

Abstract
Resistance to human immunodeficiency virus type 1 protease (HIV PR) inhibitors results primarily from the selection of multiple mutations in the protease region. Because many of these mutations are selected for the ability to decrease inhibitor binding in the active site, they also affect substrate binding and potentially substrate specificity. This work investigates the substrate specificity of a panel of clinically derived protease inhibitor-resistant HIV PR variants. To compare protease specificity, we have used positional-scanning, synthetic combinatorial peptide libraries as well as a select number of individual substrates. The subsite preferences of wild-type HIV PR determined by using the substrate libraries are consistent with prior reports, validating the use of these libraries to compare specificity among a panel of HIV PR variants. Five out of seven protease variants demonstrated subtle differences in specificity that may have significant impacts on their abilities to function in viral maturation. Of these, four variants demonstrated up to fourfold changes in the preference for valine relative to alanine at position P2 when tested on individual peptide substrates. This change correlated with a common mutation in the viral NC/p1 cleavage site. These mutations may represent a mechanism by which severely compromised, drug-resistant viral strains can increase fitness levels. Understanding the altered substrate specificity of drug-resistant HIV PR should be valuable in the design of future generations of protease inhibitors as well as in elucidating the molecular basis of regulation of proteolysis in HIV.
Human immunodeficiency virus type 1 (HIV-1) encodes a protease that is essential for processing of the Gag and Gag-Pol polyproteins and for maturation of infectious virus particles (18, 20). Because of its essential role in viral maturation, HIV protease (HIV PR) has been a major focus for drug development efforts. To date, six protease inhibitors have been approved for the treatment of HIV infection, and several more are currently in clinical trials. The ultimate long-term success of these drugs has been diminished, however, through the in vivo selection of drug-resistant strains of HIV (6).
Phenotypic resistance has been shown to result primarily from the accumulation of multiple mutations in the protease (8). Primary mutations, one or more of which are necessary for protease inhibitor resistance, include D30N, G48V, V82A/F/T, I84V, and L90M (for a review, see reference 6). Other, compensatory mutations are selected for increased enzyme activity and/or stability and include L10I/F, M36I, M46I, I54V, L63P, and A71T/V. The absence of a single common evolutionary pathway toward resistance precludes clear elucidation of the individual contributions of these mutations to the altered properties of a given drug-resistant protease variant.
In addition to mutations in the protease region, several groups have observed mutations in Gag cleavage sites that have been selected in response to saquinavir, ritonavir, indinavir, lopinavir, and other experimental inhibitors (11, 23, 46, 47). Mutations have been observed in all viral cleavage sites, and their occurrence correlates specifically with protease inhibitor therapy (9). However, two mutations occur at a significantly higher frequency. These are a Val→Ala substitution in the P2 position of the NC/p1 cleavage site and a Leu→Phe substitution at the P1′ position of the p1/p6 cleavage site (for an explanation of subsite nomenclature, see Materials and Methods). The frequency of the NC/p1 cleavage site mutation has been measured to be 16% in a study of 300 patients (B. A. B. Larder, K. Hertogs, C. Van den Eynde, and R. Pauwels, presented at the 2nd International Workshop on HIV Drug Resistance Treatment and Strategies, 1998). Both mutations have been shown to increase the fitness of protease inhibitor-resistant viruses (7, 11, 23, 46). Notably, cleavage at the NC/p1 site is among the slowest processing events in Gag (10, 28, 41, 44); for this reason, the NC/p1 cleavage has been suggested to be a rate-limiting step in Gag processing (9). Mutations in the p1/p6 cleavage site have been postulated to create a new frameshift site that could increase translation of the Pol polyproteins (12), thereby increasing the concentration of viral enzymes. However, the selection of mutations in both cleavage sites raises the possibility of simultaneous changes in protease specificity and a potential role of altered specificity in the development of drug resistance.
It is reasonable to expect that some HIV PR mutations will affect substrate specificity, since resistance mutations are primarily selected to alter inhibitor binding. Furthermore, it has been shown that various drug-resistant viruses produce different patterns of accumulated, partially processed polyproteins (46). This observation suggests that particular mutations in the protease can differentially affect cleavage site recognition and, by association, the substrate preference of the protease. Various mutations in the protease are associated with the NC/p1 cleavage site mutation in Gag, including M46I/L, V47A, and V82A/F/T (3, 7, 19), suggesting that these mutations may correlate to a change in substrate specificity. Moreover, in viruses with mutations in both the NC/p1 and p1/p6 cleavage sites, the protease mutations M46L, I54V, and V82A have been shown to increase the rate of viral replication (47). Changes in protease specificity may contribute to viral resistance (13) and/or to compensatory increases in the fitness of resistant viruses (7).
The precise determinants of HIV PR specificity are not well understood. To date, most studies have been conducted by comparison of related retroviral proteases (e.g., simian immunodeficiency virus, HIV-2, Rous sarcoma virus, murine leukemia virus) and the creation of chimeras that have elements of multiple proteases. These studies have identified many amino acids both in the active site (including residues 30, 32, 46, 47, 50, and others) and outside of the active site (residues 31 to 37 and 78 to 85) that are involved in determining the size and selectivity of the substrate binding pocket (22, 24, 38, 40). In one study, single mutations at residues 8, 32, 82, and 84 and double mutations at residues 48 and 90 or 82 and 84 did not have a large effect on substrate specificity (33). However, in another study, mutations G48V and L90M differentially affected the relative cleavage of peptide substrates with different amino acid sequences, suggesting that these mutations do have an effect on specificity (14). While many residues have been identified by these studies as being important for ligand specificity, the complexity of the data prevents the definition of contributions from individual residues or mutations.
Because of the difficulty in assigning changes in substrate specificity to specific mutations, the specificity of drug-resistant proteases must eventually be defined through the enzymatic characterization of individual variants. Traditionally, protease substrate specificity has been studied using series of closely related individual substrates. The advent of combinatorial peptide chemistry has led to the development of substrate libraries, which can be designed to maximize the amount of information gathered in a short time. Positional-scanning, synthetic combinatorial libraries (PS-SCL) of peptide substrates that probe the specificities of the P1 to P4 subsites have been used to assay a wide variety of serine and cysteine proteases (2, 15). These libraries have the potential to facilitate analysis of resistant HIV PR variants.
In this work, we have assembled a panel of seven clinically derived drug-resistant protease variants from viruses encoding different Gag cleavage site sequences. The variants contain multiple mutations and include most of the common resistance mutations. We compared the specificities of these proteases to that of wild-type HIV PR by using combinatorial libraries of peptide substrates. We show that, in certain cases, the accumulation of mutations in the protease results in subtle changes in substrate specificity. These changes are accompanied by substitutions in the NC/p1 cleavage site of the corresponding virus. This correlation between altered substrate specificity and an altered NC/p1 cleavage site strongly suggests a need to maintain efficient cleavage of the NC/p1 cleavage site, one of the poorest Gag substrates.
