

Abstract
The unique N-terminal region of the parvovirus VP1 capsid protein is required for infectivity by the capsids but is not required for capsid assembly. The VP1 N terminus contains a number of groups of basic amino acids which resemble classical nuclear localization sequences, including a conserved sequence near the N terminus comprised of four basic amino acids, which in a peptide can act to transport other proteins into the cell nucleus. Testing with a monoclonal antibody recognizing residues 2 to 13 of VP1 (anti-VP1-2-13) and with a rabbit polyclonal serum against the entire VP1 unique region showed that the VP1 unique region was not exposed on purified capsids but that it became exposed after treatment of the capsids with heat (55 to 75°C), or urea (3 to 5 M). A high concentration of anti-VP1-2-13 neutralized canine parvovirus (CPV) when it was incubated with the virus prior to inoculation of cells. Both antibodies blocked infection when injected into cells prior to virus inoculation, but neither prevented infection by coinjected infectious plasmid DNA. The VP1 unique region could be detected 4 and 8 h after the virus capsids were injected into cells, and that sequence exposure appeared to be correlated with nuclear transport of the capsids. To examine the role of the VP1 N terminus in infection, we altered that sequence in CPV, and some of those changes made the capsids inefficient at cell infection.
Virus infection of cells is a multistep process that requires the particle to bind to a receptor and then enter the cytoplasm either directly through the plasma membrane or after receptor-mediated endocytosis. For many viruses infection requires that viral proteins undergo conformational changes induced by interacting with cells, such as binding to a receptor or to a coreceptor, or by exposure to low pH or to proteases within the endosome. The resulting conformational changes may include exposure of membrane fusion sequences, dissociation or loss of viral components, exposure of buried sequences to the outside of the capsid, or activation of viral enzymes required for infection (18, 22, 23, 29).
The canine parvovirus (CPV) capsid is a 25-nm-diameter icosahedron assembled from 60 copies of the overlapping VP1 and VP2 proteins; VP1 contains the complete sequence of VP2, as well as a 143-residue unique N-terminal sequence. Ninety percent of the protein in the newly produced capsid is VP2, and about 10% is VP1. In full capsids the N-terminal 19 to 20 amino acids of some VP2 molecules are exposed on the outside of the virion (31, 49), probably by passing through pores at the fivefold axes of icosahedral symmetry (52). Those VP2 N termini may be removed by proteolytic digestion, and antibodies against that sequence can neutralize viral infectivity (4, 17, 49). About 24 nucleotides of the 5′ ends of viral single-stranded DNA (ssDNA) genomes are also exposed on the outside of the capsid, and that sequence has the large nonstructural protein (NS1) attached when the virus is first produced (9, 48). However, that extraparticle viral DNA can apparently be cleaved off without affecting infectivity. There are loops in the capsid structure which can vary significantly in conformation, and in CPV the structure of one of those variable regions is correlated with coordination of two or three divalent ions, most likely calcium ions (39).
Cell infection by parvoviruses is a complex process that is tightly regulated by both the cell and the virus. Although CPV can efficiently infect feline and canine cells, mutants containing only one or two amino acid sequence substitutions within the capsid protein are reduced in specific infectivity for canine cells by up to 106-fold (5, 34). CPV capsids bind the transferrin receptor on feline cells and then enter the cells by clathrin-mediated endocytosis, followed by trafficking through endosomal pathways, and they are retained for long periods in cellular vesicles (32, 46). The capsids appear to enter the cytoplasm and travel to the nuclear pore and the nucleus in a process which can be blocked by injection of antiviral antibody into the cytoplasm up to several hours after virus inoculation (33, 47). When CPV capsids were injected into the cytoplasm of cells, nuclear transport appeared to involve two steps: initial transport of a small proportion of the capsids into the nucleus and transport of the remaining virus over the next several hours. The slower form of nuclear transport was prevented by depolymerization of the cellular microtubules with nocodazole (47).
