


Abstract
EDEN-BP (embryo deadenylation element-binding protein) binds specifically to the EDEN motif in the 3′-untranslated regions of maternal mRNAs and targets these mRNAs for deadenylation and translational repression in Xenopus laevis embryos. EDEN-BP contains three RNA recognition motifs (RRMs) and is related to the elav family of RNA-binding proteins. In the present study we show that the two N-terminal RRMs of EDEN-BP are necessary for the interaction with EDEN as well as a part of the linker region (between RRM2 and RRM3). Using a band shift assay we show that two different complexes are formed according to the size and, therefore, the functional nature of the EDEN motif. Finally, we show that EDEN-BP can form a dimer in a two-hybrid assay. Accordingly, we suggest that the functional configuration of EDEN-BP is a dimer.
INTRODUCTION
In most species transcription is silent during the first hours of embryonic development. Accordingly, during this period the post-transcriptional regulation of gene expression is especially important. A number of studies have shown that the recruitment or release of many maternal mRNAs into or from the polysomes is correlated, respectively, with the lengthening or shortening of the poly(A) tail (reviewed in 1).
Several RNA elements that control poly(A) tail length have been identified, both in somatic cells and in the oocytes and embryos of several species. In Xenopus the cytoplasmic polyadenylation element (CPE) (2,3) or in the mouse the adenylation control element (ACE) (4) is required for polyadenylation and polysome recruitment during oocyte maturation. The CPE is also involved in translational regulation of mRNA in neurones (5,6). The AU-rich element (ARE) promotes the rapid deadenylation of many short-lived mRNAs in somatic cells (7,8) and in Xenopus embryos (9,10). The embryo deadenylation element (EDEN) confers a deadenylation behaviour on host RNAs in Xenopus embryos (11,12) and probably in somatic cells (13). This element acts as a translational repressor in Drosophila oocytes and in Drosophila and Xenopus embryos (13,14). The trans-acting factor, EDEN-binding protein (EDEN-BP), that binds to this motif has been identified (15) and cloned (12). EDEN-BP is a 53 kDa protein necessary for the EDEN-dependent deadenylation process in Xenopus embryo cell-free extracts (12).
EDEN-BP contains three RNA recognition motifs (RRMs) (16–18). The most highly conserved motifs in these 80–90 amino acid domains are the ribonucleoprotein 1 and 2 (RNP1 and RNP2) sequences, which have been shown to interact directly and specifically with RNA (19–21). The disposition of the three RRMs in EDEN-BP is similar to that found in RNA-binding proteins of the elav family, i.e. two consecutive RRMs (RRM1 and RRM2) situated in the N-terminal region followed by a linker region and the third RRM (RRM3) close to the C-terminus of the protein. Recently, Good et al. (22) identified a phylogenetically related sub-population of the elav-related proteins. This is the family of bruno-related RNA-binding proteins that includes EDEN-BP, also named Xenopus laevis bruno-like protein-2 (Xl BruL 2).
Within the framework of our ongoing programme to characterise the functionally active EDEN-BP complex we have determined the domains of this protein that are required for EDEN-specific binding. This data highlighted a need for a portion of the linker region, suggesting dimerisation of EDEN-BP on the cognate RNA. To substantiate this hypothesis we show that the number of U/purine repeats affects the electrophoretic mobility of the RNA–protein complex and that EDEN-BP can form a dimer in a yeast two-hybrid assay. We propose that EDEN-BP is a dimer in the deadenylation-proficient complex targeted by the EDEN motif.
MATERIALS AND METHODS
Cloning of recombinant proteins
The plasmid vectors expressing full-length EDEN-BP (protein F) and the deletion mutants (proteins A–E, 3 and 5) were constructed by PCR amplification of the appropriate regions of EDEN-BP cDNA (12). Pfu polymerase and the oligonucleotide primers listed below were used. After denaturation for 1 min at 94°C, the primers were annealed for 1 min at the following temperatures: E, 43°C; B–D and 5, 57°C; A and 3, 67°C; F, 74°C. Thirty elongation cycles were performed for 100 s at 72°C. To add an adenosine residue at each 3′ extremity the PCR products were incubated for 30 min at 72°C in the presence of Taq polymerase. Each PCR product was cloned in the PinPoint Xa 1-T vector (Promega). All the plasmid constructions were confirmed by sequencing of the complete open reading frames (ORFs).
The PCR primers were designed to add a histidine tag at the C-terminus of the recombinant proteins and to place the N-terminal extremity of the recombinant proteins in-frame with the biotinylable peptide encoded by the vector. The forward primer for A–F was 5′-TAA TGG CAC AAT GGA CCA CCC AGA CCA T-3′. The reverse primers were: A, 5′-TTA GTG ATG GTG ATG GTG ATG ACT AGT GAA AGT AAT AAA ACA GCA TCC-3′; B, 5′-TTA GTG ATG GTG ATG GTG ATG ACT AGT CGT AAC GAA TGC ACA TCC-3′; C, 5′-TTA GTG ATG GTG ATG GTG ATG ACT AGT CCC AGA GGA GGC GGT CTG-3′; D, 5′-TTA GTG ATG GTG ATG GTG ATG ACT AGT AGC TCC TAG GGA TGC CAT-3′; E, 5′-TTA GTG ATG GTG ATG GTG ATG ACT AGT GTT GGC TCC TTC TGG GCC-3′; F, 5′-TTA GTG ATG GTG ATG GTG ATG TAC GTA GGG TTT GCT GTC ATT-3′. The forward primer used to clone N-terminal deletion mutants of EDEN-BP (proteins 3 and 5) was 5′-TTA GTG ATG GTG ATG GTG ATG TAC GTA GGG TTT GCT GTC ATT-3′. The reverse primers were: 3, 5′-GGG GGC TAG CAA CCT CAA CTC CCT AAG T-3′; 5, 5′-GGG GGC TAG CTA CAC AAG AAA AGC TGC G-3′.