MATERIALS AND METHODS
Nomenclature.
Residues in substrates are designated Pn (for those N-terminal to the scissile bond) and Pn′ (for those C-terminal to the scissile bond) as per the notation defined by Schecter and Berger (37). For residues flanking the scissile bond, n = 1, and for residues farther away, the count increases by increments of 1. This nomenclature should not be confused with the terms p6, p2, and p1, which refer to specific Gag proteins.
Cloning.
Two different variants of wild-type HIV PR were cloned and characterized. The protease referred to in this paper as WT was expressed from a synthetic gene that encoded the Q7K mutation, which stabilizes the protease by reducing autoproteolysis (35), and the polymorphisms K14R, R41K, L63P, and I64V. The variant referred to as pNL4-3 has the NL4-3 wild-type consensus sequence. pNL4-3 and all drug-resistant variants were amplified from patient plasma using reverse transcription-PCR and cloned into resistance test vectors (26). Phenotypic analysis was conducted using a single-infection cycle assay as described previously (26).
For expression and purification of individual variants, the protease genes were amplified from the respective resistance test vectors using the primers 5′-GCACGTCCATGGCACCTCAGATCACTCTTTGGC-3′ and 5′-CCAGGACTCGAGGTCGACTAGAAATTTAAAG-3′. The former primer anneals to the beginning of the protease region and contains an NcoI site (underlined). The latter primer anneals in the reverse direction to the end of the protease region and contains an XhoI site (underlined). In the case of HIV PR mdr, the same primers were used to amplify the protease region from a PCR product that had been reverse transcribed from patient serum. The preparation of the PCR product has been described previously (16). Amplification products were cloned into the TOPO TA2.1 vector (Stratagene, La Jolla, Calif.) and then digested out of the TA2.1 vector using the restriction enzymes NcoI and XhoI. Fragments were subcloned through a pBS (Stratagene) shuttle vector into the pTacTac expression vector for expression in X90 cells as described previously (36). All positive clones were verified using DNA sequencing.
The mutant clone 945/A82V was constructed by PCR overlap extension. Initial PCRs were based on the pBS/945 template and the following pairs of primers: (i) CheY207.F (5′-GAAAACAATTCGTGCGGATGG-3′) and M945A82V.R (5′-GTTGACAGGTGTAGGTCTACTAATACTG-3′) and (ii) T7 (5′-AATACGACTCACTATAG-3′) and M945A82V.F (5′-GGACCTACACCTGTCAACATAATTGGAAGAAATCTG-3′) (mutations are shown in boldface). The resulting products were gel purified and then used as templates for a second PCR using the two outer primers CheY207.F and T7. The PCR product from this reaction was gel purified and cloned into the TOPO TA2.1 vector. Then the mutated protease gene was subcloned into the pTacTac expression vector as described above for all other protease clones. Positive clones were verified by sequencing.
Protein expression and purification.
All protease variants were expressed and purified using cation exchange and affinity columns as described previously (36). Proteins were purified to more than 95% homogeneity, as analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Following purification, all proteases were concentrated to a final concentration of 2 to 10 μM using Centricon-10 concentration units (Millipore, Bedford, Mass.).
Synthesis of ACC peptides and libraries.
Library assays and individual substrate assays used N-terminally acetylated tetra- and hexapeptides, respectively, with the modified coumarin leaving group 7-amino-4-carbamoylmethylcoumarin (ACC) (15). Libraries and substrates were synthesized using solid-phase synthesis (22a). Libraries were synthesized using isokinetic mixtures of amino acids to ensure that resulting mixtures of peptides would be equimolar (17, 25). Analysis of each library with a variety of proteases of differing specificities has confirmed the lack of any major bias (data not shown).
Activity assays.
All assays were run in assay buffer [100 mM 2-(N-morpholino)ethanesulfonic acid (MES; pH 6.0), 200 mM NaCl, 1 mM EDTA, 20% glycerol, 0.01% Tween-20]. Activity was monitored in microtiter plates using the ABI SpectraMax Gemini plate reader and SOFTmax software (Molecular Devices, Sunnyvale, Calif.). Assays for active-site titration used 75 μM concentrations of the substrate anthranilyl-Thr-Ile-norleucine (Nle)-Leu∼p-nitrophenylalanine-Gln-Arg-NH2 (Bachem, King of Prussia, Pa.) and were monitored as described by Toth and Marshall (39). The enzyme was titrated using the tight-binding (low- to mid-picomolar concentrations) active-site inhibitor JE-2147 (45). Inhibitor concentrations were varied, and the resulting rates were fit to the equation given below in order to determine total enzyme concentration. All rates were measured at 37°C in duplicate, and all titrations were repeated at least twice.
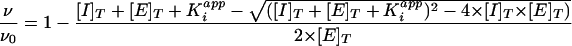 |
All assays on coumarin substrates were monitored at an excitation wavelength of 380 nm and an emission wavelength of 450 nm with a filter at 435 nm. At this combination of wavelengths, free ACC has a 900-fold-higher fluorescent yield than peptide-bound ACC (15). Thus, the increase in fluorescent signal reflects only proteolysis in the expected register. Internal cleavage has been previously observed in library assays through the resulting depletion of residues at non-P1 positions (15). We have not observed any evidence of internal cleavage by HIV PR in the library assays.
In the P1-diverse library, the total concentration of substrates was maintained at 0.16 mM, which resulted in an average substrate concentration of 0.02 μM for each of the 6,859 individual substrates per well. This value is well below the Km of any substrate measured in this study. Because HIV PR, which has a relatively narrow specificity, was unlikely to turn over more than 1% of the peptides per well, the total concentration of potential substrates remained at or below 1.6 μM. These conditions ensured that all substrates were analyzed in the kcat/Km range, in which initial rates are proportional to substrate concentrations. Because of the extremely low concentrations of individual substrates and the poor binding ability of this class of substrates, a large concentration of protease (10 μM) was required in order for activity to be observed. Results are reported as initial rates in arbitrary units.
P1-fixed libraries are referred to individually as P1-Xaa, where Xaa is the amino acid residue fixed at the P1 position. In the P1-fixed libraries there were 361 compounds per well, with an average individual substrate concentration of 0.25 μM (for a total concentration of 0.1 mM). With an estimate that the protease could bind only 10% or less of the peptides present, the total concentration of potential substrates was kept below 10 μM, safely below the Km range. Final protease concentrations used for these assays were 215 nM for P1-Met (pNL4-3 PR), 140 nM for P1-Nle (pNL4-3 PR), and 1 μM for P1-Leu (WT). All library assays were run at 37°C. Assays were run at least twice and are reported as average rates relative to the best rate in P3. For two variants, HIV PR 309 and HIV PR 1049, the P1-Met library was assayed only once and thus error was not determined. For all other experiments, error was ≤15% of reported values shown in Fig. 1 and 3.
