

Abstract
Transfer RNA structure involves complex folding interactions of the TΨC domain with the D domain. However, the role of the highly conserved nucleoside modifications in the TΨC domain, rT54, Ψ55 and m5C49, in tertiary folding is not understood. To determine whether these modified nucleosides have a role in tRNA folding, the association of variously modified yeast tRNAPhe T-half molecules (nucleosides 40–72) with the corresponding unmodified D-half molecule (nucleosides 1–30) was detected and quantified using a native polyacrylamide gel mobility shift assay. Mg2+ was required for formation and maintenance of all complexes. The modified T-half folding interactions with the D-half resulted in Kds (rT54 = 6 ± 2, m5C49 = 11 ± 2, Ψ55 = 14 ± 5, and rT54,Ψ55 = 11 ± 3 µM) significantly lower than that of the unmodified T-half (40 ± 10 µM). However, the global folds of the unmodified and modified complexes were comparable to each other and to that of an unmodified yeast tRNAPhe and native yeast tRNAPhe, as determined by lead cleavage patterns at U17 and nucleoside substitutions disrupting the Levitt base pair. Thus, conserved modifications of tRNA’s TΨC domain enhanced the affinity between the two half-molecules without altering the global conformation indicating an enhanced stability to the complex and/or an altered folding pathway.
INTRODUCTION
All species of cytoplasmic tRNA appear to have the same basic L-shaped tertiary structure. The anticodon stem and loop and the helical aminoacyl stem of the cloverleaf secondary structure remain independent of the global tertiary fold. However, the loop containing dihydrouridine (D-loop), and that containing ribothymidine and pseudouridine (TΨC-loop) form the junction between these two physically separate domains (Fig. 1). Though tRNA nucleotide sequences are not well conserved across kingdoms, some of tRNA’s post-transcriptional modifications are highly conserved throughout all cytoplasmic tRNA species (1). Modifications in tRNA’s TΨC domain represent some of the most highly conserved RNA modifications. Pseudouridine at position 55, Ψ55, occurs at a frequency of 90%. Ribothymidine, or 5-methyluridine (rT54), occurs at position 54 in 60% of all sequenced tRNAs, and m5C at positions 48, 49 or both in 22%, 11% and 6%, respectively (1). Conservation of the nucleoside modifications in the TΨC domain suggests that they may play an important role in a function common to all tRNAs or in the folding of tRNA into its conserved tertiary structure for proper function.
Figure 1.

(A) Cloverleaf secondary structure of yeast tRNAPhe color-coded by domain. Red is the aminoacyl stem; blue is the T-stem–loop; orange is the variable loop; green is the anticodon stem–loop; and black is the D-stem–loop. (B) Crystal structure of yeast tRNAPhe (4) showing tertiary interactions between the T-loop and D-loop folding tRNA into its native L structure. Color coding corresponds to the secondary domains indicated in (A). The placement of Mg2+ ions was determined by X-ray diffraction as reported previously and is indicated by pink spheres. (C) The unmodified sequence of chemically synthesized constructs that mimic the T domain and D domain in yeast tRNAPhe. Positions of incorporated modifications are boxed; m5C49, m5U54 (rT54) and Ψ55 were singly or doubly incorporated into the T-half molecule. The Levitt base pair in yeast tRNAPhe occurs between G15 and C48 and is indicated by a dashed line. Constructs altering the potential for a Levitt base pair contained a U at position 15 in the D-half molecule (D-half G15U), or a U at position 48 in the T-half molecule (T-half C48U).
tRNA’s complex folding, the result of long-range, tertiary structure hydrogen bonding and metal ion coordination between the D-, TΨC- and variable-loops, occurs soon after transcription and before processing of the tRNA is complete (2,3). The folding interactions of the conserved L-shaped structure of cytoplasmic tRNAs are characterized by hydrogen bonding of G19 in the D domain with C56 in the TΨC domain and by a G18 base pair to Ψ55 (4). The tRNA molecule does not need to be in its native, fully processed, mature size for modification enzymes to produce rT54 and Ψ55, yet some structure of the RNA molecule is required (5–7). While neither rT54 nor Ψ55 is required for tRNA aminoacylation, elongation factor recognition or codon recognition on the ribosome in vitro (8), Escherichia coli lacking the enzyme for Ψ55 synthesis are at a disadvantage in competing with wild type cells (9). In addition, E.coli lacking the gene for rT54 synthesis are not viable (10). However, the rT54 synthesis activity does not appear to be the cause, since cells with inactive enzymes are viable (11).
tRNA’s tertiary folding interactions in vitro are observed with transcripts completely devoid of modifications (12,13). Yet, thermal denaturation of the fully modified, native tRNA occurs at a higher Tm than the corresponding unmodified transcript (12,14,15). Incorporation of rT54 increased the Tm of a 17mer analog of the yeast tRNAPhe TΨC stem and loop domain with a corresponding decrease in ΔG, whereas Ψ55 had no measurable effect on the melting temperature or free energy (7). Though the base-stacking contributions of Ψ compared with uridine were significant in a base-paired region of tRNA (16–19), hydrogen bonding through pseudouridine’s N1 may be favored in tertiary interactions between loops. Therefore, rT54 increases the thermal stability of tRNA at both the secondary (7,20) and tertiary level (21) and Ψ55 provides base-stacking and/or hydrogen bonding contributions to the tertiary interaction with the D domain. However, the contributions of the individual modified nucleosides for the folding process, and not the resulting functional structure, are unclear. Perhaps the modifications affect the kinetics of tRNA’s folding interactions and/or the number of folding pathways.
Empirical data on the contributions of individual modified nucleosides to the folding pathways for tRNA have been limited because experimental systems either utilize completely modified, native tRNA or completely unmodified transcripts (12,14,15). A structural study of the isolated stem and loop of the TΨC domain suggested a modification-dependent folding intermediate (20). The 17mer with the rT54 modification adopted a structure similar to that of the same domain within the native tRNA’s crystal structure, but distinctly different from the completely unmodified 17mer RNA (22,23). This result, in addition to the folding interaction of Ψ55 with the D domain, suggests that modifications in the TΨC domain may enhance the natural folding process of tRNAs.
The folding of RNA’s highly negative phosphate backbone requires the presence of counterions, typically Mg2+. Similarly, tRNA folding depends on the presence of counterions (12), but any contributions of rT54 and Ψ55 to this counterion dependence have yet to be determined. The counterion-dependent folding of native yeast tRNAPhe and the unmodified tRNAPhe transcript suggests that several folding pathways exist in which distinct, stable intermediate conformations can be observed (15,24). However, the site-specific coordination of Mg2+ ions in tRNA has been challenged (25). Although Mg2+ is coordinated between the D- and TΨC-loops within the crystal structure of yeast tRNAPhe (4,26,27), recent results with monovalent ions have suggested that electrostatic shielding of the phosphate backbone alone localizes Mg2+ to certain regions of the tRNA without coordination to specific ligands (25). Modified nucleosides have been implicated in the specific coordination of Mg2+ by native tRNAs (28,29) and in lowering the concentration of Mg2+ required for tertiary structure interactions (28). In contrast to native tRNA, tertiary structure interactions of unmodified transcripts have an absolute requirement for either high millimolar concentrations of Mg2+ (12–14) or molar concentrations of monovalent ion (15). However, the specific effect of rT54 and Ψ55 on Mg2+ coordination in the tertiary structure has not been determined (13,30).