Expression and purification of mutant EDEN-BP
Bacteria (Sure; Stratagene) were transformed with the different EDEN-BP-derived constructs. Cells were grown in LB medium containing ampicillin (200 µg/ml) at 37°C to an A600 of 0.4, induced with isopropyl-β-d-thiogalactopyranoside (5 mM) and harvested 2 h later. The cells were suspended in 10 mM HEPES (pH 7.5), 300 mM NaCl, 50 mM sucrose, 10 mM imidazole, 1% Triton X-100 and protease inhibitors (leupeptin, chymostatin and pepstatin at 0.1 mg/ml final concentration), lysed by sonication and centrifuged for 30 min at 14 600 g. The supernatant was loaded onto a HiTrap chelating column (Pharmacia) activated with nickel cations. The nickel columns were washed with 50 ml of loading buffer (lysis buffer without Triton X-100). Bound proteins were eluted with an imidazole concentration gradient from 0 to 500 mM and collected on ice. Each fraction was conserved at –20°C with glycerol (50%), BSA (10 µg/ml final) and protease inhibitors (leupeptin, pepstatin and chymostatin at 0.1 mg/ml each). The quality and quantity of the recombinant proteins were verified by 12% SDS–PAGE and Coomassie blue staining.
Xenopus egg extracts
Egg extracts were prepared essentially as described by Murray and Kirschner (23) using versilube F-50 oil (General Electric) to remove the buffer solution before crushing the biological material by centrifugation. The cytosol layer was clarified by a further centrifugation at 10 000 g for 15 min and stored at –70°C.
RNA synthesis
The transcription template used to obtain the RNA containing the c-mos EDEN used in the UV-induced label transfer assays was generated by PCR using the oligonucleotides T7EDENmos (5′-GCGTAATACG ACTCACTATA GGG CGATATA TGTATGTGTT GTTT-3′) and SP6EDENmos (5′-GCGATTTAGG TGACACTATA GAATACAGCA CACACACACA CATA-3′) and the plasmid pGbORF/mosEDEN (12) as PCR template. The PCR product contains both T7 and SP6 promoters that allow the synthesis of c-mos EDEN RNA and antisense c-mos EDEN RNA, respectively. Oligonucleotide transcription templates were used to synthesise the RNAs used in the RNA gel shift assay [N36, N14-(UGUA)4-N14, N14-(UGUA)9-N14 c-mos, Eg5 and Eg5S3]. They were produced by hybridising an oligonucleotide containing the T7 promoter sequence (T7pr) with an oligonucleotide containing the sequence to be transcribed (24). The oligonucleotides T7pr, Eg5 and Eg5S3 have been described previously (12). The other oligonucleotide sequences were as follows: N36, 5′-(N)36CTATAGTGAG-3′; (UGUA)4, 5′-(N)10(TACA)4(N)10CTATAGTGAG-3′; (UGUA)9, 5′-(TACA)9CTATAGTGAG-3′; MOS, 5′-AGAA GAGCAC ACACACACAC ATAAAACAAC ACATA CATAT AAAACACTCT ATAGTGAG-3′. Briefly, the oligonucleotide T7pr was phosphorylated and hybridised to a template oligonucleotide. Then, the oligonucleotides were ligated, phenol extracted, precipitated and used as transcription templates.
32P-labelled uncapped transcripts were made in vitro with the Promega Riboprobe transcription kit as previously described (15). The RNAs were phenol/chloroform extracted and ethanol precipitated.
UV-induced label transfer and western blot analyses
For UV-induced label transfer or UV crosslinking, egg extracts were diluted 10-fold in reaction buffer (10 mM HEPES pH 7.5, 1 mM MgCl2, 0.1 mM CaCl2 and 100 mM KCl) as described previously (15) with the addition of heparin (10 mg/ml), tRNA (1 mg/ml) and dithiothreitol (5 mM). The different purified recombinant proteins (∼40 ng/sample) were incubated under the same conditions.
32P-labelled RNAs (50 fmol) containing the c-mos EDEN RNA motif or antisense EDEN motif were added to these reaction solutions, which were processed for UV-induced label transfer as previously described (15). The radiolabelled proteins were separated by electrophoresis on 12% denaturing polyacrylamide gels. After migration, the gels were transferred to nitrocellulose membranes. Western analysis was performed using anti EDEN-BP antibodies (12) and revealed with anti-rabbit alkaline phosphatase antibody (1/30 000; Sigma) and NBT/BCIP reagent. The membranes were then exposed for autoradiography.
RNA gel shift
Aliquots of 100 fmol in vitro synthesised, radiolabelled RNA were incubated at 25°C for 30 min in XB buffer (10 mM HEPES–KOH pH 7, 100 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2) with energy mix (7.5 mM creatine phosphate, 1 mM ATP, 1 mM MgCl2, 0.1 mM EGTA, pH 7.7), 5 mM DTT, 4 U/µl RNAsin, 10 mg/ml heparin, 5 mg/ml yeast tRNA with or without 0.5 µl of 4 h post-fertilisation embryo extract. Samples were resolved on a 5% polyacrylamide gel (acrylylamide/bisacrylamide 60:1) in 0.25× TBE. The gel was dried and autoradiographed.