FIG. 1.

Substrate preferences for HIV PR. (A) Initial rates for HIV PR with the P1-diverse library indicate the relative preferences for residues at P1. Rates are expressed as relative fluorescent units per second. (B) Preferences at P2 through P4 were assayed using three P1-fixed libraries. Data bars are colored as specified in the key in the P2 panel. All values are reported as initial rates relative to either P3-Leu (for the P1-Met and P1-Nle libraries) or P3-Trp (for the P1-Leu library). Since a single experiment collects data on all three subsites simultaneously, the y-axis scale is consistent among the three panels.
FIG. 3.

The substrate preferences of HIV PR and drug-resistant protease variants were compared using P1-Met library results. All experiments have been normalized to Nle at P3 in order to facilitate comparison.
Individual kinetic constants for isolated peptides were determined using 20 to 2,000 nM protease and substrate concentrations ranging from 50 to 2,000 μM. The enzyme concentration was always held at least 100-fold lower than the lowest substrate concentration in order to satisfy steady-state assumptions. Rates were measured at 37°C and fit to the Michaelis-Menten equation for steady-state kinetics in order to determine the specificity constant kcat/Km. All individual rates were determined in duplicate, and all experiments were repeated at least twice.
Computational modeling.
The K45Q and D30N mutations were modeled using the InsightII software package (Molecular Simulations, Inc., San Diego, Calif.) on an Indigo II workstation (Silicon Graphics, Mountain View, Calif.). Structural coordinates from the 7hvp.pdb and 1hvr.pdb files were overlaid using the conserved catalytic residues 24 to 26 in both monomers. Distances were then measured from the side chain atoms of Asp30 and Lys45 of the 1hvr structure to the carbonyl of the P4 acetyl group in the bound ligand of the 7hvp structure.
RESULTS
Specificity of HIV PR.
The specificity of wild-type HIV PR was determined using two types of PS-SCL. To examine specificity at P1, a P1-diverse library was used. In this library, 20 amino acids (representing all natural residues except for cysteine and with the addition of Nle) were arrayed at the P1 position of a tetrapeptide coumarin substrate while the same amino acids (excluding methionine) were kept in mixtures of equimolar concentrations at positions P2 to P4. There were a total of 6,859 peptides per well, each of which was at a concentration well below the expected Km for HIV PR. This ensured that all substrates were analyzed in the kcat/Km range, in which initial rates are proportional to substrate concentrations (see Materials and Methods). Analysis of HIV PR revealed a strong preference for Met, followed, in order of decreasing preference, by Nle, Phe, and Leu (Fig. 1A). In previous studies on the substrate specificity of HIV PR, the preferred residues at P1 have generally been Met, Tyr, Nle, Phe, and Leu, although the order of preference has depended on the residue in P1′ (4, 21, 31).
Based on the P1 preferences of HIV PR, three libraries were selected for further analysis of subsite specificity. These libraries each had a single amino acid fixed at P1 (Met, Nle, or Leu) and are referred to as P1-fixed libraries. Each library contained three sublibraries (P2 to P4). In each sublibrary, 19 amino acids were arrayed at one position while all amino acids were in mixtures at equimolar concentrations at the remaining two positions, resulting in 361 peptides per well. As before, individual substrates were held at concentrations well below the expected Km. Results obtained for HIV PR on the three different P1-fixed libraries indicated cooperativity between subsite preferences, i.e., relative preferences at subsites P2, P3, and P4 depended on the identity of the residue at P1 (Fig. 1B).
At P2, there was a strong preference for the hydrophobic residues Val and Ile and, to a lesser extent, Ala and Glu. Other studies have reported a preference for Val and Ile, with varying levels of other hydrophobic residues, as well as Asn, for substrates that have hydrophobic residues at P1 or P1′ (4, 21, 30). The ability of HIV PR to turn over substrates with Glu at P2, observed with the PS-SCL, has not been reported in previous studies. All other observations based on the PS-SCL were consistent with published data.
At P3, there was a slightly broader preference for the larger, hydrophobic residues Leu, Trp, Tyr, and Nle; the strongest preference was Leu when P1 was Met or Nle, and Trp when P1 was Leu or Nle. Ridky et al. also observed significant cooperativity between the P1 and P3 sites, suggesting that the size of each subsite is dependent on the residue bound in the cis adjacent site (32). In general, previous studies reported P3 preference for a wide variety of mostly hydrophobic residues, the relative ranking of which depended on the extended sequence of the substrate (5, 21, 43).
P4 was the subsite with the broadest specificity and the greatest number of differences between libraries. However, all three libraries showed a strong tendency toward small residues. Similarly, a preference for small amino acids such as serine in this position has been reported (42).
Because library results measured cleavage of pools of substrates, each of the resulting rates was an average over the whole pool. Any cooperativity between subsites would result in a disparity between library-based predictions and specificity constants determined for individual substrates. Cooperativity between subsites of HIV PR was tested by the synthesis and assay of individual peptide substrates that were designed to test preferences measured using the libraries (Table 1). Substrate 6, with the sequence Ac-Arg-Lys-Asp-Leu-Asp-Met∼ACC, was synthesized to confirm that the inclusion of Asp at P2 would prevent the peptide from being cleaved, as predicted by P1-Met library results. All remaining peptides were, as predicted, cleaved by HIV PR. In every case, the Km was estimated to be in the range of 1 mM or higher; thus, the kcat and Km constants were not measured separately. The range of specificity constants for the ACC peptides was between 26 and 477 M−1 s−1. The best of these substrates, Ac-Arg-Lys-Ser-Leu-Ile-Met∼ACC, had a specificity constant of 477 M−1 s−1. This value was nearly 3 orders of magnitude lower than that for the substrate anthranilyl-Thr-Ile-Nle-Leu∼p-nitrophenylalanine-Gln-Arg-NH2, a hexapeptide that bound in P3-P3′ and had a specificity constant of 2.8 × 105 M−1 s−1. In general, HIV PR requires extensive interactions with substrates on both sides of the scissile bond. The diminished capability to cleave ACC substrates likely resulted from the fact that the peptide portion of the substrate was bound only on the non-prime side of the scissile bond. With no amino acid residues bound on the prime side, only the ACC group was available for interaction with prime side determinants, resulting in the measurable but low level of turnover by HIV PR.
TABLE 1.