To better understand the role of modified nucleosides in the ion-dependent folding interactions of tRNA, we have developed an experimental bimolecular model system. The model system allows us to site-specifically incorporate the naturally occurring, highly conserved modified nucleosides and monitor a bimolecular interaction of two RNA fragments mimicking the TΨC domain and the D domain of yeast tRNAPhe. The interaction of chemically synthesized half molecules, corresponding to nucleosides 1–30 (D-half) with nucleosides 40–72 (T-half) of yeast tRNAPhe, was monitored in a quantifiable polyacrylamide gel mobility shift assay. Here, we report that formation of the complex by the two half molecules required Mg2+ and introduction of the highly conserved modifications rT54 and Ψ55 and the commonly occurring m5C49 enhanced the affinity of the unmodified D-half molecule for the T-half molecule, but did not alter the global fold of the complex, which was similar to native yeast tRNAPhe.
MATERIALS AND METHODS
RNA synthesis, purification and analysis
tRNA half molecules were chemically synthesized with the nucleotide sequence of yeast tRNAPhe (Fig. 1C) using 2′-bis(acetoxyethoxy)-methyl ether (ACE; Dharmacon, Inc., Boulder, CO) (31), 2′-O-triisopropylsilyl-oxy-methyl (TOM; Xeragon, AG, Zurich, Switzerland) (32) and standard phosphoramidite chemistries (NCSU Nucleic Acids Facility, Raleigh, NC) (33,34). The unmodified D-half molecule RNAs corresponded to the 30mer sequence of nucleotides 1–30 in yeast tRNAPhe and the variously modified T-half RNAs corresponded to the 33mer sequence of nucleotides 40–72 in yeast tRNAPhe (Fig. 1). The 1–72 nt unmodified sequence of yeast tRNAPhe was chemically synthesized (Xeragon, AG) to mimic the completely unmodified yeast tRNAPhe transcript. RNAs were 3′-end labeled with [5′-32P]Cp using RNA ligase (35) and then diluted with known quantities of unlabeled half molecule. Radiolabeled [5′-32P]Cp was produced from [γ-32P] ATP (Amersham Biosciences, Piscataway, NJ) and CMP using polynucleotide kinase (35).
Gel mobility-shift assay
Gel mobility-shift assays were conducted with 15% native polyacrylamide gels composed of a Tris–borate buffer (TB; 89 mM Tris–HCl, 89 mM boric acid, pH 8.1). Mg2+, present only in the gel-forming reaction and in TB running buffer, was at a concentration of 3 mM, unless noted. The electrophoresis apparatus was surrounded by ice to maintain a temperature <25°C. All 3′-end-labeled T-half molecules were purified by a 7 M urea denaturing polyacrylamide gel electrophoresis (PAGE) (15%) prior to reaction with the D-half molecule in the native mobility-shift assay. Purified T-half molecules, eluted from gels using denaturing elution buffer (0.5 M ammonium acetate, 1% SDS, 0.1 mM ethylenediamine-tetraacetic acid), were reacted with the unmodified D-half in 15 µl of native loading buffer (30% glycerol in 89 mM Tris–HCl, 89 mM boric acid, pH 8.1). A fixed concentration of the T-half molecule was titrated with increasing amounts of the D-half molecule in individual reaction mixtures as described in the figure legends. The reaction mixtures were heated at 65°C for 10 min (in the absence of Mg2+), then allowed to cool to room temperature prior to loading on the gel. After electrophoresis, the gel was dried and visualized by phosphorimaging (36). T-half molecules free and within the T-half/D-half complex were quantified by analyzing the volume of the phosphorimage using ImageQuant software (Molecular Dynamics, Amersham Biosciences). Only the two prominent bands, free and bound, were analyzed. The material between the two bands was not included in the analysis of free or bound material (see Fig. 5). A lane containing no RNA was used for a background subtraction from all lanes with bound and free RNA. The percent complex formation was determined from the volumes and then normalized to 100% for relative Kd determination. The data were fitted to a one-site binding model using Prism software and errors were based on the deviations from the least squares fit (GraphPad Software, San Diego, CA).
Figure 5.
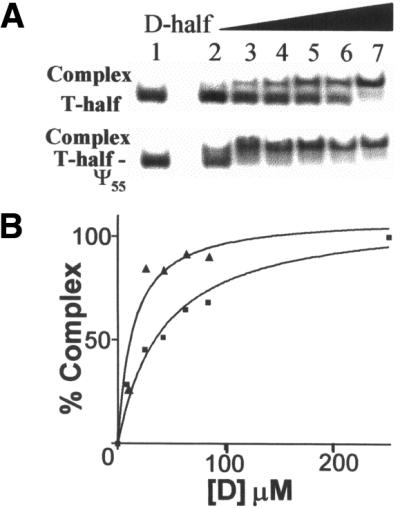
(A) Mobility-shift assays of 32P-end-labeled, unmodified and modified, T-half molecules with increasing amounts of unmodified D-half molecule. In the native PAGE (15%, 3.0 mM Mg2+) at the top of the figure, complex formation is detected when the unmodified, 32P-labeled T-half molecule interacts with the unmodified D-half molecule. Below, a complex between the Ψ55-containing T-half molecule (T-half-Ψ55) with the unmodified D-half molecule is detected. All lanes contained 84.2 µM T-half molecule. Lanes 2–7 of each gel contained increasing amounts of unmodified D-half molecule as follows: 8.42, 25.3, 42.1, 63.2, 84.2 and 253 µM, respectively. (B) Percent complex formed versus concentration of the D-half molecule for the unmodified D-half molecule with unmodified T-half molecule (closed square) or with the Ψ55-containing T-half molecule (closed triangle). Data were normalized to 100% complex formation and curves were fitted to the data with a non-linear regression assuming one-site binding. The percent complex was determined by quantifying the phosphorimages from the PAGE in (A) using ImageQuant software (Molecular Dynamics) and errors were determined as deviations from the least squares fit.
Ribonuclease T1 and lead cleavage reactions
The 3′-end-labeled RNAs and RNA complexes (D- and T-half molecule complexes) were purified by PAGE (15%). The RNAs were identified on the gel by ethidium bromide staining. Individual RNAs were eluted from the gel in denaturing elution buffer (0.5 M ammonium acetate, 1% SDS, 0.1 mM EDTA), whereas complexes of the D- and T-half molecules were eluted with native elution buffer (20 mM HEPES, 250 mM KCl and 5 mM MgCl2). The eluted RNAs were ethanol precipitated, dried and resuspended to a concentration of 20 µM RNA in water. RNAs that were not in complexes were denatured and renatured by heating to 70°C for 5 min and slowly cooled to room temperature prior to use.
