Reinberg,D. (1996) The general transcription factors of RNA polymerase II. Genes Dev., 10, 2657–2683. [DOI] [PubMed] [Google Scholar]
- 5.Giardina C., and Lis,J.T. (1993) DNA melting on yeast RNA polymerase II promoters. Science, 261, 759–762. [DOI] [PubMed] [Google Scholar]
- 6.Struhl K., (1987) Promoters, activator proteins, and the mechanism of transcriptional initiation in yeast. Cell, 49, 295–297. [DOI] [PubMed] [Google Scholar]
- 7.Guarente L., (1987) Regulatory proteins in yeast. Annu. Rev. Genet., 21, 425–452. [DOI] [PubMed] [Google Scholar]
- 8.Ha I., Roberts,S., Maldonado,E., Sun,X., Kim,L.U., Green,M. and Reinberg,D. (1993) Multiple functional domains of human transcription factor IIB: distinct interactions with two general transcription factors and RNA polymerase II. Genes Dev., 7, 1021–1032. [DOI] [PubMed] [Google Scholar]
- 9.Berroteran R.W., Ware,D.E. and Hampsey,M. (1994) The sua8 suppressors of Saccharomyces cerevisiae encode replacements of conserved residues within the largest subunit of RNA polymerase II and affect transcription start site selection similarly to sua7 (TFIIB) mutations. Mol. Cell. Biol., 14, 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Flanagan,P.M., Tschochner,H. and Kornberg,R.D. (1994) RNA polymerase II initiation factor interactions and transcription start site selection. Science, 263, 805–807. [DOI] [PubMed] [Google Scholar]
- 11.Pinto I., Ware,D.E. and Hampsey,M. (1992) The yeast SUA7 gene encodes a homolog of human transcription factor TFIIB and is required for normal start site selection in vivo. Cell, 68, 977–988. [DOI] [PubMed] [Google Scholar]
- 12.Bangur C.S., Pardee,T.S. and Ponticelli,A.S. (1997) Mutational analysis of the D1/E1 core helices and the conserved N-terminal region of yeast transcription factor IIB (TFIIB): identification of an N-terminal mutant that stabilizes TATA-binding protein–TFIIB–DNA complexes. Mol. Cell. Biol., 17, 6784–6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinto I., Wu,W.H., Na,J.G. and Hampsey,M. (1994) Characterization of sua7 mutations defines a domain of TFIIB involved in transcription start site selection in yeast. J. Biol. Chem., 269, 30569–30573. [PubMed] [Google Scholar]
- 14.Hawkes N.A., and Roberts,S.G. (1999) The role of human TFIIB in transcription start site selection in vitro and in vivo. J. Biol. Chem., 274, 14337–14343. [DOI] [PubMed] [Google Scholar]
- 15.Pardee T.S., Bangur,C.S. and Ponticelli,A.S. (1998) The N-terminal region of yeast TFIIB contains two adjacent functional domains involved in stable RNA polymerase II binding and transcription start site selection. J. Biol. Chem., 273, 17859–17864. [DOI] [PubMed] [Google Scholar]
- 16.Faitar S.L., Brodie,S.A. and Ponticelli,A.S. (2001) Promoter-specific shifts in transcription initiation conferred by yeast TFIIB mutations are determined by the sequence in the immediate vicinity of the start sites. Mol. Cell. Biol., 21, 4427–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu W.H., Pinto,I., Chen,B.S. and Hampsey,M. (1999) Mutational analysis of yeast TFIIB. A functional relationship between Ssu72 and Sub1/Tsp1 defined by allele-specific interactions with TFIIB. Genetics, 153, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bushnell D.A., Bamdad,C. and Kornberg,R.D. (1996) A minimal set of RNA polymerase II transcription protein interactions. J. Biol. Chem., 271, 20170–20174. [DOI] [PubMed] [Google Scholar]
- 19.Awrey D.E., Weilbaecher,R.G., Hemming,S.A., Orlicky,S.M., Kane,C.M. and Edwards,A.M. (1997) Transcription elongation through DNA arrest sites. A multistep process involving both RNA polymerase II subunit RPB9 and TFIIS. J. Biol. Chem., 272, 14747–14754. [DOI] [PubMed] [Google Scholar]