Specificity constants for wild-type HIV PR with peptide substrates
| Substrate no. | Compositiona | kcat/Km (M−1 s−1) |
|---|---|---|
| 1 | Abz-TIn∼F (NO2) QR-NH2 | 280,000 ± 40,000 |
| 2 | Ac-RKSLIL∼ACC | 33.8 ± 7.1 |
| 3 | Ac-RKSLIM∼ACC | 477 ± 124 |
| 4 | Ac-RKDLIM∼ACC | 103 ± 7.2 |
| 5 | Ac-RKDWIM∼ACC | 104 ± 26.7 |
| 6 | Ac-RKDLDM∼ACC | < 0.2 |
| 7 | Ac-RKDLIn∼ACC | 26.1 ± 1.4 |
| 8 | Ac-RKSLIn∼ACC | 220 ± 1.7 |
| 9 | Ac-RKSLAn∼ACC | 53.1 ± 28.1 |
| 10 | Ac-RKSLVn∼ACC | 296 ± 83.4 |
Abz, anthranilyl; n, norleucine.
ACC peptide substrates constructed to test preferences for Met, Nle, and Leu at P1 showed a strong correlation with library results; however, substrates that varied only at P2, P3, or P4 did not always agree with library predictions. For example, specificity constants for substrates 4 and 5, which vary only at P3, as well as constants for substrates 8 to 10, which vary only at P2, reflect overall preferences from library predictions but differ somewhat. Differences between library results and results from individual substrates were most likely due to cooperativity between subsites, for which there are strong precedents in the published literature on retroviral proteases (29, 32).
Characterization of the panel of drug-resistant HIV PRs.
A panel of seven proteases cloned from clinical isolates was assembled to investigate potential changes in substrate specificity. These variants harbored a variety of mutations in both the protease and the Gag cleavage sites NC/p1 and p1/p6 (Fig. 2A). Mutations in the protease sequence included the commonly observed resistance-associated mutations L10I, D30N, M36I, M46I, G48V, I54V, L63P, A71T/V, V82A/I/T, I84V, N88D/S, and L90M. Mutations M46I, I54V, and V82A/T, implicated in affecting specificity (47), were present in several permutations. The viral genome that encoded HIV PR mdr encoded wild-type sequences at both the NC/p1 and p1/p6 cleavage sites. Viruses from which variants 945, 958, and 1049 were cloned also encoded an Ala→Val substitution in P2 of the NC/p1 cleavage site. Viruses encoding the three variants 309, 769, and 954 encoded a Leu→Phe or Leu→Pro substitution in P1′ of the p1/p6 cleavage site. Data on the protease inhibitors to which the viruses had been exposed in the patient, as well as phenotypic drug resistance profiles, are shown in Fig. 2B. Several of the variants were from multiple-drug-resistant viruses. Notably, HIV PR mdr was cloned from a virus that was transmitted to a new host despite resistance to several different protease and reverse transcriptase inhibitors (16).
FIG. 2.

(A) Alignment of all protease sequences used in this study. All sequences include NC/p1 and p1/p6 cleavage site sequences except for WT, the synthetic wild-type variant. Mutations associated with protease inhibitor resistance are shown against a green background; polymorphic mutations are shown against a blue background. The Q7K mutation in WT, highlighted in gray, was engineered to stabilize the protease to autoproteolysis. (B) In vivo exposure to protease inhibitors and phenotypic resistance profiles are shown for the drug-resistant protease panel. Inhibitor susceptibilities are expressed as 50% inhibitory concentrations relative to that of a reference control.
Drug-resistant protease variants were screened using only the P1-Met fixed PS-SCL, on which HIV PR is most active. A comparison of all proteases assayed indicated that overall specificity is conserved among variants (Fig. 3). Two of the proteases assayed, HIV PR mdr and HIV PR 309, had specificities identical to that of HIV PR in all subsites. This was particularly striking because these clones had 12 and 11 mutations, respectively, in the 99-amino-acid protease monomer, representing changes in more than 10% of the total protein sequence. To further test for differences, HIV PR mdr was assayed with the P1-Leu and P1-Nle libraries. Results from all three P1-fixed libraries confirmed that HIV PR mdr had a substrate specificity identical to that of wild-type HIV PR (data not shown). Given that the protease must cleave at nine sequentially distinct sites during maturation, this conservation of specificity is evidence of the close regulation of proteolytic activity.
Despite a general conservation of specificity, several variants demonstrated significant changes. HIV PR 954 had the same specificity as wild-type HIV PR at P2 and P3 but demonstrated a sharp decrease in hydrolysis of peptides with Asp and Glu at P4 (Fig. 4). This change was accompanied by an increase in hydrophobic preferences at that subsite. HIV PR 954 harbored two mutations, K45Q and D30N, which are not found in any other protease from the panel. Both of these residues lie within hydrogen-bonding distance (<3.5 Å) of the substrate P4 residue. The decrease in acidic preference is most likely due to the loss of the lysine at position 45.
FIG. 4.

Comparison of P1-Met data for the WT, mdr, and 954 proteases at position P4. Rates are shown only for residues with altered preferences.
Changes in Val/Ala preference at P2.
The most significant variations in specificity were observed in the relative preferences for valine and alanine at P2. This change in specificity was measured as the ratio of the initial rate of valine cleavage at P2 to the initial rate of alanine cleavage at P2. Based on library results, this ratio was in the range of 1.7 to 2.8 for HIV PR as well as several drug-resistant variants, indicating that, on average, substrates with valine at P2 were cleaved twice as well as those with alanine at P2 (Table 2). The Val/Ala ratio was significantly altered for three variants; it was higher for HIV PRs 945 and 958 (with ratios of 5.0 and 6.1, respectively), and it was lower for HIV PR 769 (with a ratio of 0.8).
TABLE 2.