Lead cleavage reactions (10 µl) contained 4 µM RNA (40 mM NaCl, 10 mM Tris–HCl, pH 7.2, 10 mM MgCl2) and 0.5 mM Pb(II) acetate. Reaction mixtures were stored on ice, initiated by the addition of Pb(II) acetate and incubated at 20°C for 15 min. The lead cleavage sites were mapped by generating an alkaline hydrolysis ladder. Alkaline hydrolysis was conducted by heating reaction mixtures (10 µl) containing 2 µM RNA and formamide (1:5 by volume in 50 mM NaHCO3/Na2CO3 pH 9.1) at 100°C for 10 min. In addition, the three Gs (G18, G19 and G20) in the D-loop that are 3′ to the expected lead cleavage site at U17, were mapped with RNase T1 digestion (Promega, Madison, WI). RNase T1 partial digestions were performed in reaction mixtures (10 µl) containing 2 µM RNA (50 mM sodium citrate, 4.5 M urea) with 5 U RNase T1 at 55°C for 20 min. Cleavage reactions were stopped by addition of 10 µl formamide loading dye (100% deionized formamide, 10 mM EDTA, 0.25% bromophenol blue and 0.25% xylene cyanol FF). Radiolabeled products of the cleavage reactions were analyzed by 7 M urea–PAGE (15%) and visualized by phosphorimaging.
RESULTS
Design of a bimolecular system to mimic tRNA folding
A bimolecular experimental model was designed to determine the contributions of modified nucleosides to the folding interaction of tRNA’s TΨC domain with the D domain. The experimental system consisted of a 33 nt T-half molecule and a 30 nt D-half molecule (Fig. 1C). The T-half molecule was synthesized unmodified or with the modified nucleosides rT54, Ψ55, m5C49 or with both rT54 and Ψ55 (Fig. 2). While a previous study had used a similar bimolecular model to determine the minimal fragments of yeast tRNAPhe required for leadzyme activity (37), we extended the lengths of our half molecules to include the aminoacyl stem and anticodon stem base-paired regions (Fig. 1C). Though a 26 nt D-half formed a complex with a 29 nt T-half molecule, an extension to slightly larger RNAs was necessary to achieve complete T- and D-half molecule complex formation as a gel mobility shift under the native PAGE conditions used in this study (Fig. 3). Mobilities of RNAs in native gels are apparently sensitive to molecular conformation and Mg2+ concentrations (38). In the presence of 3 mM Mg2+ and increasing concentrations of the D-half molecule, the intensity of the faster moving band of individual half molecules decreased with a concomitant increase in the intensity of the slower moving complex (Fig. 3A, lanes 2–5). An increase in the intensity of the faster moving band was observed with addition of excess D-half molecule (Fig. 3A, lanes 6–9). Complex formation required the presence of both half molecules. No gel shift was evident with either half molecule alone, even at 10-fold higher concentrations indicating that the individual half molecules did not aggregate (Fig. 3B).
Figure 2.

The chemical structure and hydrogen bonding of the conserved modifications, rT54, Ψ55 and m5C49. Modified T-half molecule constructs were chemically synthesized with the modified nucleosides rT54, Ψ55 and m5C49 individually, or with both rT54 and Ψ55. The hydrogen bonding of each of these modified nucleosides as found in the X-ray crystallographic tertiary structure yeast tRNAPhe is shown in the figure. Ribothymidine or 5-methyluridine (rT54) forms a tertiary structure base pair with m1A58. However, our T-half molecules are constructed with the unmodified A58. Methyl-5-cytidine forms a canonical Watson–Crick base pair with G65.
Figure 3.
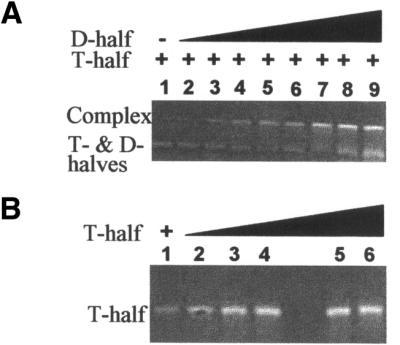
Complex formation between the unmodified D- and T-half molecules was detected in a native PAGE (15%, 3.0 mM Mg2+) with ethidium bromide staining. Free D- and T-half molecules migrated similarly, and stained less well than the slower migrating complex. (A) Lanes 1–9 contained 5.6 µM unmodified T-half molecule. Lanes 2–9 contained increasing amounts of unmodified D-half molecule with the following µM concentrations: 0.56, 1.68, 2.81, 4.21, 5.61, 16.8, 56.1 and 112. (B) Increasing amounts of unmodified T-half molecule alone in a native PAGE (15%, 3.0 mM Mg2+) demonstrated that the half molecule does not aggregate. Lanes 1–6 contained unmodified T-half with the following µM concentrations: 1.6, 3.0, 4.2, 5.6, 7.0 and 8.4.
Complex formation also required Mg2+ in the gel and running buffer. Mg2+ in the sample alone was not sufficient to demonstrate complex formation by a gel shift (Fig. 4A). At least 0.75 mM Mg2+ was required for a complex to form whether the T-half was unmodified or had Ψ55 (Fig. 4B). A 3 mM concentration of Mg2+ was determined to be at saturation for complex formation (Fig. 4B). Since Ψ55, the most highly conserved of the three T-half modifications, did not lower the requirement for Mg2+ (Fig. 4B), experiments were routinely conducted with 3 mM Mg2+ to ensure that the effects of modifications, and not of Mg2+, were being measured.
Figure 4.

(A) The absence of Mg2+ in both the running buffer and gel precluded complex formation. A native polyacrylamide gel (15%) was polymerized in the absense of Mg2+. The running buffer (TB) also lacked Mg2+. RNA was detected with ethidium bromide staining. Samples for lanes 1 and 2 of the gel contained 84.2 µM unmodified T-half and D-half, respectively, but the D-half did not stain well with ethidium bromide, due to lack of secondary structure (John Stuart and Paul F. Agris, personal communication). Lane 3 contained 84.2 µM unmodified T-half and 84.2 µM D-half. (B) A Mg2+ concentration >0.75 mM is required for complex formation. Complex formation was analyzed under different Mg2+ concentrations from 0 to 5 mM. Samples of 84.2 µM unmodified (lanes 1, 3, 5, 7, 9 and 11), or Ψ-containing T-half (lanes 2, 4, 6, 8, 10 and 12) and 84.2 µM D-half molecules were subjected to native PAGE in which the gel, sample and running buffers contained the same concentration of Mg2+. The composite of gels depicts examples of the results used to produce the graph below (unmodified T-half, open circles, solid line; Ψ-containing T-half, cross, dotted line). (C) The left-hand native gel demonstrates a lack of complex formation for the unmodified T-half molecule with increasing concentrations of the D-half molecule in the presence of only 0.5 mM Mg2+. Lane 1 contained 84.2 µM unmodified T-half molecule. Lanes 2–9 contained 84.2 µM unmodified T-half with the following µM concentrations of D-half: 8.42, 25.3, 42.1, 63.2, 84.2, 253, 842 and 1684 µM. The right-hand native gel demonstrates a lack of complex formation for the Ψ55-containing T-half molecule in the presence of 0.5 mM Mg2+. Lane 1 contains 84.2 µM Ψ55 T-half alone. Lanes 2–9 contain 84.2 µM Ψ55 T-half with the following µM concentrations of D-half: 8.42, 25.3, 42.1, 63.2, 84.2, 253, 842 and 1684 µM.