In CPV and minute virus of mice (MVM) the unique N-terminal sequence of VP1 is enclosed within the capsid, and it can be exposed by heat or urea treatment without complete capsid disintegration (8, 49). The VP1 unique region contains several basic sequences which resemble classical nuclear localization signals (NLS), and one of those (MAPPAKRARRGLV) at the N terminus of VP1 can mediate the transport of bovine serum albumin (BSA) into the nuclei of cells (45). In the case of MVM a basic amino acid rich sequence in VP2 (KGKLTMRAKLR) has been associated with nuclear transport in a conformation-dependent manner (21). However, the equivalent sequence in CPV did not mediate nuclear import when fused to a heterologous protein, and mutations of that sequence affected capsid assembly but not nuclear transport (45, 56). A phospholipase A2 (PLA2) activity has been associated with the VP1 N terminal sequences of several parvoviruses, and that was suggested to be involved in release of virus from the endosomes during infection (57).
Nuclear transport of most proteins involves classical NLS which interact with importin-α, and then the complex associates with Ran proteins to mediate transport through the nuclear pore (6, 7, 13, 14, 25-27, 50). The nuclear pore had an effective internal diameter of about 27 nm when gold particles conjugated to NLS-containing peptides were studied in Xenopus oocytes (10), similar to the diameter of the parvovirus particle, suggesting that the more-or-less intact viral particle may enter the nucleus during the infectious process (47).
Many other viruses replicating in the nucleus have specific mechanisms for trafficking within the cytoplasm and for transporting the viral genome and some viral proteins to the nucleus (50). Examples include influenza virus, where the RNA and nucleoprotein are apparently transported rapidly into the nucleus (3, 24), and simian virus 40 particles, which enter the cell through a caveolae pathway and are transported through the nuclear pore in a slow process that appears to require conformational changes of the capsids (28, 30, 36). The large capsids of adenoviruses or herpesviruses are transported at least in part by a microtubule-dependent pathway to the vicinity of the nuclear pore, where they release their DNA genomes and viral proteins that mediate nuclear transport (12, 40, 42).
Here we further examined the location of the N terminus of the VP1 unique region of CPV in the capsid and defined its role in infection of cells and viral replication. That sequence was normally buried but became exposed after some treatments of the capsids, and specific antibodies to that sequence could neutralize the virus both outside the cell and in the cell cytoplasm. Some mutations within the first N-terminal basic sequence of VP1 greatly reduced production of infectious virus.
MATERIALS AND METHODS
Viruses and cells.
CPV type 2 (CPV-d) was grown in Norden laboratory feline kidney (NLFK) cells, and aliquots were stored frozen at −70°C (35). Full CPV particles were purified, quantities were determined as described previously, and then the particles were stored in 10 mM Tris-HCl (pH 7.5) at 4°C (1). NLFK cells were grown and maintained in a 1:1 mixture of McCoy's 5A and Leibovitz L15 media with 5% fetal bovine serum. Virus stocks were titrated by 50% tissue culture infectious dose (TCID50) assay as described previously (35).
Antibodies.
A monoclonal antibody (MAb) (anti-VP1-2-13) was prepared against a 13-amino-acid peptide containing residues 2 to 13 of the CPV VP1 along with a C-terminal cysteine for conjugation (APPAKRARRGLV-C). That peptide was conjugated with keyhole limpet hemocyanin, mice were immunized three times with the peptide conjugate, and hybridomas were produced by fusion with SP2/0 mouse myeloma cells. Anti-VP1-2-13 recognized both the peptide conjugated to BSA and denatured CPV or feline panleukopenia virus capsids. MAb 8 recognizes the intact CPV capsid (41, 49). A rabbit polyclonal antibody against the VP1 N-terminal unique region of MVM (VP1 residues 1 to 141) was obtained from Susan Cotmore and Peter Tattersall, Yale University. In antibody microinjection studies, normal mouse immunoglobulin G (IgG) (Sigma, St. Louis, Mo.) was used as a control.