Specificity constants and P2 Val/Ala ratios for wild-type and drug-resistant HIV PR variants
| HIV PR | Library-based Val/Ala ratio | Ac-RKSLVn∼ACC kcat/Km (M−1 s−1) | Ac-RKSLAn∼ACC kcat/Km (M−1 s−1) | Substrate-based Val/Ala ratio |
|---|---|---|---|---|
| WT | 1.7 ± 0.6 | 296 ± 83.4 | 53.1 ± 28.1 | 5.6 ± 0.6 |
| mdr | 2.8 ± 0.9 | 63.4 ± 2.4 | 9.4 ± 0.2 | 6.8 ± 0.04 |
| 309 | 2.3 | 13.9 ± 0.6 | 1.4 ± 0.2 | 9.9 ± 0.1 |
| 769 | 0.8 ± 0.04 | 37 ± 14 | 7.1 ± 1.4 | 5.3 ± 0.4 |
| 945 | 5.0 ± 1.2 | 14.0 ± 2.0 | 1.0 ± 0.3 | 14.4 ± 0.4 |
| 958 | 6.1 ± 0.3 | 5.1 ± 1.1 | 0.2 ± 0.1 | 22.8 ± 0.5 |
| 1049 | 2.0 | 80.7 ± 7.2 | 4.8 ± 0.2 | 17.0 ± 0.1 |
| 945/A82V | 3.4 ± 0.2 | 38.0 ± 2.9 | 2.4 ± 1.0 | 16.0 ± 0.4 |
To further investigate this potential change in substrate specificity, individual peptide substrates were synthesized. Substrates representing the natural cleavage site sequences, Ac-Glu-Arg-Gln-Ala-Asn∼ACC and Ac-Glu-Arg-Gln-Val-Asn∼ACC, were not cleaved by wild-type HIV PR even at concentrations up to 10 μM protease and 1 mM substrate (data not shown). This result was consistent with studies showing that the representative decapeptide was poorly cleaved (11). To increase the sensitivity of the assay, proteases were assayed with substrates based on the optimal substrate that resulted from profiling the wild-type protease, Ac-Arg-Lys-Ser-Ile-Met∼ACC. To reduce the risk of oxidation, norleucine was substituted for methionine at P1. The preference at P2 was then assayed using substrates with either alanine or valine at P2: Ac-Arg-Lys-Ser-Leu-Ala-Nle∼ACC and Ac-Arg-Lys-Ser-Leu-Val-Nle∼ACC. Specificity constants were measured for HIV PR and six drug-resistant protease variants (Table 2). For all drug-resistant proteases except HIV PR mdr, the level of activity on the substrate with alanine at P2 was less than 10% that for HIV PR. Replacement of the alanine at P2 with valine resulted in a better substrate in every case. Val/Ala ratios measured with these substrates differed from those measured with the P1-Met library because the library results were averages over hundreds of peptides. Nonetheless, HIV PR 945 and HIV PR 958, as well as HIV PR 1049, maintained ratios that were significantly higher (14.4, 22.8, and 17.0, respectively) than that of wild-type HIV PR (5.6).
Determinants of changes in specificity: Val82Ala.
All three drug-resistant variants with increased Val/Ala ratios at P2 had reductive mutations in active-site residues. HIV PR 958 had an I84V substitution, and HIV PRs 945 and 1049 each had a V82A substitution. Although residues 82 and 84 form part of the S1 pocket and not the S2 pocket, a reductive mutation at either position could cause a shift in bound substrate that would favor larger residues at the adjacent site, S2. In order to test the effect of the V82A substitution on specificity at P2, position 82 in HIV PR 945 was reverted to the wild-type residue, valine. The resulting variant, HIV PR 945/A82V, was assayed using both the P1-Met library and the individual substrates described in the preceding section. The specificity constants of HIV PR 945/A82V were three times greater than those of HIV PR 945, which is consistent with the reconstitution of a wild-type-like active site. In addition, the library-based Val/Ala ratio for the revertant was 3.4, which is midway between that of the parent HIV PR 945 (5.0) and that of wild-type HIV PR (1.7). Although the individual-substrate-based valine/alanine ratio for HIV PR 945/A82V did not shift toward the wild-type value, the library results remain a compelling indicator of altered specificity because they reflect proteolysis of a large pool of peptide substrates. The observation that the A82V reversion mutation seems to affect specificity but does not completely return the protease to wild-type-like specificity is indicative of the complexity of retroviral protease substrate specificity.
Correlation of altered specificity with viral cleavage sites.
The shift of relative preferences for valine and alanine at P2 is particularly intriguing because of its potential relevance to the resistance mutation in which Val is substituted for Ala at the P2 position of the NC/p1 cleavage site. Of the seven drug-resistant variants examined here, three were derived from viruses that harbored this mutation (Fig. 2A). These are the same three variants, 945, 958, and 1049, that demonstrated an increased preference for Val relative to Ala at P2. HIV PR 769, which preferred Ala to Val at that position, was derived from a virus that harbored the wild-type cleavage site with Ala at P2. This suggests that four out of seven viruses have evolved, through varying pathways, toward increased efficiency of the NC/p1 cleavage.
DISCUSSION
The substrate specificity of HIV PR has been studied previously using series of individual peptide substrates that differ only by one or two residues. It is prohibitively laborious to conduct a complete analysis of all residues at every subsite using this method. We have chosen to use PS-SCLs of tetrapeptide substrates, which allow simultaneous analysis of an unbiased set of thousands of substrates in a short period of time. In this work, we have compared the non-prime-side specificity of HIV PR with that of a panel of clinically derived drug-resistant HIV PR variants.
Given the strict requirements of aspartyl proteases for extended substrate interactions, it is unusual that HIV PR is able to catalyze proteolysis of the tetrapeptide ACC substrates, in which only the coumarin binds on the prime side. Indeed, the specificity constants for the ACC substrates are at least 3 orders of magnitude lower than those for longer substrates. That ACC proteolysis is measurable runs contrary to the findings of prior studies that have shown the minimal length of an HIV PR substrate to be P4 to P3′ (10, 42). However, specificity constants this low would be extremely difficult to measure using the detection methods typically used with HIV PR, which often involve high-pressure liquid chromatographic analysis and/or the use of a less-sensitive chromogenic or fluorogenic reporter group. Because the free ACC group fluoresces strongly, proteolysis of these substrates may be detected at rates that are below previous detection limits. The lack of extended peptide binding on the prime side does not appear to affect non-prime-side specificity, since the substrate specificity of HIV PR determined using the PS-SCLs is consistent with that reported previously for longer peptide substrates. We are therefore confident that the comparisons between wild-type HIV PR and drug-resistant HIV PR that are based on the ACC libraries reflect true changes in specificity.
Because resistance mutations are selected for decreased inhibitor binding and potentially alter the shape of the active site, it follows that the same mutations may affect substrate specificity. To the contrary, all clinically derived proteases in this study demonstrated a general conservation of wild-type specificity. Moreover, both HIV PR 309 and HIV PR mdr had specificities indistinguishable from that of wild-type HIV PR despite 11 and 12 mutations, respectively, with respect to the wild-type sequence. Notably, HIV PR mdr harbors the mutation L90M, which as a single mutation has been shown to alter the specificity of WT protease in a previous study (14). Taken together, these data suggest that compensatory mutations, in addition to being selected for increased activity, may also be selected for maintenance of specificity. This preservation of specificity is consistent with the role of the protease in the viral life cycle, in which it must cleave nine different sites in a specific, well-regulated order.
There were a few exceptions in which we observed changes in protease specificity. In the most dramatic case, HIV PR 954 demonstrated greatly reduced cleavage of acidic residues at P4. Because this is likely the result of losing a charge-charge interaction, we are able to predict with some certainty the amino acid residues responsible for the change. This protease variant has two mutations that are unique within the panel of variants studied here, namely, D30N and K45Q. The reduced preference for acidic residues is likely due to the loss of the basic lysine residue at position 45. The D30N mutation may also be involved in the concomitant increase in hydrophobic preference for this protease. Mutations at Asp30 as well as Lys45 have been observed in drug-resistant proteases.