- 20.Hemming S.A., Jansma,D.B., Macgregor,P.F., Goryachev,A., Friesen,J.D. and Edwards,A.M. (2000) RNA polymerase II subunit Rpb9 regulates transcription elongation in vivo. J. Biol. Chem., 275, 35506–35511. [DOI] [PubMed] [Google Scholar]
- 21.Zhang D.Y., Dorsey,M.J., Voth,W.P., Carson,D.J., Zeng,X., Stillman,D.J. and Ma,J. (2000) Intramolecular interaction of yeast TFIIB in transcription control. Nucleic Acids Res., 28, 1913–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boeke J.D., Trueheart,J., Natsoulis,G. and Fink,G.R. (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol., 154, 164–175. [DOI] [PubMed] [Google Scholar]
- 23.Hull M.W., McKune,K. and Woychik,N.A. (1995) RNA polymerase II subunit RPB9 is required for accurate start site selection. Genes Dev., 9, 481–490. [DOI] [PubMed] [Google Scholar]
- 24.Ostapenko D., and Gileadi,O. (2000) Rad25p, a DNA helicase subunit of yeast transcription factor TFIIH, is required for promoter escape in vivo. Gene, 245, 109–117. [DOI] [PubMed] [Google Scholar]
- 25.Furter-Graves E.M., Furter,R. and Hall,B.D. (1991) SHI, a new yeast gene affecting the spacing between TATA and transcription initiation sites. Mol. Cell. Biol., 11, 4121–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furter-Graves E.M., Hall,B.D. and Furter,R. (1994) Role of a small RNA pol II subunit in TATA to transcription start site spacing. Nucleic Acids Res., 22, 4932–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barberis A., Pearlberg,J., Simkovich,N., Farrell,S., Reinagel,P., Bamdad,C., Sigal,G. and Ptashne,M. (1995) Contact with a component of the polymerase II holoenzyme suffices for gene activation. Cell, 81, 359–368. [DOI] [PubMed] [Google Scholar]
- 28.Gaudreau L., Adam,M. and Ptashne,M. (1998) Activation of transcription in vitro by recruitment of the yeast RNA polymerase II holoenzyme. Mol. Cell, 1, 913–916. [DOI] [PubMed] [Google Scholar]
- 29.Farrell S., Simkovich,N., Wu,Y., Barberis,A. and Ptashne,M. (1996) Gene activation by recruitment of the RNA polymerase II holoenzyme. Genes Dev., 10, 2359–2367. [DOI] [PubMed] [Google Scholar]
- 30.Gaudreau L., Keaveney,M., Nevado,J., Zaman,Z., Bryant,G.O., Struhl,K. and Ptashne,M. (1999) Transcriptional activation by artificial recruitment in yeast is influenced by promoter architecture and downstream sequences. Proc. Natl Acad. Sci. USA, 96, 2668–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chatterjee S., and Struhl,K. (1995) Connecting a promoter-bound protein to TBP bypasses the need for a transcriptional activation domain. Nature, 374, 820–822. [DOI] [PubMed] [Google Scholar]
- 32.Ryan M.P., Stafford,G.A., Yu,L. and Morse,R.H. (2000) Artificially recruited TATA-binding protein fails to remodel chromatin and does not activate three promoters that require chromatin remodeling. Mol. Cell. Biol., 20, 5847–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez-Couto E., Klages,N. and Strubin,M. (1997) Synergistic and promoter-selective activation of transcription by recruitment of transcription factors TFIID and TFIIB. Proc. Natl Acad. Sci. USA, 94, 8036–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tachibana H., and Ishihama,A. (1985) Correlation between the rate of productive transcription initiation and the strand-melting property of Escherichia coli promoters. Nucleic Acids Res., 13, 9031–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leuther K.K., Bushnell,D.A. and Kornberg,R.D. (1996) Two-dimensional crystallography of TFIIB– and IIE–RNA polymerase II complexes: implications for start site selection and initiation complex formation. Cell, 85, 773–779. [DOI] [PubMed] [Google Scholar]
- 36.Zawel L., Kumar,K.P. and Reinberg,D. (1995) Recycling of the general transcription factors during RNA polymerase II transcription. Genes Dev., 9, 1479–1490. [DOI] [PubMed] [Google Scholar]