A gel mobility shift, indicating complex formation of the T- and D-half molecules, was observable with ethidium bromide staining (3 mM Mg2+, pH 8.1, 25°C). The individual T- and D-half molecules, though of different lengths and sequences, exhibited very similar migrations under native PAGE conditions. However, labeling either half molecule with 32P allowed us to observe that half molecule as free or as participating in the complex (Fig. 5). The complex was detected independently of which half molecule was labeled, but the T-half molecule was routinely labeled for all quantified experiments. The integrity of the complex could be maintained during extraction from the gel in the presence of Mg2+. Upon removal of Mg2+, the complex dissociated into the individual D- and T-half molecules (data not shown).
TΨC domain modifications enhanced affinity of the D-half molecule for the T-half molecule
The binding affinities of the unmodified and variously modified T-half molecules for the completely unmodified D-half molecule were compared. Binding affinities for the interacting half molecules were determined by titrating the 3′-32P-labeled T-half molecules with increasing concentrations of the unlabeled D-half molecule (Fig. 5). Free and complexed T-half molecules were separated by native PAGE and quantified by phosphorimaging, as described in Materials and Methods. Binding affinity, calculated as dissociation constants (Kd), varied with the modification present in the T-half molecule (Table 1). All Kd values were determined under saturating conditions although, for clarity, binding isotherms are not shown to saturation (Fig. 5). While all of the naturally occurring TΨC domain modifications that we investigated, m5C49, T54 and Ψ55, enhanced the affinity of the half molecules, the rT54-containing T-half molecule exhibited the highest affinity for the unmodified D-half molecule (Table 1). The T-half molecule with rT site-specifically incorporated at its naturally occurring position, 54, exhibited an affinity for the D-half molecule, 7-fold higher (Kd = 6 ± 2 µM) than that of the unmodified T-half molecule (Kd = 40 ± 10 µM). Pseudouridine at position 55 increased the binding affinity of the T-half molecule ∼3-fold (Kd = 14 ± 5 µM), compared with the unmodified T-half molecule. The m5C49 modification increased the affinity of the T-half almost 4-fold (Kd = 11 ± 2 µM) compared with that of the unmodified T-half molecule. A doubly modified T-half molecule containing both rT54 and Ψ55 exhibited an affinity (Kd = 11 ± 3 µM) comparable with that of the m5C49-containing T-half molecule and ∼4-fold higher than that of the completely unmodified T-half molecule. The affinity of the rT54 and Ψ55 modified T-half molecule for the D-half was intermediate to that of the rT54 or Ψ55 modification alone.
Table 1. Binding affinities and free energy for complex formation between the unmodified D-half and variously modified T-half molecules.
| T-half moleculea | Kd (µM)b | ΔG37c |
|---|---|---|
| Unmodified | 40 ± 10 | –6.2 ± 1.6 |
| RT54 | 6 ± 2 | –7.4 ± 2.0 |
| m5C49 | 11 ± 2 | –7.0 ± 1.3 |
| Ψ55 | 14 ± 5 | –6.9 ± 2.5 |
| RT54;Ψ55 | 11 ± 3 | –7.0 ± 1.9 |
aT-half molecule constructs containing the site-specific incorporations of the indicated modified nucleosides.
bBinding constants were determined by quantifying mobility shift assays (Materials and Methods), plotting % complex formation versus concen tration of D-half molecule, and fitting data with non-linear regression.
cΔG37 (kcal/mol at 37°C) was calculated from the association constants (ΔG = –RT ln K).
Modifications do not lower the Mg2+ requirement for complex formation
Binding affinities were determined by mobility-shift assays conducted under excess Mg2+ concentrations to minimize any contributions the modified nucleosides may have had in coordinating Mg2+. In order to determine if the most highly conserved modification in tRNA (1), Ψ55, altered the Mg2+ requirement, native PAGE mobility-shift assays were conducted at various concentrations of Mg2+ (0–5 mM). The unmodified T-half and Ψ55-containing T-half molecules were titrated with the unmodified D-half molecule. The minimum Mg2+ concentration required for the observable interaction between the two unmodified half molecules was ∼0.75 mM (Fig. 4B). In the presence of 0.5 mM Mg2+, even a 20-fold concentration of D-half to T-half molecule failed to generate an observable complex formation (Fig. 4C). Incorporation of Ψ55 into the T-half molecule did not lower the Mg2+ concentration required to observe the mobility-shift (Fig. 4B and C). Additions of very small amounts of Mg2+ readily alter the conformation of tRNAPhe, as revealed by hydroxy radical cleavage patterns (39). Therefore, while the conserved modifications enhanced the affinity of the T-half molecule for the D-half molecule, they did not lower the Mg2+ requirement for the interaction of the two half molecules under the conditions of the assay.
Comparison of global folds of the D and TΨC complex and native yeast tRNAPhe
Yeast tRNAPhe has a G15:C48 tertiary structure base pair in common with some 65% of all tRNAs (1,40). This base pair, known as the Levitt pair, occurs between G15, in the D-loop, and C48, in the variable loop, in yeast tRNAPhe (Fig. 1C). To ensure that complex formation observed under native PAGE conditions was a result of known tertiary structure interactions between D- and T-half molecules, and not simply a result of canonical base-pairing between the aminoacyl and anticodon stems, RNAs were synthesized to eliminate the possibility of the G15:C48 Levitt tertiary structure base pair. D- and T-half molecules with U substitutions in both positions 15 (D-half G15U) and 48 (T-half C48U) would not be expected to form a complex, and none was observed (Fig. 6). No complex was observed when the completely unmodified, native sequence T-half molecule was combined with the D-half G15U, since a U15:C48 base pair would have to form (Fig. 6). However, when the unmodified, but native sequence D-half molecule was incubated with the T-half C48U molecule, a small amount of RNA with the mobility of the native sequence D-half/T-half complex and a faster moving, diffuse band of RNA in the gel were observed. The diffuse band of RNA in the gel could have resulted from a G15:U48 tertiary interaction plus tertiary structure mis-pairings that yielded complexes with many conformations (Fig. 6). Observation of a mobility-shift that required the two half-molecule constructs, the Levitt base pair and Mg2+ was indicative of the complex having a global fold similar to that of native yeast tRNAPhe. To confirm that the overall structure of the complex composed of the T- and D-half molecules was indeed similar to that of native tRNAPhe, we took advantage of the tRNA’s ability to self-cleave with Pb2+ only when in the native fold (41). A T-half/D-half complex with a fold similar to that of native tRNAPhe should exhibit the well-characterized self-cleavage reaction catalyzed by lead (37). When correctly folded native tRNAPhe acts as a leadzyme, a specific and strong, single site cleavage of the D-loop between D17 (or U17) and G18 results in identifiable fragments (Fig. 7A). RNase T1 digestions, under denaturing conditions, and alkaline hydrolysis ladders confirmed the location of the lead cleavage (Fig. 7A). The location of the lead cleavage produced by an unmodified, chemically synthesized and slightly truncated tRNAPhe (nucleotides 1–72) was identical to that of the native tRNA (Fig. 7A), as reported previously for an unmodified transcript (37). A specific self-cleavage by lead between U17 and G18 was achieved with the complex composed of the unmodified half molecules indicating that the global fold of the complex was similar to that of native tRNAPhe (Fig. 7B). Lead cleavage of the D-half molecule alone revealed random cleavages quite similar to that of an alkaline hydrolysis ladder, and dramatically different to the site-specific cleavage when complexed with the T-half molecule (Fig. 7B). Since the TΨC domain modifications enhanced the interaction of the T-half molecule for the unmodified D-half molecule, the modifications may also alter the structure of the resulting complex. Therefore, we performed the lead cleavage analysis to determine if modifications influenced the overall tertiary conformation of the complex. The strong, site-specific lead cleavage of a complex composed of the unmodified D-half and a rT54,Ψ55-containing T-half molecule was similar to that of the unmodified D-half and T-half complex (Fig. 7B). Thus, the lead cleavage patterns of the complexes, native yeast tRNAPhe, and the unmodified analog of yeast tRNAPhe, were all similar.