The most intriguing alteration in specificity is the deviation in the preference for valine relative to alanine at P2. Several protease variants, i.e., HIV PRs 945, 958, and 1049, demonstrate an increase in preference for valine relative to alanine. The altered specificity, observed with the libraries, is corroborated by specificity constants for individual substrates. By one or both measures, the ratios of valine turnover to alanine turnover for HIV PRs 945, 958, and 1049 are approximately twice that for wild-type HIV PR. All variants with an increased Val/Ala ratio contain a reductive mutation at either position 82 or position 84, both situated in the active site.
The reversion of position 82 to Val in HIV PR 945 resulted in a partial but not complete return to wild-type specificity. The residue at position 82 contributes directly to the S1 binding pocket and does not come in contact with the P2 residue (33). Because the substrate binds in an extended, β-strand conformation, a mutation that makes the S1 subsite larger may result in a shift in the position of the substrate backbone toward the S1 pocket. This would result in a preference for larger amino acids at P2. The mutation at position 82 seems to be partially responsible for changing the specificity, but it is clearly insufficient by itself. Several previous studies have noted the correlation of the altered NC/p1 site with mutations at position 82, in particular V82A (3, 9). In addition, studies on NC/p1 cleavage site mutations have implicated protease mutations at V32, M46, I47, and I54, all of which are in the vicinity of the S2 subsite (7, 19, 40, 47). The interplay among the determinants of substrate specificity, which include these mutations and others yet to be identified, is clearly complex.
Among the viruses examined in this study, the increased preference for valine over alanine at P2 corresponds in every case to a NC/p1 cleavage site mutation in which valine has replaced alanine at the P2 position. The result of the cleavage site mutation is a more efficient substrate. The additional selection for an increased preference for valine suggests that there may be further selective pressure to increase proteolysis of the NC/p1 cleavage site. Furthermore, HIV PR 769 demonstrated a marked increase in the preference for alanine relative to valine, even preferring alanine over valine at the P2 position. Based on this result, we would predict that HIV PR 769 could cut the wild-type cleavage site, which has alanine at P2, more efficiently than the mutated sequence. Indeed, the corresponding virus encodes a Gag polypeptide with the wild-type NC/p1 cleavage site. We postulate that the different clinical isolates from which the panel of variant proteases was obtained have developed resistance to protease inhibitors and maintained efficiency of cleavage of the NC/p1 site through multiple evolutionary mechanisms.
Such consistent evolution is likely to be relevant in the context of viral subspecies competing in a highly selective environment. Under such conditions, it is plausible that the rate of viral maturation is limited by single molecular events, such as cleavage of a specific Gag site. This hypothesis of a “choke point” in viral fitness is consistent with our previous demonstration that there is a minimum threshold of proteolytic activity of 2 to 5% (1, 34), which is approximately the level of activity of the resistant HIV PR variants in our panel. Below this level of activity, infectious particles are no longer produced. Because the protease cuts the cleavage sites within Gag and Gag-Pol with different efficiencies, this threshold is most likely defined by one or a few sites rather than by all nine cleavage sites. When the protease is a poor catalyst, which often happens as a consequence of acquiring resistance to protease inhibitors, HIV may be under selective pressure to increase the efficiency of the slowest cleavage events. In vitro, the order of Gag cleavage has been shown to be as follows: p2/NC, MA/CA and p1/p6, NC/p1, and CA/p2 (28). Cleavage of CA/p2 and cleavage of NC/p1 are the two slowest cleavage events and are thus good candidates for threshold-defining events.
The order of cleavage of isolated peptide substrates representing the Gag sites differs from the order of cleavage in the context of full-length Gag. Specifically, in the absence of the entire Gag polyprotein, the CA/p2 site is cleaved approximately as efficiently as the MA/CA site, which is one of the best Gag substrates (41). In the context of the complete Gag polyprotein, cleavage at the CA/p2 site is reduced as a result of the p2/NC cleavage (27). Thus, the CA/p2 cleavage event is regulated by more than the interaction of the substrate with the active site of the protease, and selection for improved cleavage at this site would likely not be observable in vitro as a simple change in substrate specificity. To the contrary, the inefficiency of NC/p1 cleavage is completely consistent with poor catalysis of the corresponding decapeptide substrate. Thus, there is great potential for enhancement of this cleavage event through an evolution of the protease-substrate interaction. The coevolution of protease specificity and cleavage site sequence toward a more-efficient cleavage of the NC/p1 cleavage site suggests that cleavage of this site becomes limiting in the context of impaired, drug-resistant protease.
In this work, we have compared the substrate specificities of a panel of drug-resistant protease clones from patients. While previous studies have looked at the effects of individual mutations, subtle changes may have been missed because the effect is dependent on multiple mutations. By studying multiply mutated clones from HIV-positive patients, we have been able to compare relevant sets of mutations and their roles in determining protease specificity. Identification of mutations or sets of mutations that change specificity, such as K45Q, may help to guide the design of future generations of protease inhibitors. In addition, variations in preference for valine versus alanine at P2 have illuminated the role of the NC/p1 cleavage as a regulatory checkpoint in viral maturation. Regulation of this cleavage event may be eased when the virus is under antiproteolytic pressure. Further studies to correlate altered specificity and cleavage site mutations with viral fitness are expected in order to further refine this model, identify other regulatory checkpoints, and ultimately provide a better understanding of the regulation of viral maturation.
Acknowledgments
C.S.C. was funded by NIH grant GM56531 and the Daiichi Research Center. D.S.D. was funded by UARP grants D99-SF-011 and D00-SF-076. J.A.E. and D.J.M. were funded by NIH grant GM54051.
We thank Agouron for the tight-binding inhibitor JE-2147 and R. Grant (San Francisco General Hospital) for the gift of the PCR product encoding HIV PR mdr. We also thank I. D. Kuntz (University of California, San Francisco), S. Todd (Incyte Pharmaceuticals), and T. Wrin (ViroLogic, Inc.) for many helpful conversations as well as critical evaluation of the manuscript.