In coupled UV-induced label transfer/RNA gel shift experiments the gel shift samples were UV irradiated before resolution on a native polyacrylamide gel. After electrophoresis the RNA–protein complexes were localised by autoradiography of the wet gel and the appropriate regions excised. The gel slabs were soaked for 30 min at 20°C in a RNase A solution (5 µg/µl) in 10 mM Tris–HCl pH 8, 1 mM EDTA and then inserted into the wells of a denaturing SDS–polyacrylamide gel. After electrophoresis the gel was dried and autoradiographed.
Yeast two-hybrid assay
pAD/IRP, a plasmid expressing the rabbit iron regulatory protein fused to the Gal4 activation domain was a gift from M. Wickens (University of Madison, Madison, WI) (25); the parent vector was pACT2. pGBT9/EDEN-BP was produced by subcloning the entire ORF of EDEN-BP (12) into the EcoRI and SalI sites of pGBT9. pAD/EDEN-BP was produced by subcloning the EDEN-BP ORF, isolated from pGBT9/EDEN-BP by restriction digestion of this plasmid with EcoRI followed by Klenow treatment and a second digestion with SalI. The insert was subcloned into the pACT2 vector first digested with XmaI and Klenow treated and then digested with XhoI. Sequences were confirmed by sequencing. Cells of the strain HF7c [MATa ura3-52 his3-200 ade2-101 lys2-801 trp1-901 leu2-3,112 gal4-542 gal80-538 LYS2::GAL1-HIS3 URA3::(GAL4 17mers)3-CYC1-lacZ] (26) were transformed with pGBT9/EDEN-BP. Transformants were selected and transformed with pAD/IRP or pAD/EDEN-BP. Cells bearing both pGBT9/EDEN-BP and pAD/IRP or pGBT9/EDEN-BP and pAD/EDEN-BP were selected on medium lacking leucine and tryptophan. Individual clones were picked and analysed for their ability to activate His3 expression in the presence of several concentrations of 3-aminotriazole (3-AT) at 30°C. Plates were photographed.
RESULTS
Domain mapping of EDEN-BP shows sequence-specific binding
To map the region of EDEN-BP that is required for sequence-specific binding to the RNA motif EDEN, the complete reading frame and five recombinant proteins with C-terminal deletions and two recombinant proteins with N-terminal deletions (Fig. 1A) were produced in bacteria. These proteins were obtained by cloning the appropriate PCR products in the PinPoint Xa 1-T vector as described in Materials and Methods. The C-terminal deletions consist of consecutive 83–88 amino acid deletions without separating the RNP1/RNP2 motifs of any RRM (see Fig. 1A). The N-terminal deleted recombinant proteins (proteins 3 and 5 in Fig. 1A) contained, respectively, amino acids 238–489 and 65–489 of EDEN-BP. All of these proteins possess a biotinylable tag (13 kDa) at the N-terminal extremity and a His6 tag (0.84 kDa) at the C-terminus (Fig. 1A).
Figure 1.

Analysis of the RNA binding specificity of full-length, N-terminal and C-terminal deletion mutants of EDEN-BP. (A) Schematic representation of mutant recombinant EDEN-BP. All constructs were tagged at the N-terminus with a biotinylable peptide (Bio-Pep) and at the C-terminus with a histidine tag (6×His). For simplicity these tags are only shown for protein F. The predicted molecular weight and name of each of these tagged proteins are indicated on the right. The positions of the RRMs are indicated (R1, R2 and R3) above the schematic proteins as well as the number of the boundary amino acids relative to the wild-type protein without the biotinylable peptide. The individual RNP motifs are represented by dark boxes. (B) UV-induced label transfer of the recombinant proteins and wild-type EDEN-BP to RNA containing (+) or devoid of (–) the EDEN motif. After irradiation with UV light and treatment with RNase A, the proteins were resolved by electrophoresis in the presence of SDS. Proteins were transferred to nitrocellulose membranes and the radiolabelled proteins were revealed by autoradiography. The asterisk indicates the position of the radioactive signal. (C) The same membranes were probed with purified anti-EDEN-BP antibodies. Lane indications and numbers are as for (B). The positions of the molecular weight markers (kDa) are indicated on the left of each panel. The asterisk indicates the position of the recombinant EDEN-BP protein.
Specific binding of these recombinant proteins to the EDEN motif was analysed by UV-induced label transfer. The purified recombinant proteins were incubated with [32P]UTP-labelled in vitro transcripts containing either the c-mos EDEN or the antisense RNA sequence and processed for UV-induced label transfer analysis as described in Materials and Methods.
During purification of the recombinant proteins we observed that they were relatively unstable and were rapidly degraded in certain preparations. Addition of carrier protein, such as BSA, to the purified protein did not systematically alleviate this problem. Therefore, in all the experiments described here the integrity of the proteins after UV-induced label transfer was verified by western blotting. After separation by SDS–PAGE the UV-irradiated proteins were transferred to nitrocellulose membrane and revealed with anti-EDEN-BP antibodies (Fig. 1C). The membrane was then exposed for autoradiography (Fig. 1B). As a control, the signals obtained with the recombinant proteins (Fig. 1B, lanes 3–18) were compared with that produced by maternal Xenopus EDEN-BP present in activated egg extracts (Fig. 1B, lanes 1 and 2).
As expected, the maternal EDEN-BP only produced a UV-induced radiolabelling signal with the sense EDEN motif (Fig. 1B, compare lanes 1 and 2). The full-length recombinant protein F (Fig. 1B, lanes 3 and 4) showed the same pattern of the UV-induced radiolabelling signal. The western blot data (Fig. 1C) confirm that the absence of UV-induced radiolabelling signal for the maternal EDEN-BP or recombinant protein F incubated with the antisense EDEN transcript was not due to an absence of protein.