Figure 6.
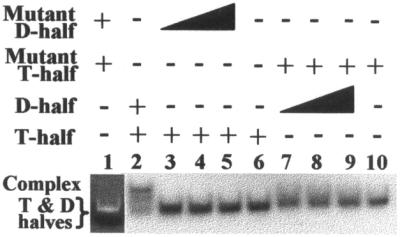
Absence of complex formation by T- and D-half molecules without the potential for Levitt base-pair. Lane 1 contained 84.2 µM of unlabeled T-half C48U and 84.2 µM D-half G15U. The single RNA band, visualized with ethidium bromide staining, is that of the free T-half C48U and D-half G15U molecules that migrate similarly in the gel. A mobility shift indicating complex formation was not detectable. Lane 2 contained the positive control of unmodified T-half molecule (84.2 µM) with unmodified D-half (84.2 µM). The major band was the mobility-shifted, 32P-end-labeled T-half molecule in complex with D-half molecule. The RNA was visualized by phosphorimaging. Some free T-half molecule is also observed. Lanes 3–5 contained 32P-end-labeled unmodified T-half molecule (84.2 µM) with an increasing concentration of the D-half G15U, 8.42, 84.2 and 253 µM, respectively. A Levitt base pair interaction by the half molecules in lanes 3–5 would require a U15C48 base pair to occur. Lane 6 contained 32P-end- labeled, unmodified T-half molecule (84.2 µM) only. Lanes 7–9 contained the T-half C48U molecule (84.2 µM) with increasing amounts of unmodified D-half molecule, 8.42, 84.2 and 253 µM, respectively. A Levitt base pair interaction by these half molecules would require formation of a G15U48 base pair. Lane 10 contained 84.2 µM 32P-end-labeled C48U T-half molecule, alone. Mobility of the free T-half molecule varied slightly across the gel.
Figure 7.

Lead, Pb(II), cleavage of native tRNAPhe and the D-half molecule in the presence of the unmodified and the rT54 and Ψ55 modified T-half molecules produced a distinct cleavage pattern not observed in the D-half molecule alone. (A) Native tRNAPhe and unmodified tRNAPhe 72mer, and (B) free D-half molecule, the complex from unmodified D- and T-half molecules and that from the unmodified D-half with the rT54 and Ψ55 modified T-half molecule were 32P-3′-end-labeled and subjected to: (–) no treatment, (OH) alkaline hydrolysis, (T1) T1 nuclease and (Pb) 0.5 mM Pb2+. Alkaline hydrolysis and T1 nuclease treatments were used to confirm cleavage sites. All reactions are described in Materials and Methods. Reaction products were separated by 10% PAGE–7 M urea and visualized by phosphorimaging. The positions of the nucleotides sensitive to T1 cleavage (G18–20) and Pb2+ cleavage (between U16 and U17) are indicated in the figure. The heavy bands at the top of the lanes in (A) and (B) are the full-length native tRNAPhe and the unmodified, 72mer analog.
DISCUSSION
The contributions of some highly conserved modifications to the folding of tRNA have not been well documented, although the Mg2+-dependent folding of both unmodified and fully modified tRNAs has been studied (12,13,24). With tRNA containing the largest and most variant number of modified nucleosides, there may be a role for modifications in the folding process of tRNA by either stabilizing intermediates or altering local conformations for the overall folding of the tRNA into its native tertiary structure. In particular, site-specifically conserved modifications, such as rT54 and Ψ55, may be important for tRNA to efficiently and effectively achieve functional conformation under physiological conditions. To assess the roles of highly conserved, modified nucleosides and Mg2+ in the folding interactions of tRNA’s domains, we developed an experimental bimolecular model. Tertiary interactions of the TΨC domain with the D domain in the unimolecular yeast tRNAPhe were mimicked by the interactions of half-molecules. Incorporation of the highly conserved modifications rT54 and Ψ55, or the commonly occurring m5C49, enhanced the affinity of the T-half molecule with the unmodified D-half. However, Ψ55 did not lower the Mg2+ requirement for this interaction. Modifications in the T-half molecule did not affect the global conformation of the resulting complexes. Taken together, the enhanced affinity of the modified T-half molecule for the unmodified D-half and the early events of rT54 and Ψ55 syntheses in pre-tRNA (3) indicate that these conserved modifications contribute to the native TΨC and D domain folding interactions.
The TΨC domain folding interaction with the D domain required both the Levitt tertiary structure base pair present in the native yeast tRNAPhe and the presence of Mg2+. No complex was observed in our gel mobility shift analysis when base substitutions completely negated the Levitt pair. Disruption of the Levitt base pair, as well as all D- to T-loop tertiary structure interactions, in an effort to define a functional, minimal sequence tRNALeu analog, completely negated aminoacylation by the E.coli leucyl-tRNA synthetase (42). Thus, formation of the Levitt pair may define one of the lowest energy conformations in the RNA’s folding landscape (43) on the path to the native global fold. In tRNA’s three-dimensional structure (4,26), the highly charged phosphate backbone folds sharply at the TΨC and D domain interface. The requirement for Mg2+ could be explained by neutralizing electrostatic repulsion (44,45), in particular the large local negative charge at this interdomain interface (24,25). Though high concentrations of Mg2+ were required, the unmodified yeast tRNAPhe transcript exhibited, in NMR spectra, the tertiary and secondary structure hydrogen bonding similar to that of the native tRNA and expected of a folding interaction between the TΨC and D domains (13). Results of Levitt base pair substitutions and site-specific lead cleavages reported here are consistent with the NMR investigation. Cleavage of native tRNAPhe, an unmodified analog of tRNAPhe, the complex formed from the unmodified D- and T-half molecules and the complex formed from the unmodified D-half molecule with variously modified T-half molecules all produced similar fragmentation patterns (Fig. 7). A Mg2+ requirement for the bimolecular folding interaction in our model system is also consistent with electrostatic interactions dominating the free energy of RNA folding (46). The result is that stable tertiary structures of RNAs are observed only in the presence of divalent metal ions or with very high concentrations of monovalent ions (46).