REFERENCES
- 1.Babé, L. M., J. Rosé, and C. S. Craik. 1995. Trans-dominant inhibitory human immunodeficiency virus type 1 protease monomers prevent protease activation and virion maturation. Proc. Natl. Acad. Sci. USA 92:10069–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backes, B. J., J. L. Harris, F. Leonetti, C. S. Craik, and J. A. Ellman. 2000. Synthesis of positional-scanning libraries of fluorogenic peptide substrates to define the extended substrate specificity of plasmin and thrombin. Nat. Biotechnol. 18:187–193. [DOI] [PubMed] [Google Scholar]
- 3.Bally, F., R. Martinez, S. Peters, P. Sudre, and A. Telenti. 2000. Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res. Hum. Retrovir. 16:1209–1213. [DOI] [PubMed] [Google Scholar]
- 4.Beck, Z. Q., L. Hervio, P. E. Dawson, J. H. Elder, and E. L. Madison. 2000. Identification of efficiently cleaved substrates for HIV-1 protease using a phage display library and use in inhibitor development. Virology 274:391–401. [DOI] [PubMed] [Google Scholar]
- 5.Billich, A., and G. Winkler. 1991. Analysis of subsite preferences of HIV-1 proteinase using MA/CA junction peptides substituted at the P3–P1′ positions. Arch. Biochem. Biophys. 290:186–190. [DOI] [PubMed] [Google Scholar]
- 6.Boden, D., and M. Markowitz. 1998. Resistance to human immunodeficiency virus type 1 protease inhibitors. Antimicrob. Agents Chemother. 42:2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrillo, A., K. D. Stewart, H. L. Sham, D. W. Norbeck, W. E. Kohlbrenner, J. M. Leonard, D. J. Kempf, and A. Molla. 1998. In vitro selection and characterization of human immunodeficiency virus type 1 variants with increased resistance to ABT-378, a novel protease inhibitor. J. Virol. 72:7532–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Condra, J. H., D. J. Holder, W. A. Schleif, O. M. Blahy, R. M. Danovich, L. J. Gabryelski, D. J. Graham, D. Laird, J. C. Quintero, A. Rhodes, H. L. Robbins, E. Roth, M. Shivaprakash, T. Yang, J. A. Chodakewitz, P. J. Deutsch, R. Y. Leavitt, F. E. Massari, J. W. Mellors, K. E. Squires, R. T. Steigbigel, H. Teppler, and E. A. Emini. 1996. Genetic correlates of in vivo viral resistance to indinavir, a human immunodeficiency virus type 1 protease inhibitor. J. Virol. 70:8270–8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cote, H. C. F., Z. L. Brumme, and P. R. Harrigan. 2001. Human immunodeficiency virus type 1 protease cleavage site mutations associated with protease inhibitor cross-resistance selected by indinavir, ritonavir, and/or saquinavir. J. Virol. 75:589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darke, P. L., R. F. Nutt, S. F. Brady, V. M. Garsky, T. M. Ciccarone, C. T. Leu, P. K. Lumma, R. M. Freidinger, D. F. Veber, and I. S. Sigal. 1988. HIV-1 protease specificity of peptide cleavage is sufficient for processing of gag and pol polyproteins. Biochem. Biophys. Res. Commun. 156:297–303. [DOI] [PubMed] [Google Scholar]
- 11.Doyon, L., G. Croteau, D. Thibeault, F. Poulin, L. Pilote, and D. Lamarre. 1996. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 70:3763–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doyon, L., C. Payant, L. Brakier-Gingras, and D. Lamarre. 1998. Novel Gag-Pol frameshift site in human immunodeficiency virus type 1 variants resistant to protease inhibitors. J. Virol. 72:6146–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erickson, J. W., and S. K. Burt. 1996. Structural mechanisms of HIV drug resistance. Annu. Rev. Pharmacol. Toxicol. 36:545–571. [DOI] [PubMed] [Google Scholar]
- 14.Ermolieff, J., X. L. Lin, and J. Tang. 1997. Kinetic properties of saquinavir-resistant mutants of human immunodeficiency virus type 1 protease and their implications in drug resistance in vivo. Biochemistry 36:12364–12370. [DOI] [PubMed] [Google Scholar]
- 15.Harris, J. L., B. J. Backes, F. Leonetti, S. Mahrus, J. A. Ellman, and C. S. Craik. 2000. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. USA 97:7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hecht, F. M., R. M. Grant, C. J. Petropoulos, B. Dillon, M. A. Chesney, H. Tian, N. S. Hellmann, N. I. Bandrapalli, L. Digilio, B. Branson, and J. O. Kahn. 1998. Sexual transmission of an HIV-1 variant resistant to multiple reverse-transcriptase and protease inhibitors. N. Engl. J. Med. 339:307–311. [DOI] [PubMed] [Google Scholar]
- 17.Houghten, R. A., C. Pinilla, J. R. Appel, S. E. Blondelle, C. T. Dooley, J. Eichler, A. Nefzi, and J. M. Ostresh. 1999. Mixture-based synthetic combinatorial libraries. J. Med. Chem. 42:3743–3778. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan, A. H., J. A. Zack, M. Knigge, D. A. Paul, D. J. Kempf, D. W. Norbeck, and R. Swanstrom. 1993. Partial inhibition of the human immunodeficiency virus type 1 protease results in aberrant virus assembly and the formation of noninfectious particles. J. Virol. 67:4050–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koch, N., N. Yahi, J. Fantini, and C. Tamalet. 2001. Mutations in HIV-1 gag cleavage sites and their association with protease mutations. AIDS 15:526–528. [DOI] [PubMed] [Google Scholar]
- 20.Kohl, N. E., E. A. Emini, W. A. Schleif, L. J. Davis, J. C. Heimbach, R. A. Dixon, E. M. Scolnick, and I. S. Sigal. 1988. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 85:4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konvalinka, J., P. Strop, J. Velek, V. Cerna, V. Kostka, L. H. Phylip, A. D. Richards, B. M. Dunn, and J. Kay. 1990. Sub-site preferences of the aspartic proteinase from the human immunodeficiency virus, HIV-1. FEBS Lett. 268:35–38. [DOI] [PubMed] [Google Scholar]
- 22.Lin, Y. C., Z. Beck, T. Lee, V. D. Le, G. M. Morris, A. J. Olson, C. H. Wong, and J. H. Elder. 2000. Alteration of substrate and inhibitor specificity of feline immunodeficiency virus protease. J. Virol. 74:4710–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22a.Maly, D. J., F. Leonetti, B. J. Backes, D. S. Dauber, J. L. Harris, C. S. Craik, and J. A. Ellman. Expedient solid-phase synthesis of fluorogenic protease substrates using the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. J. Organic Chem., in press. [DOI] [PubMed]