Protein E, from which essentially all of RRM3 is deleted, still binds specifically to the EDEN motif (Fig. 1B, lanes 5 and 6). Sequence-specific binding was also retained in protein D (lanes 7 and 8), from which ∼80 amino acids of the 251 amino acid linker region had been removed. However, further shortening of the recombinant proteins (proteins A–C) abrogated RNA binding (Fig. 1B, lanes 9–14). Neither of the N-terminal deleted proteins (proteins 3 and 5) produced a radiolabelled product with either the sense or the antisense EDEN (Fig. 1B, lanes 15–18).
The western blot data obtained using EDEN-specific antibodies (Fig. 1C) shows that the lack of radioactive product for proteins A–C, 3 and 5 was due to a loss of RNA binding and not to a degradation of the recombinant proteins during the UV-induced label transfer.
The epitopes of the anti-EDEN-BP polyclonal antibodies used for these western analyses are not known. Therefore, it is not possible to make a quantitative comparison between the recombinant proteins. Indeed, these antibodies produced only a weak signal for proteins A and B (see Fig. 1C, lanes 11–14), whereas Coomassie Blue staining of these same samples on a parallel SDS–PAGE (data not shown) showed that the amount of recombinant protein in these samples was greater than that for the other proteins analysed in this experiment.
Therefore these data show that binding to the c-mos EDEN requires both of the N-terminal RRM of EDEN-BP and a major part of the linker region.
RNA gel shift analysis of EDEN-BP binding
We have previously shown that contiguous UGUA repeats can constitute a functional synthetic EDEN so long as they contain a sufficient number of repetitions (27). Four (16 nt) and six (24 nt) repeats were ineffective in conferring deadenylation on a RNA; nine repetitions (36 nt) conferred efficient deadenylation. Furthermore, the two fully characterised natural EDENs (those of the Eg5 and c-mos mRNA) clearly contain two distinct U/purine domains (see Fig. 2A). However, the crystal structure of the RNA-binding domains of HuD and sexlethal with their associated RNA show that the two tandem RRMs of these proteins only occupy 11 and 12 nt, respectively (28,29). These considerations, in addition to the need for a portion of the linker region in the binding experiments, suggested that EDEN-BP might form a dimer when active. If this were the case then the size of the EDEN-BP/EDEN complex should depend on the length of the sequence to which EDEN-BP is bound.
Figure 2.

Analysis of the EDEN-BP/UGUA repeat complex. (A) Sequence of the RNA molecules. U/purine dinucleotides are in bold. The minimal c-mos EDEN that is efficient in deadenylation is underlined. (B) RNA gel shift analysis. The indicated radiolabelled RNAs were incubated with (+) or without (–) 4 h post-fertilisation embryo extract. The complexes formed were analysed by native PAGE. The position of the free probes are indicated as well as the position of the two complexes formed: Upper (U) and Lower (L). (C) Coupled UV-induced label transfer and RNA gel shift. Lanes 1 and 2, UV-induced label transfer analysis of Eg5 and the mutant Eg5S3 RNAs incubated in embryo extracts. Lanes 3 and 4, gel slabs corresponding to the positions of arrows U and L in lane 5 were excised, treated with RNase A and deposited as indicated in the wells of a denaturing SDS–polyacrylamide gel. After electrophoresis the gel was dried and the radiolabelled proteins revealed by autoradiography. Lane 5, The Eg5 RNA was incubated in embryo extracts and UV irradiated prior to separation of the protein–RNA complexes on a native polyacrylamide gel and autoradiography of the unfixed wet gel. At the bottom of the figure, D indicates denaturing gel and N indicates native gel.
Accordingly, band shift experiments were performed using cell-free embryo extracts and radiolabelled RNAs corresponding to synthetic EDENs of different size as well as the c-mos- and Eg5-derived EDENs (Fig. 2A). In order to be able to directly compare the size of the complexes formed, all the RNAs contained approximately the same number of nucleotides. We have previously shown that UV-induced label transfer using Eg5 and c-mos EDEN gives a unique radioactive signal, that of EDEN-BP (12).
The results of the band shift experiment given in Figure 2B show that the c-mos EDEN (lanes 7 and 8) forms a unique complex (U) when incubated in cytoplasmic embryo extracts. The functional synthetic EDEN, (UGUA)9, forms a complex of identical size (lanes 5 and 6). The shorter non-functional synthetic EDEN, (UGUA)4 (lanes 3 and 4), produces a faster moving complex (L). Both complexes U and L are observed with the Eg5-derived EDEN.
To confirm that these complexes contain EDEN-BP, coupled UV-induced label transfer/RNA gel shift experiments were performed using Eg5, as this RNA produced both complexes U and L in the RNA gel shift assay. As shown in Figure 2C (lane 1), UV irradiation of embryo extracts containing 32P-labelled Eg5 RNA yielded a single radioactive protein at 53 kDa. This protein, which was previously identified as EDEN-BP (12), was absent when the Eg5S3 RNA was used (Fig. 2C, lane 2). The Eg5S3 RNA contains a mutation in the EDEN motif that abrogates EDEN-BP binding (12). For the coupled UV-induced label transfer/RNA gel shift experiments the embryo extracts containing the 32P-labelled Eg5 RNA were UV irradiated prior to separation on a native polyacrylamide gel (Fig. 2C, lane 5). This treatment did not modify migration of the RNA–protein complexes (data not shown). In these experiments the separation of the protein–RNA complexes (Fig. 2C, lane 5) was less apparent, for technical reasons, than that shown in Figure 2B (lane 10). First, more radioactivity had to be loaded in order to retain a detectable signal after the second electrophoretic separation and, secondly, autoradiography of the wet gel led to a loss of resolution. Irrespective of this loss of separation the RNA probe is clearly shifted to an extended region similar to that covered by complexes U and L (Fig. 2B, lane 10). Bands corresponding to the upper (U) and lower (L) parts of this extended region were excised and treated as described in Materials and Methods. The data given in Figure 2C (lanes 3 and 4) show that protein–RNA complexes within both parts of this extended region contained a radiolabelled protein that co-migrated with EDEN-BP.