Our bimolecular model system allows investigation of the contributions of individual modifications to interdomain contacts and folding interactions within tRNA. As with other RNAs, the folding process of tRNA involves a series of early and late folding events (46,47), secondary and tertiary interactions (48), as it is being transcribed in the cell and is then processed as pre-tRNA to the modified and sized, functioning molecule. Initiation of the early, secondary interactions in the tRNA folding process is likely to occur in a time scale (10–100 µs) approximately 1000 times more rapid than the late-occurring, tertiary interactions (49,50). Support that our model system is reporting the late occurring, specific tertiary interactions, is that the ΔG values observed in our model system (Table 1) are of a similar magnitude for interdomain contacts in other RNAs (1.7 ± 2 to 7.6 ± 1.2 kcal/mol) (51). In addition, the specific lead cleavage pattern and the presence of the Levitt base-pair in the completely unmodified D- and T-half molecule suggests that the contributions of modifications are to stabilize, rather than to form, the tertiary contacts.
Modifications in the TΨC domain possibly enhance the correct tertiary structure contacts for tRNA folding interactions. Thus, unproductive pathways and stable, but mis-folded, conformations that have been observed in unmodified RNAs (52) are thwarted. The m1A9-dependent folding of a mitochondrial tRNA that has a small D domain loop (53) may be an example of a modified nucleoside required for a late folding event to the native structure. However, one of the most common modifications, rT54, probably contributes to an early secondary structure alteration of the TΨC domain conformation such that the native folding interaction with the D domain would be affected. rT54’s ability to stabilize and affect the local conformation of the T stem and loop (7,20), and the tRNAfMet species from E.coli and Thermus thermophilus (21), indicates that enhanced secondary structure interactions could be responsible for our observation of the higher affinity of the modified T-half molecule than the unmodified for the D-half. The fact that catalytically inactive yeast m5U54-tRNA methyltransferase is required for stabilizing at least one of the cell’s tRNAs, tRNASerCGA (11), indicates that the modification enzyme may play a role in tRNA folding prior to catalyzing formation of rT54. After methylation of U54, the resulting folded structure would be retained through the stabilizing contributions of the modification.
In contrast to rT54, Ψ55 may directly favor tertiary interactions between the TΨC domain with the D domain without contributing secondary interactions to the TΨC domain alone. The presence of Ψ55 in the otherwise unmodified 17mer TΨC domain did not further stabilize the RNA (7), but the presence of Ψ in base-paired regions provides stability to RNA (16–19,54). Ψ55 may not contribute to local stability of the TΨC domain, as does T54. However, Ψ’s additional hydrogen bonding and/or base stacking capabilities (16–19) would understandably favor tertiary interactions between the TΨC domain and the D domain. Compared with the unmodified T-half molecule, the T-half molecule with Ψ55 had a higher affinity for the D-half molecule. Introduction of Ψ55 into the T-half molecule resulted in an additional –0.7 kcal/mol of free energy for the association of the two half molecules. Similarly, the introduction of the conserved Ψ in snRNA stabilized that RNA’s interaction with an intron branch point by –0.7 kcal/mol (55). These results, together with our finding that the unmodified D-half molecule’s affinity for the doubly modified T-half rT54;Ψ55 (Kd = 11 ± 3 µM) was less than that for the singly modified T-half (rT54, Kd = 6 ± 2 µM), yet greater than that of the Ψ55-modified T-half (Kd = 14 ± 5 µM) and the unmodified T-half (Kd = 40 ± 10 µM), suggest that rT54 and Ψ55 could play different roles in stabilizing the tertiary interactions in tRNA. The contributions of these individual modifications were not additive in our experimental model. When the other, adjacent, modification was present, each could influence the contributions of the other to the RNAs’ folding interactions.
Recent theoretical and empirical approaches to folding describe the folding interactions of RNA molecules with Mg2+ as one of loose and diffusely bound hydrated Mg2+ ions participating in a rapid, non-specific collapse of the RNA into an intermediate that then finds, within this restricted conformational space, its stable native conformation (56,57). The enhanced folding interaction observed when conserved modifications are present in the TΨC domain may be indicative of modifications reducing conformational space and thus affecting the folding pathway and/or kinetics of reaching the native structure. We have observed by NMR that additions of a modification to an already modified yeast tRNAPhe anticodon stem and loop domain restricted its conformational space accessible in the direction of the native structure (J.W.Stuart and P.F.Agris, unpublished results). Although we have as yet to identify a stable folding intermediate by native PAGE under conditions of low Mg2+ concentration, the application of urea titrations (14,15) and circular dichroism studies to our bimolecular approach may yield information about modified nucleoside contributions to the formation of specific intermediates in the tertiary folding of tRNA. Because tRNA processing enzymes and the translation machinery of the cell are sensitive to the tertiary fold of tRNA, effects of modification on the pathway and rate of tRNA folding and unfolding could determine an organism’s ability to survive (58,59).
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Drs Salman Ashraf and Charles Hardin for their helpful suggestions and discussions, Winnell Newman, as director of the NCSU Nucleic Acids Facility, for her synthesis of the unmodified T- and D-half molecules, and Susanna Smith for her assistance in the lead cleavage experiments. This research was supported in part by NSF Grant MCB 9986011 and NIH Grant GM-23027 to P.F.A. and an NSF REU to P.F.A. for K.N.N.
REFERENCES
- 1.Sprinzl M., Horn,C., Brown,M., Ioudovitch,A. and Steinberg,S. (1998) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res., 26, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McClain W.H. and Seidman,J.G. (1975) Genetic perturbations that reveal tertiary conformation of tRNA precursor molecules. Nature, 257, 106–110. [DOI] [PubMed] [Google Scholar]
- 3.Bjork G.R. (1995) Biosynthesis and function of modified nucleosides. In Soll,D. and RajBhandary,U.L. (eds), tRNA: Structure, Biosynthesis and Function. ASM, Washington DC, pp. 165–205.