- 23.Mammano, F., C. Petit, and F. Clavel. 1998. Resistance-associated loss of viral fitness in human immunodeficiency virus type 1: phenotypic analysis of protease and gag coevolution in protease inhibitor-treated patients. J. Virol. 72:7632–7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menéndez-Arias, L., I. T. Weber, and S. Oroszlan. 1995. Mutational analysis of the substrate binding pocket of murine leukemia virus protease and comparison with human immunodeficiency virus proteases. J. Biol. Chem. 270:29162–29168. [DOI] [PubMed] [Google Scholar]
- 25.Ostresh, J. M., J. H. Winkle, V. T. Hamashin, and R. A. Houghten. 1994. Peptide libraries: determination of relative reaction rates of protected amino acids in competitive couplings. Biopolymers 34:1681–1689. [DOI] [PubMed] [Google Scholar]
- 26.Petropoulos, C. J., N. T. Parkin, K. L. Limoli, Y. S. Lie, T. Wrin, W. Huang, H. Tian, D. Smith, G. A. Winslow, D. J. Capon, and J. M. Whitcomb. 2000. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 44:920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pettit, S. C., M. D. Moody, R. S. Wehbie, A. H. Kaplan, P. V. Nantermet, C. A. Klein, and R. Swanstrom. 1994. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 68:8017–8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettit, S. C., N. Sheng, R. Tritch, S. Erickson-Viitanen, and R. Swanstrom. 1998. The regulation of sequential processing of HIV-1 Gag by the viral protease. Adv. Exp. Med. Biol. 436:15–25. [DOI] [PubMed] [Google Scholar]
- 29.Pettit, S. C., J. Simsic, D. D. Loeb, L. Everitt, C. A. Hutchison III, and R. Swanstrom. 1991. Analysis of retroviral protease cleavage sites reveals two types of cleavage sites and the structural requirements of the P1 amino acid. J. Biol. Chem. 266:14539–14547. [PubMed] [Google Scholar]
- 30.Phylip, L. H., A. D. Richards, J. Kay, J. Kovalinka, P. Strop, I. Blaha, J. Velek, V. Kostka, A. J. Ritchie, A. V. Broadhurst, et al. 1990. Hydrolysis of synthetic chromogenic substrates by HIV-1 and HIV-2 proteinases. Biochem. Biophys. Res. Commun. 171:439–444. [DOI] [PubMed] [Google Scholar]
- 31.Richards, A. D., L. H. Phylip, W. G. Farmerie, P. E. Scarborough, A. Alvarez, B. M. Dunn, P. H. Hirel, J. Konvalinka, P. Strop, L. Pavlickova, et al. 1990. Sensitive, soluble chromogenic substrates for HIV-1 proteinase. J. Biol. Chem. 265:7733–7736. [PubMed] [Google Scholar]
- 32.Ridky, T. W., C. E. Cameron, J. Cameron, J. Leis, T. Copeland, A. Wlodawer, I. T. Weber, and R. W. Harrison. 1996. Human immunodeficiency virus, type 1 protease substrate specificity is limited by interactions between substrate amino acids bound in adjacent enzyme subsites. J. Biol. Chem. 271:4709–4717. [DOI] [PubMed] [Google Scholar]
- 33.Ridky, T. W., A. Kikonyogo, J. Leis, S. Gulnik, T. Copeland, J. Erickson, A. Wlodawer, I. Kurinov, R. W. Harrison, and I. T. Weber. 1998. Drug-resistant HIV-1 proteases identify enzyme residues important for substrate selection and catalytic rate. Biochemistry 37:13835–13845. [DOI] [PubMed] [Google Scholar]
- 34.Rosé, J. R., L. M. Babé, and C. S. Craik. 1995. Defining the level of human immunodeficiency virus type 1 (HIV-1) protease activity required for HIV-1 particle maturation and infectivity. J. Virol. 69:2751–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosé, J. R., R. Salto, and C. S. Craik. 1993. Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem. 268:11939–11945. [PubMed] [Google Scholar]
- 36.Rozzelle, J. E., D. S. Dauber, S. Todd, R. Kelley, and C. S. Craik. 2000. Macromolecular inhibitors of HIV-1 protease. Characterization of designed heterodimers. J. Biol. Chem. 275:7080–7086. [DOI] [PubMed] [Google Scholar]
- 37.Schechter, I., and A. Berger. 1968. On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochem. Biophys. Res. Commun. 32:898–902. [DOI] [PubMed] [Google Scholar]
- 38.Stebbins, J., E. M. Towler, M. G. Tennant, I. C. Deckman, and C. Debouck. 1997. The 80’s loop (residues 78 to 85) is important for the differential activity of retroviral proteases. J. Mol. Biol. 267:467–475. [DOI] [PubMed] [Google Scholar]
- 39.Toth, M. V., and G. R. Marshall. 1990. A simple, continuous fluorometric assay for HIV protease. Int. J. Peptide Protein Res. 36:544–550. [DOI] [PubMed] [Google Scholar]
- 40.Towler, E. M., S. K. Thompson, T. Tomaszek, and C. Debouck. 1997. Identification of a loop outside the active site cavity of the human immunodeficiency virus proteases which confers inhibitor specificity. Biochemistry 36:5128–5133. [DOI] [PubMed] [Google Scholar]
- 41.Tözsér, J., I. Bláha, T. D. Copeland, E. M. Wondrak, and S. Oroszlan. 1991. Comparison of the HIV-1 and HIV-2 proteinases using oligopeptide substrates representing cleavage sites in Gag and Gag-Pol polyproteins. FEBS Lett. 281:77–80. [DOI] [PubMed] [Google Scholar]
- 42.Tözsér, J., A. Gustchina, I. T. Weber, I. Blaha, E. M. Wondrak, and S. Oroszlan. 1991. Studies on the role of the S4 substrate binding site of HIV proteinases. FEBS Lett. 279:356–360. [DOI] [PubMed] [Google Scholar]
- 43.Tözsér, J., I. T. Weber, A. Gustchina, I. Bláha, T. D. Copeland, J. M. Louis, and S. Oroszlan. 1992. Kinetic and modeling studies of S3–S3′ subsites of HIV proteinases. Biochemistry 31:4793–4800. [DOI] [PubMed] [Google Scholar]
- 44.Wondrak, E. M., J. M. Louis, H. de Rocquigny, J. C. Chermann, and B. P. Roques. 1993. The gag precursor contains a specific HIV-1 protease cleavage site between the NC (P7) and P1 proteins. FEBS Lett. 333:21–24. [DOI] [PubMed] [Google Scholar]
- 45.Yoshimura, K., R. Kato, K. Yusa, M. F. Kavlick, V. Maroun, A. Nguyen, T. Mimoto, T. Ueno, M. Shintani, J. Falloon, H. Masur, H. Hayashi, J. Erickson, and H. Mitsuya. 1999. JE-2147: a dipeptide protease inhibitor (PI) that potently inhibits multi-PI-resistant HIV-1. Proc. Natl. Acad. Sci. USA 96:8675–8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zennou, V., F. Mammano, S. Paulous, D. Mathez, and F. Clavel. 1998. Loss of viral fitness associated with multiple Gag and Gag-Pol processing defects in human immunodeficiency virus type 1 variants selected for resistance to protease inhibitors in vivo. J. Virol. 72:3300–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, Y. M., H. Imamichi, T. Imamichi, H. C. Lane, J. Falloon, M. B. Vasudevachari, and N. P. Salzman. 1997. Drug resistance during indinavir therapy is caused by mutations in the protease gene and in its Gag substrate cleavage sites. J. Virol. 71:6662–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]