In contrast to the c-mos EDEN, the two parts of the Eg5-derived EDEN are not comparable in U/purine richness (see Fig. 2A). The c-mos EDEN contains two blocks of six and eight U/purine repeats whereas the Eg5 EDEN has a 5′ section of seven U/purine repeats and a 3′ section with only four U/purine repeats. We interpret the RNA gel shift data as indicating that the (UGUA)4 RNA binds one molecule of EDEN-BP to give the lower complex L. When the EDEN is lengthened to (UGUA)9, two EDEN-BP molecules can associate with the same RNA to give complex U. This complex is also observed with the c-mos EDEN, that is similar in size to the (UGUA)9 RNA (Fig. 2B). We propose that the unbalanced nature of the Eg5 EDEN leads to the formation of both complex U (dimer) and L (monomer). As (UGUA)4 is not functional whereas the (UGUA)9 EDEN is capable of conferring a deadenylation behaviour this interpretation implies that the functional form of EDEN-BP is a dimer.
EDEN-BP can dimerise in a two-hybrid assay
In order to determine whether EDEN-BP can form a dimer, a two-hybrid screen protocol was set up. Two-hybrid analysis allows the strength of a protein–protein interaction to be assessed by the reconstitution of a bipartite transcription factor. This bipartite transcription factor is formed by two proteins: one is a fusion between the first protein to be tested and the GAL4 DNA-binding domain; the second protein is composed of the other protein to be tested (here EDEN-BP) fused to the GAL4 activation domain. When the two tested proteins interact, a functional transcription factor is formed and the activity of reporter gene (HIS3) can be determined by its capacity to permit growth of yeast cells on medium lacking histidine. The strength of the interaction can be evaluated by adding 3-AT, a competitive inhibitor of the HIS3 gene. The concentration of 3-AT still allowing growth of the yeast cells is an indication of the strength of the interaction.
HF7c yeast cells expressing the DNA-binding domain/EDEN-BP fusion protein (pGBT9/EDEN-BP) were transformed with either the activating domain/EDEN-BP fusion protein (pAD/EDEN-BP) or the activating domain/iron regulatory fusion protein (pAD/IRP) as a control. When plated on agar plates lacking leucine and tryptophan to select for the presence of both plasmids PGBT9/EDENBP and pAD/EDEN-BP or pAD/IRP, both sets of yeast cells grew (Fig. 3, –LW). In the absence of histidine, leucine and tryptophan (–HLW), which tests for transcriptional activation of the HIS3 gene, growth was detected only for the yeast cells expressing both forms of the EDEN-BP fusion protein. This indicates that the two EDEN-BP-containing fusion proteins interact. In order to assess the strength of this interaction cells were plated on medium containing increasing concentrations of 3-AT. Growth was still observed with 10 mM 3-AT, indicating a robust interaction between the two EDEN-BP-containing proteins. Growth was lost at 20 mM 3-AT.
Figure 3.
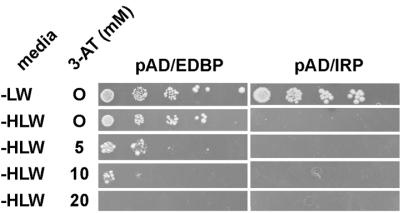
EDEN-BP dimerises in a yeast two-hybrid assay. HF7c cells that had been transformed by pGBT9/EDEN-BP (DNA-binding domain/EDEN-BP fusion protein) were subsequently transformed with either pAD/EDEN-BP (activating domain/EDEN-BP fusion protein) or pAD/IRP (activating domain/iron regulatory fusion protein) as indicated at the top of the figure. Serial dilutions (1/10) of individual clones were replica plated on the indicated medium (–LW, medium lacking leucine and tryptophan; –HLW, medium lacking histidine, leucine and tryptophan). When used, the concentration of 3-AT was as indicated (5, 10 and 20 mM).
DISCUSSION
We report here experiments demonstrating that sequence-specific binding of the Xenopus protein EDEN-BP requires the two N-terminal RRMs and at least the first 167 amino acids of the linker region. This need for a portion of the linker, combined with the previous observation that a 16 nt U/purine repeat was insufficient to constitute a functional EDEN (27) and that an 11 or 12 nt RNA is associated, respectively, with the two tandem RRMs of HuD and sexlethal (28,29), suggested that the active configuration of EDEN-BP may be a dimer. In agreement with this hypothesis we observed, using a RNA gel shift assay, that two complexes are formed on U/purine repeats of different size; functional EDENs [c-mos, Eg5 and (UGUA)9] produce a complex with a reduced electrophoretic mobility when compared to the complex formed on the short (non-functional) U/purine repeats. Lastly, EDEN-BP was shown to form a dimer in a two-hybrid assay.
We interpret the two complexes formed in the gel shift assay as representing monomers and dimers of the full-length EDEN-BP. This would appear to be incompatible with the initial hypothesis that the loss of UV-induced label transfer, for proteins shortened 5′ to amino acid 320, may be due to the inability of these proteins to form dimers. However, these two experimental approaches do not probe the same structural features of the RNA–protein complex. In the UV-induced label transfer the RNA is of optimum length (c-mos EDEN) and the proteins are of variable size. In contrast, in the gel shift assay the protein contains its full complement of RRMs and the RNAs are of variable length. Maintenance of the EDEN-BP monomer on the (UGUA)4 and Eg5 RNAs could be due to non-specific binding of the protein to the RNA, for instance via RRM3, such that the binding of the monomer is sufficiently reinforced to allow it to be visualised in the denaturing gel. This non-specific binding would be minimalised with the C-terminal deleted proteins.