- 4.Kim S.H., Quigley,G.J., Suddath,F.L., McPherson,A., Sneden,D., Kim,J.J., Weinzierl,J. and Rich,A. (1973) Three-dimensional structure of yeast phenylalanine transfer RNA: folding of the polynucleotide chain. Science, 179, 285–288. [DOI] [PubMed] [Google Scholar]
- 5.Gu X.R. and Santi,D.V. (1991) The T-arm of tRNA is a substrate for tRNA (m5U54)-methyltransferase. Biochemistry, 30, 2999–3002. [DOI] [PubMed] [Google Scholar]
- 6.Gu X., Yu,M., Ivanetick,K.M. and Santi,D.V. (1998) Molecular recognition of tRNA by tRNA pseudouridine 55 synthase. Biochemistry, 37, 339–343. [DOI] [PubMed] [Google Scholar]
- 7.Sengupta S., Saulius,V., Yarian,C., Sochacka,E., Malkiewicz,A., Guenther,R., Koshlap,K.M. and Agris,P.F. (2000) Modified constructs of the tRNA TΨC domain to probe substrate conformational requirements of m(1)A(58) and m(5)U(54) tRNA methyltransferases. Nucleic Acids Res., 28, 1374–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrington K.M., Nazarenko,I.A., Dix,D.B., Thompson,R.C. and Uhlenbeck,O.C. (1993) In vitro analysis of translational rate and accuracy with an unmodified tRNA. Biochemistry, 32, 7617–7622. [DOI] [PubMed] [Google Scholar]
- 9.Gutgsell N., Englund,N., Niu,L., Kaya,Y., Lane,B.G. and Ofengand,J. (2000) Deletion of the Escherichia coli pseudouridine synthase gene truB blocks formation of pseudouridine 55 in tRNA in vivo, does not affect exponential growth, but confers a strong selective disadvantage in competition with wild-type cells. RNA, 6, 1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Persson B.C., Gustafsson,C., Berg,D.E. and Bjork,G.R. (1992) The gene for a tRNA modifying enzyme, m5U54-methyltransferase, is essential for viability in Escherichia coli. Proc. Natl Acad. Sci. USA, 89, 3995–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johansson M.J.O. and Byström,A.S. (2002) Dual function of the tRNA(m5U54)-methyltransferase in tRNA maturation. RNA, 8, 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maglott E.J., Deo,S.S., Przykorska,A. and Glick,G.D. (1998) Conformational transitions of an unmodified tRNA: implications for RNA folding. Biochemistry, 37, 16349–16359. [DOI] [PubMed] [Google Scholar]
- 13.Hall K.B., Sampson,J.R., Uhlenbeck,O.C. and Redfield,A.G. (1989) Structure of an unmodified tRNA molecule. Biochemistry, 28, 5794–5801. [DOI] [PubMed] [Google Scholar]
- 14.Shelton V.M., Sosnick,T.R. and Pan,T. (1999) Applicability of urea in the thermodynamic analysis of secondary and tertiary RNA folding. Biochemistry, 38, 16831–16839. [DOI] [PubMed] [Google Scholar]
- 15.Shelton V.M., Sosnick,T.R. and Pan,T. (2001) Altering the intermediate in the equilibrium folding of unmodified yeast tRNAPhe with monovalent and divalent cations. Biochemistry, 40, 3629–3638. [DOI] [PubMed] [Google Scholar]
- 16.Yarian C.S., Basti,M.M., Cain,R., Ansari,G., Guenther,R.H., Sochacka,E., Malkiewicz,A. and Agris,P.F. (1999) Structural and functional roles of the N1- and N3-protons of Ψ at tRNA’s position 39. Nucleic Acids Res., 27, 3543–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yarian C., Marszalek,M., Sochacka,E., Malkiewicz,M., Guenther,R., Miskiewicz,A. and Agris,P.F. (2000) Modified nucleoside dependent Watson–Crick and wobble codon binding by tRNALysUUU species. Biochemistry, 39, 13390–13395. [DOI] [PubMed] [Google Scholar]
- 18.Durant P.C. and Davis,D.R. (1999) Stabilization of the anticodon stem–loop of tRNALys,3 by an A+-C base-pair and by pseudouridine. J. Mol. Biol., 285, 115–131. [DOI] [PubMed] [Google Scholar]
- 19.Agris P.F., Guenther,R., Sochacka,E., Newman,W., Czerwinska,G. and Malkiewicz,A. (1999) Thermodynamic contribution of nucleoside modifications to yeast tRNA(Phe) anticodon stem loop analogs. Acta Biochim. Pol., 46, 163–172. [PubMed] [Google Scholar]
- 20.Koshlap K.M., Guenther,R., Sochacka,E., Malkiewicz,A. and Agris,P.F. (1999) A distinctive RNA fold: the solution structure of an analogue of the yeast tRNAPhe TΨC domain. Biochemistry, 38, 8647–8656. [DOI] [PubMed] [Google Scholar]
- 21.Davanloo P., Sprinzl,M., Watanabe,K., Albani,M. and Kersten,H. (1979) Role of ribothymidine in the thermal stability of transfer RNA as monitored by proton magnetic resonance. Nucleic Acids Res., 6, 1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao L.J., James,T.L., Kealey,J.T., Santi,D.V. and Schmitz,U. (1997) The dynamic NMR structure of the TΨC-loop: implications for the specificity of tRNA methylation. J. Biomol. NMR, 9, 229–244. [DOI] [PubMed] [Google Scholar]
- 23.Schmitz U., Donati,A., James,T.L., Ulyanov,N.B. and Yao,L. (1998) Small structural ensembles for a 17-nucleotide mimic of the tRNA TΨC-loop via fitting dipolar relaxation rates with the quadratic programming algorithm. Biopolymers, 46, 329–342. [DOI] [PubMed] [Google Scholar]
- 24.Serebrov V., Clarke,R.J., Gross,H.J. and Kisselev,L. (2001) Mg2+-induced tRNA folding. Biochemistry, 40, 6688–6698. [DOI] [PubMed] [Google Scholar]
- 25.Misra V.K. and Draper,D.E. (2000) Mg(2+) binding to tRNA revisited: the nonlinear Poisson–Boltzmann model. J. Mol. Biol. 299, 813–825. [DOI] [PubMed] [Google Scholar]
- 26.Shi H. and Moore,P.B. (2000) The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: a classic structure revisited. RNA, 6, 1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jovine L., Djordjevic,S. and Rhodes,D. (2000) The crystal structure of yeast phenylalanine tRNA at 2.0 Å resolution: cleavage by Mg(2+) in 15-year old crystals. J. Mol. Biol. 301, 401–414. [DOI] [PubMed] [Google Scholar]
- 28.Agris P.F. (1996) The importance of being modified: roles of modified nucleosides and Mg2+ in RNA structure and function. Prog. Nucleic Acid Res. Mol. Biol., 53, 79–129. [DOI] [PubMed] [Google Scholar]
- 29.Stuart J.W., Basti,M.M., Smith,W.S., Forrest,B., Guenther,R., Sierzputowska-Gracz,H., Nawrot,A., Malkiewicz,A. and Agris,P.F. (1996) Structure of the trinucleotide D-acp3U-A with coordinated Mg2+ demonstrates that modified nucleosides contribute to regional conformations of RNA. Nucl. Nucl., 15, 1009–1029. [Google Scholar]
- 30.Chow C.S., Behlen,L.S.,Uhlenbeck,O.C. and Barton,J.K. (1992) Recognition or tertiary structure in tRNAs by Rh(phen)2phi3+, a new reagent for RNA structure-function mapping. Biochemistry, 31, 972–982. [DOI] [PubMed] [Google Scholar]