- 37.Cho E.J., and Buratowski,S. (1999) Evidence that transcription factor IIB is required for a post-assembly step in transcription initiation. J. Biol. Chem., 274, 25807–25813. [DOI] [PubMed] [Google Scholar]
- 38.Ranish J.A., Yudkovsky,N. and Hahn,S. (1999) Intermediates in formation and activity of the RNA polymerase II preinitiation complex: holoenzyme recruitment and a postrecruitment role for the TATA box and TFIIB. Genes Dev., 13, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y., Bjorklund,S., Kim,Y.J. and Kornberg,R.D. (1996) Yeast RNA polymerase II holoenzyme. Methods Enzymol., 273, 172–175. [DOI] [PubMed] [Google Scholar]
- 40.Wade P.A., Werel,W., Fentzke,R.C., Thompson,N.E., Leykam,J.F., Burgess,R.R., Jaehning,J.A. and Burton,Z.F. (1996) A novel collection of accessory factors associated with yeast RNA polymerase II. Protein Expr. Purif., 8, 85–90. [DOI] [PubMed] [Google Scholar]
- 41.Kimura H., Tao,Y., Roeder,R.G. and Cook,P.R. (1999) Quantitation of RNA polymerase II and its transcription factors in an HeLa cell: little soluble holoenzyme but significant amounts of polymerases attached to the nuclear substructure. Mol. Cell. Biol., 19, 5383–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koleske A.J., and Young,R.A. (1994) An RNA polymerase II holoenzyme responsive to activators. Nature, 368, 466–469. [DOI] [PubMed] [Google Scholar]
- 43.Koleske A.J., and Young,R.A. (1995) The RNA polymerase II holoenzyme and its implications for gene regulation. Trends Biochem. Sci., 20, 113–116. [DOI] [PubMed] [Google Scholar]
- 44.Hemming S.A., and Edwards,A.M. (2000) Yeast RNA polymerase II subunit RPB9. Mapping of domains required for transcription elongation. J. Biol. Chem., 275, 2288–2294. [DOI] [PubMed] [Google Scholar]
- 45.Cramer P., Bushnell,D.A. and Kornberg,R.D. (2001) Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science, 292, 1863–1876. [DOI] [PubMed] [Google Scholar]
- 46.Gnatt A.L., Cramer,P., Fu,J., Bushnell,D.A. and Kornberg,R.D. (2001) Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 Å resolution. Science, 292, 1876–1882. [DOI] [PubMed] [Google Scholar]
- 47.Healy A.M., and Zitomer,R.S. (1990) A sequence that directs transcriptional initiation in yeast. Curr. Genet., 18, 105–109. [DOI] [PubMed] [Google Scholar]
- 48.Nagawa F., and Fink,G.R. (1985) The relationship between the TATA sequence and transcription initiation sites at the HIS4 gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 82, 8557–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hahn S., Hoar,E.T. and Guarente,L. (1985) Each of three TATA elements specifies a subset of the transcription initiation sites at the CYC-1 promoter of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 82, 8562–8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu W.H., and Hampsey,M. (1999) An activation-specific role for transcription factor TFIIB in vivo. Proc. Natl Acad. Sci. USA, 96, 2764–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts S.G., and Green,M.R. (1994) Activator-induced conformational change in general transcription factor TFIIB. Nature, 371, 717–720. [DOI] [PubMed] [Google Scholar]
- 52.Bangur C.S., Faitar,S.L., Folster,J.P. and Ponticelli,A.S. (1999) An interaction between the N-terminal region and the core domain of yeast TFIIB promotes the formation of TATA-binding protein–TFIIB–DNA complexes. J. Biol. Chem., 274, 23203–23209. [DOI] [PubMed] [Google Scholar]
- 53.Hawkes N.A., Evans,R. and Roberts,S.G. (2000) The conformation of the transcription factor TFIIB modulates the response to transcriptional activators in vivo. Curr. Biol., 10, 273–276. [DOI] [PubMed] [Google Scholar]
- 54.Buratowski S., and Zhou,H. (1993) Functional domains of transcription factor TFIIB. Proc. Natl Acad. Sci. USA, 90, 5633–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]