EDEN-BP has the same overall organisation as the RNA-binding proteins of the elav family. A number of the human and mouse Hu RNA-BPs, which are related to elav by sequence homology, have been analysed with respect to the role of the different RRMs in RNA binding. In most cases, specific RNA binding required RRM1 and all or part of RRM2 (30–33). Evidence has also been reported that RRM1 is mainly responsible for binding and RRM2 enhances the interaction (30,31). This functional pattern for the different RRMs in the Hu family of elav-related proteins is compatible with the results described here for EDEN-BP.
Recently, Good et al. (22) identified by sequence comparison a family of RNA-binding proteins, of which EDEN-BP is a member, that are more closely related to the Drosophila protein bruno than to the Hu elav-related proteins. The binding specificity of the RRMs for several of these proteins has been reported. Binding of the Drosophila protein bruno (49% identity with EDEN-BP in the overlapping region) to the bruno response element was observed for a fragment containing RRM3 and the distal half of the linker (34). For the human CUG-BP, which is more closely related to EDEN-BP (87% identity), Timchenko et al. (35) reported that RRM1 and RRM2 were necessary and sufficient for binding to a (CUG)8 motif. Using a three-hybrid assay, Takahashi et al. (36) showed that this same protein bound much more strongly to UG than to CUG repeats. RRM1 and RRM3 were dispensable for binding to the RNA containing 36 UG repeats. The binding to this RNA was partially reduced by deleting RRM2 and most seriously affected by deleting the linker region. The Zebrafish protein Brul, which is 81% identical to EDEN-BP, also shows a preference for UG-rich RNAs (37). However, using a RNA containing eight contiguous U/purine dinucleotides these authors observed that RRM1 and RRM2 together or RRM3 alone could bind specifically to this RNA.
The divergence between the previously published results for bruno, CUG-BP and Brul and the data reported here for EDEN-BP resides mainly in the role of the third RRM and the linker region. There are several dissimilarities in these experiments that could, individually or together, lead to the observed differences.
First, only the UV-induced label transfer data reported for bruno (34) and that reported here were obtained with a naturally occurring sequence element. In addition, the other RNAs are of insufficient length to allow the formation of a dimer. This may possibly explain why a dependence on the linker region was not observed in other studies with the exception of the three-hybrid analysis (36).
Secondly, the cDNA used to produce the recombinant EDEN-BP was of maternal origin (12) whereas CUG-BP was cloned from human adult tissue cDNA libraries (38) and Brul was cloned from a 20–28 h post-fertilisation Zebrafish library (39). We have identified several isoforms of EDEN-BP in Xenopus embryos that show different patterns of expression during development (Y. Audic, unpublished data). Therefore, the cloned Brul cDNA may well correspond to a zygotic rather than a maternal isoform.
Although these proteins show almost identical binding specificity in vitro, their function may be different in vivo. Indeed, we have shown that EDEN-BP is a deadenylation specificity factor (12) whereas the only identified functions for CUG-BP and Brul are in regulating splicing or translation independently of the poly(A) tail (37,40–43). These differences in function could require different associations of the RNA-binding protein with the target mRNA. Although bruno is also of maternal origin and acts as a translational repressor (44) it is only 49% identical to EDEN-BP in the overlapping region and there is no evidence to date that it acts as a deadenylation factor.
The human orthologue of EDEN-BP, the protein CUG-BP, has been shown to be a phosphoprotein (45) and we have evidence (L.Detivaud, G.Pascreau, H.B.Osborne and J.Kubiak, submitted for publication) that EDEN-BP is also phosphorylated. As the full-length recombinant protein (protein F) used in the present study was expressed in bacteria we can assume that phosphorylation is not required for sequence-specific binding to the EDEN motif. These post-translational modifications may, however, be important for the transactivation of deadenylation by EDEN-BP.
We have previously shown that 8 or 12 U/purine repeats (16 or 24 nt) are ineffective in conferring deadenylation on a reporter RNA whereas a 36 nt U/purine motif is much more efficient (27). The proposed dimerisation of EDEN-BP could explain this length effect of the EDEN motif. For instance, the dimer may have a greater affinity for the RNA element than the sum of the individual affinities. Alternatively, the EDEN-BP dimer may form an interface that is required for interaction with necessary cofactors. Eventually the association of the EDEN-BP dimer with the RNA could induce a conformational change in the protein that unmasks the interaction interface. A precedent for such a scenario is the functional complex formed by two U1A proteins and the cognate RNA (46). A consequence of this dimer scenario is that the different isoforms of EDEN-BP could form a variety of heterodimers. It will be of interest to determine whether different heterodimers favour one or the other of the processes in which the homologues of EDEN-BP have been implicated.
Acknowledgments
ACKNOWLEDGEMENTS
This work has been supported by the Centre National de la Recherche Scientifique and by Research Grants from the European Union (contract QLK3-CT-2000-00721) and the Association pour la Recherche sur le Cancer (ARC no. 9529). S.B.-C. was supported by a stipend from the Conseil Regional de Bretagne.