- 31.Scaringe S.A. (2001) RNA oligonucleotide synthesis via 5′-silyl-2′-orthoester chemistry. Methods, 23, 206–217. [DOI] [PubMed] [Google Scholar]
- 32.Pitsch S., Weiss,P.A., Wu,X., Ackermann,D. and Honegger,T. (1999) Fast and reliable automated synthesis of RNA and partially 2′-O-protected precursors (‘Caged RNA’) based on two novel, orthogonal 2′-O-protecting groups. Helv. Chim. Acta, 82, 1753–1761. [Google Scholar]
- 33.Ogilvie K.K., Usman,N., Nicoghosian,K. and Cedergren,R.J. (1988) Total chemical synthesis of a 77-nucleotide-long RNA sequence having methionine-acceptance activity. Proc. Natl Acad. Sci. USA, 85, 5764–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agris P.F., Malkiewicz,A., Kraszewski,A., Everett,K., Nawrot,B., Sochacka,E., Jankowska,J. and Guenther,R. (1995) Site-selected introduction of modified purine and pyrimidine ribonucleosides into RNA by automated phosphoramidite chemistry. Biochimie, 77, 125–34. [DOI] [PubMed] [Google Scholar]
- 35.England T.E. and Uhlenbeck,O.C. (1978) 3′-terminal labelling of RNA with T4 RNA ligase. Nature, 275, 560–561. [DOI] [PubMed] [Google Scholar]
- 36.Johnston R.J., Pickett,S.C. and Barker,D.L. (1990) Autoradiography using storage phosphor technology. Electrophoresis, 11, 355–360. [DOI] [PubMed] [Google Scholar]
- 37.Deng H.Y. and Termini,J. (1992) Catalytic RNA reactions of yeast tRNA(Phe) fragments. Biochemistry, 31, 10518–10528. [DOI] [PubMed] [Google Scholar]
- 38.Beuning P.J., Tessmer,M.R., Baumann,C.G., Kallick,D.A. and Musier-Forzyth,K. (1999) Sequence-dependent conformational differences of small RNAs as revealed by native gel electrophoresis. Anal. Biochem., 273, 284–290. [DOI] [PubMed] [Google Scholar]
- 39.Barciszewska M.Z., Rapp,G., Betzel.C., Erdmann,V.A. and Barciszewski,J. (2001) Structural changes of tRNA and 5S rRNA induced with magnesium and visualized with synchrotron mediated hydroxyl radical cleavage. Mol. Biol. Rep., 28, 103–110. [DOI] [PubMed] [Google Scholar]
- 40.Levitt M. (1969) Detailed molecular model for transfer ribonucleic acid. Nature 224, 759–763. [DOI] [PubMed] [Google Scholar]
- 41.Sampson J.R., Sullivan,F.X., Behlen,L.S., DiRenzo,A.B. and Uhlenbeck,O.C. (1987) Characterization of two RNA-catalyzed RNA cleavage reactions. Cold Spring Harbor Symp. Quant. Biol., 52, 267–275. [DOI] [PubMed] [Google Scholar]
- 42.Larkin D.C., Williams,A.M., Martinis,S.A. and Fox,G.E. (2002) Identification of essential domains for Escherichia coli tRNAleu aminoacylation and amino acid editing using minimalist RNA molecules. Nucleic Acids Res., 30, 2103–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russell R., Zhuang,X., Babcock,H.P., Millett,I.S., Doniach,S., Chu,S. and Herschlag,D. (2002) Exploring the folding landscape of a structured RNA. Proc. Natl Acad. Sci. USA, 99, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urbanke C., Romer,R. and Maass,G. (1975) Tertiary structure of tRNAPhe (yeast): kinetics and electrostatic repulsion. Eur. J. Biochem., 55, 439–444. [DOI] [PubMed] [Google Scholar]
- 45.Serra M.J., Baird,J.D., Dale,T., Fey,B.L., Retatagos,K. and Westhof,E. (2002) Effects of magnesium ions on the stabilization of RNA oligomers of defined structures. RNA, 8, 307–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodson S.A. (2000) Compact but disordered states of RNA. Nature Struct. Biol., 7, 349–352. [DOI] [PubMed] [Google Scholar]
- 47.Tinoco I. Jr, and Bustamante,B. (1999) How RNA folds. J. Mol. Biol., 293, 271–281. [DOI] [PubMed] [Google Scholar]
- 48.Wu M. and Tinoco,I.,Jr (1998) RNA folding causes secondary structure rearrangement. Proc. Natl Acad. Sci. USA, 95, 11555–11560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cole P.E. and Crothers,D.M. (1972) Conformational changes of transfer ribonucleic acid. Relaxation kinetics of the early melting transition of methionine transfer ribonucleic acid (Escherichia coli). Biochemistry, 11, 4368–4374. [DOI] [PubMed] [Google Scholar]
- 50.Riesner D., Maass,G., Thiebe,R., Philippsen,P. and Zachau,H.G. (1973) The conformational transitions in yeast tRNAPhe as studied with tRNAPhe fragments. Eur. J. Biochem., 36, 76–88. [DOI] [PubMed] [Google Scholar]
- 51.Ralston C.Y., He,Q., Brenowitz,M. and Chance,M.R. (2000) Stability and cooperativity of individual tertiary contacts in RNA revealed through chemical denaturation. Nature Struct. Biol., 7, 371–374. [DOI] [PubMed] [Google Scholar]
- 52.Russell R. and Herschlag,D. (2001) Probing the folding landscape of the Tetrahymena ribozyme: commitment to form the native conformation is late in the folding pathway. J. Mol. Biol., 308, 839–851. [DOI] [PubMed] [Google Scholar]
- 53.Helm M., Geige,R. and Florentz,C. (1999) A Watson–Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry, 38, 13338–13346. [DOI] [PubMed] [Google Scholar]
- 54.Meroueh M., Grohar,P.J., Qiu,J., SantaLucia,J.,Jr, Scaringe,S.A. and Chow,C.S. (2000) Unique structural and stabilizing roles for the individual pseudouridine residues in the 1920 region of Escherichia coli 23S rRNA. Nucleic Acids Res., 28, 2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newby M.I. and Greenbaum,N.L. (2001) A conserved pseudouridine modification in eukaryotic U2 snRNA induces a change in branch-site architecture. RNA, 7, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Misra V.K. and Draper,D.E. (2002) The linkage between magnesium binding and RNA folding. J. Mol. Biol., 317, 507–521. [DOI] [PubMed] [Google Scholar]
- 57.Russell R., Millett,I.S., Tate,M.W., Kwok,L.W., Nakatani,B., Gruner,S.M., Mochrie,S.G.J., Pande,V., Doniach,S., Herschlag,D. and Pollack,L. (2002) Rapid compaction during RNA folding. Proc. Natl Acad. Sci. USA, 99, 4266–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gutgsell N., England,N., Niu,L., Kaya,Y., Lane,B.G. and Ofengand,J. (2000) Deletion of the Escherichia coli pseudouridine synthase gene truB blocks formation of pseudouridine 55 in tRNA in vivo, does not affect exponential growth, but confers a strong selective disadvantage in competition with wild-type cells. RNA, 6, 1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Persson B.C., Gustafsson,C., Berg,D.E. and Bjork,G.R. (1992) The gene for a tRNA modifying enzyme, m5U54-methyltransferase, is essential for viability in Escherichia coli. Proc. Natl Acad. Sci. USA, 89, 3995–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]