REFERENCES
- 1.Stebbins-Boaz B. and Richter,J.D. (1997) Translational control during early development. Crit. Rev. Eukaryot. Gene Expr., 7, 73–94. [DOI] [PubMed] [Google Scholar]
- 2.Fox C.A., Sheets,M.D. and Wickens,M. (1989) Poly(A) addition during maturation of frog oocytes: distinct nuclear and cytoplasmic activities and regulation by the sequence UUUUUAU. Genes Dev., 3, 2151–2162. [DOI] [PubMed] [Google Scholar]
- 3.McGrew L.L., Dworkin-Rastl,E., Dworkin,M.B. and Richter,J.D. (1989) Poly(A) elongation during Xenopus oocyte maturation is required for translational recruitment and is mediated by a short sequence element. Genes Dev., 3, 803–815. [DOI] [PubMed] [Google Scholar]
- 4.Huarte J., Stutz,A., O’Connel,M.L., Gubler,P., Belin,D., Darrow,A.L., Strickland,S. and Vassali,J.-D. (1992) Transient translational silencing by reversible mRNA deadenylation. Cell, 69, 1021–1030. [DOI] [PubMed] [Google Scholar]
- 5.Wu L., Wells,D., Tay,J., Mendis,D., Abbott,M., Barnitt,A., Quinlan,E., Heynen,A., Fallon,J. and Richter,J. (1998) CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron, 5, 1129–1139. [DOI] [PubMed] [Google Scholar]
- 6.Wells D.G., Dong,X., Quinlan,E.M., Huang,Y.S., Bear,M.F., Richter,J.D. and Fallon,J.R. (2001) A role for the cytoplasmic polyadenylation element in NMDA receptor-regulated mRNA translation in neurons. J. Neurosci., 21, 9541–9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson T. and Treisman,R. (1988) Removal of poly(A) and consequent degradation of c-fos mRNA facilitated by 3′ AU-rich sequences. Nature, 336, 396–399. [DOI] [PubMed] [Google Scholar]
- 8.Wilusz C.J., Wormington,M. and Peltz,S.W. (2001) The cap-to-tail guide to mRNA turnover. Nature Rev. Mol. Cell Biol., 2, 237–246. [DOI] [PubMed] [Google Scholar]
- 9.Voeltz G., Ongkasuwan,J., Standart,N. and Steitz,J. (2001) A novel embryonic poly(A) binding protein, ePAB, regulates mRNA deadenylation in Xenopus egg extracts. Genes Dev., 15, 774–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voeltz G.K. and Steitz,J.A. (1998) AUUUA sequences direct mRNA deadenylation uncoupled from decay during Xenopus early development. Mol. Cell. Biol., 18, 7537–7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouvet P., Omilli,F., Arlot-Bonnemains,Y., Legagneux,V., Roghi,C., Bassez,T. and Osborne,H.B. (1994) The deadenylation conferred by the 3′ untranslated region of a developmentally controlled mRNA in Xenopus embryos is switched to polyadenylation by deletion of a short sequence element. Mol. Cell. Biol., 14, 1893–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paillard L., Omilli,F., Legagneux,V., Bassez,T., Maniey,D. and Osborne,H.B. (1998) EDEN and EDEN-BP, a cis element and an associated factor that mediates sequence-specific mRNA deadenylation in Xenopus embryos. EMBO J., 17, 278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paillard L., Legagneux,V., Maniey,D. and Osborne,H. (2002) c-jun ARE targets mRNA deadenylation by an EDEN-BP (Embryo deadenylation element binding protein) dependent pathway. J. Biol. Chem., 277, 3232–3235. [DOI] [PubMed] [Google Scholar]
- 14.Ezzeddine N., Paillard,L., Capri,M., Maniey,D., Bassez,T., Aït-Ahmed,O. and Osborne,B. (2002) EDEN dependent translational repression of maternal mRNAs is conserved between Xenopus and Drosophila. Proc. Natl Acad. Sci. USA, 99, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Legagneux V., Bouvet,P., Omilli,F., Chevalier,S. and Osborne,H.B. (1992) Identification of RNA-binding proteins specific to Xenopus Eg maternal mRNAs: association with the portion of Eg2 mRNA that promotes deadenylation in embryos. Development, 116, 1193–1202. [DOI] [PubMed] [Google Scholar]
- 16.Birney E., Kumar,S. and Krainer,A.R. (1993) Analysis of the RNA-recognition motif and RS and RGG domains: conservation in metazoan pre-mRNA splicing factors. Nucleic Acids Res., 21, 5803–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kenan D.J., Query,C.C. and Keene,J.D. (1991) RNA recognition: towards indentifying determinants of specificity. Trends Biochem. Sci., 16, 214–220. [DOI] [PubMed] [Google Scholar]
- 18.Burd C.G. and Dreyfuss,G. (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- 19.Merrill B.M., Stone,K.L., Cobianchi,F., Wilson,S.H. and Williams,K.R. (1988) Phenylalanines that are conserved among several RNA-binding proteins form part of a nucleic acid-binding pocket in the A1 heterogeneous nuclear ribonucleoprotein. J. Biol. Chem., 263, 3307–3313. [PubMed] [Google Scholar]
- 20.Nagai K., Oubridge,C., Ito,N., Avis,J. and Evans,P. (1995) The RNP domain: a sequence specific RNA-binding domain involved in processing and transport of RNA. Trends Biochem. Sci., 20, 235–240. [DOI] [PubMed] [Google Scholar]
- 21.Draper D.E. (1999) Themes in RNA-protein recognition. J. Mol. Biol., 293, 255–270. [DOI] [PubMed] [Google Scholar]
- 22.Good P., Chen,O., Warner,S. and Herring,D. (2000) A family of human RNA-binding proteins related to the Drosophila Bruno translational regulator. J. Biol. Chem., 275, 28583–28592. [DOI] [PubMed] [Google Scholar]
- 23.Murray A.W. and Kirschner,M.W. (1989) Cyclin synthesis drives the early embryonic cell cycle. Nature, 339, 275–280. [DOI] [PubMed] [Google Scholar]
- 24.Kohli V. and Temsamani,J. (1993) Comparison of in vitro transcriptions using various types of DNA templates. Anal. Biochem., 208, 223–227. [DOI] [PubMed] [Google Scholar]
- 25.SenGupta D., Zhang,B., Kraemer,B., Pochart,P., Fields,S. and Wickens,M. (1996) A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl Acad. Sci. USA, 93, 8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feilotter H.E., Hannon,G.J., Ruddell,C.J. and Beach,D. (1994) Construction of an improved host strain for two hybrid screening. Nucleic Acids Res., 22, 1502–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Audic Y., Omilli,F. and Osborne,H.B. (1998) Embryo deadenylation element-dependent deadenylation is enhanced by a cis element containing AUU repeats. Mol. Cell. Biol., 18, 6879–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Handa N., Nureki,O., Kurimoto,K., Kim,I., Sakamoto,H., Shimura,Y., Muto,Y. and Yokoyama,S. (1999) Structural basis for recognition of the tra mRNA precursor by the Sex-lethal protein. Nature, 398, 579–585. [DOI] [PubMed] [Google Scholar]
- 29.Wang X. and Tanaka Hall,T.M. (2001) Structural basis for recognition of AU-rich element RNA by the HuD protein. Nature Struct. Biol., 8, 141–145. [DOI] [PubMed] [Google Scholar]
- 30.Chung S., Jiang,L., Cheng,S. and Furneaux,H. (1996) Purification and properties of HuD, a neuronal RNA-binding protein. J. Biol. Chem., 271, 11518–11524. [DOI] [PubMed] [Google Scholar]
- 31.Abe R., Sakashita,E., Yamamoto,K. and Sakamoto,H. (1996) Two different RNA binding activities for the AU-rich element and the poly(A) sequence of the mouse neuronal protein mHuC. Nucleic Acids Res., 24, 4895–4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakai K., Kitagawa,Y. and Hirose,G. (1999) Analysis of the RNA recognition motifs of human neuronal ELAV-like proteins in binding to a cytokine mRNA. Biochem. Biophys. Res. Commun., 256, 263–268. [DOI] [PubMed] [Google Scholar]
- 33.Park S., Myszka,D.G., Yu,M., Littler,S.J. and Laird-Offringa,I.A. (2000) HuD RNA recognition motifs play distinct roles in the formation of a stable complex with AU-rich RNA. Mol. Cell. Biol., 20, 4765–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webster P.J., Liang,L., Berg,C.A., Lasko,P. and Macdonald,P.M. (1997) Translational repressor bruno plays mutilple roles in development and is widely conserved. Genes Dev., 11, 2510–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Timchenko N.A., Welm,A.L., Lu,X. and Timchenko,L.T. (1999) CUG repeat binding protein (CUGBP1) interacts with the 5′ region of C/EBPβ mRNA and regulates translation of C/EBPβ isoforms. Nucleic Acids Res., 27, 4517–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi N., Sasagawa,N., Suzuki,K. and Ishiura,S. (2000) The CUG-binding protein binds specifically to UG dinucleotide repeats in a yeast three-hybrid system. Biochem. Biophys. Res. Commun., 277, 518–523. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki H., Jin,Y., Otani,H., Yasuda,K. and Inoue,K. (2002) Regulation of alternative splicing of α-actinin transcript by Bruno-like proteins. Genes Cells, 7, 133–141. [DOI] [PubMed] [Google Scholar]
- 38.Timchenko L.T., Miller,J.W., Timchenko,N.A., DeVore,D.R., Datar,K.V., Lin,L., Roberts,R., Caskey,C.T. and Swanson,M.S. (1996) Idenitification of a (CUG)n triplet repeat RNA-binding protien and its expression in myotonic dystrophy. Nucleic Acids Res., 24, 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki H., Maegawa,S., Nishibu,T., Sugiyama,T., Yasuda,K. and Inoue,K. (2000) Vegetal localization of the maternal mRNA encoding an EDEN-BP/Bruno-like protein in zebrafish. Mech. Dev., 93, 205–209. [DOI] [PubMed] [Google Scholar]
- 40.Timchenko N.A., Iakova,P., Cai,Z.J., Smith,J.R. and Timchenko,L.T. (2001) Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol. Cell. Biol., 21, 6927–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Philips A.V., Timchenko,L.T. and Cooper,T.A. (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science, 280, 737–741. [DOI] [PubMed] [Google Scholar]
- 42.Poleev A., Hartmann,A. and Stamm,S. (2000) A trans-acting factor, isolated by the three-hybrid system, that influences alternative splicing of the amyloid precursor protein minigene. Eur. J. Biochem., 267, 4002–4010. [DOI] [PubMed] [Google Scholar]
- 43.Ladd A.N., Charlet,N. and Cooper,T. (2001) The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell. Biol., 21, 1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim-Ha J., Kerr,K. and Macdonald,P.M. (1995) Translational regulation of oskar mRNA by Bruno, an ovarian RNA-binding protein is essential. Cell, 81, 403–412. [DOI] [PubMed] [Google Scholar]
- 45.Roberts R., Timchenko,N., Miller,J., Reddy,S., Caskey,C., Swanson,M. and Timchenko,L. (1997) Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice. Proc. Natl Acad. Sci. USA, 94, 13221–13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klein Gunnewiek J.M., Hussein,R.I., van Aarssen,Y., Palacios,D., de Jong,R., van Venrooij,W.J. and Gunderson,S.I. (2000) Fourteen residues of the U1 snRNP-specific U1A protein are required for homodimerization, cooperative RNA binding and inhibition of polyadenylation. Mol. Cell. Biol., 20, 2209–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]