Capsid treatments and VP1 and 3"-end DNA exposure.
The exposure of VP1 was examined by treating capsids for 30 min with urea at concentrations of up to 7 M or at 37, 45, 55, 65, or 75°C. The capsids were bound to nitrocellulose membranes by using a slot blot apparatus then incubated with either anti-VP1-2-13 MAb, MAb 8, the rabbit anti-VP1 unique region serum, or a rabbit polyclonal antibody against the capsid. Antibodies were detected with goat anti-mouse IgG-horseradish peroxidase or goat anti-rabbit IgG-horseradish peroxidase, and the bound antibody was detected with Supersignal substrate and exposure to X-ray films.
The exposure of the 3′ end of the viral genome was detected by polymerase extension. Capsids treated with urea or heat as described above were washed in Microcon filter units (10-kDa retention) (Millipore, Bedford, Mass.) and then incubated with modified T7 DNA polymerase (Sequenase; Amersham Life Science, Cleveland, Ohio)-0.2 mM deoxynucleoside triphosphates-5 μCi of [32P]dATP-10 mM Tris-HCl (pH 7.5)-50 mM NaCl-2 mM MgCl2 for 45 min. The samples were electrophoresed in a 1% agarose gel, which was then dried and exposed to X-ray film.
Neutralization by antibodies outside or inside the cell.
For neutralization assays, various amounts of either anti-VP1-2-13 or the rabbit anti-VP1 unique region antibody were incubated with 105 TCID50s of CPV for 1 h at 37°C. Each virus-antibody combination was then diluted and titrated in a TCID50 assay in NLFK cells.
For microinjection into cells, IgGs were dialyzed against 10 mM Tris-HCl (pH 7.4)-120 mM KCl. Cells were injected with 0.1 to 0.5 pl of antibodies at 5 mg/ml using an Eppendorf 5246 microinjector and an Eppendorf 5171 micromanipulator. The cells were inoculated with 5 TCID50s of CPV per cell for 1 h, incubated for a further 24 h, and then fixed with 3.7% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min and treated with PBS containing 0.1% (vol/vol) Triton X-100 and 1% (wt/vol) BSA for 20 min. The cells were then stained with fluorescein isothiocyanate (FITC)-labeled goat anti-mouse IgG to detect injected cells, while virus infection was detected using Texas Red (TxR)-conjugated anti-NS1 antibody (54). Coverslips were mounted in Prolong medium (Molecular Probes, Eugene, Oreg.) and examined by fluorescence microscopy. In control studies antibodies were microinjected along with a 200-μg/ml concentration of an infectious plasmid clone of CPV (35), and then the cells were incubated and analyzed as described above. The proportions of cells that expressed NS1 after injection with anti-VP1-2-13 MAb and plasmid were compared to the proportions in cells coinjected with control mouse IgG and plasmid or in noninjected cells in the same cultures.
Capsid injection into cells: nuclear transport and VP1 exposure.
To examine VP1 N-terminal exposure on capsids within cells, full capsids at a concentration of 5 mg/ml were injected into the cytoplasm of cells and then incubated at 37°C for 1, 4, or 8 h. The cells were then fixed with 3.75% paraformaldehyde, permeabilized with 0.5% Triton X-100, and stained with rabbit anti-VP1 unique region antibody followed by TxR-conjugated anti-rabbit IgG and with MAb 8 to detect the viral capsids followed by FITC-conjugated anti-mouse IgG.
To examine the effect of anti-VP1 antibodies on nuclear transport of capsids, we injected full or empty capsids at 2.5 mg/ml along with anti-VP1-2-13, with rabbit anti-VP1, or with control mouse IgG at concentrations of 2.5 mg of IgG per ml. The cells were then incubated for 6 h before fixing and staining for the capsids with either MAb 8 or rabbit anti-capsid IgG as described above. The cells were observed by confocal microscopy, and images were collected from the centers of the cells.
Mutagenesis of VP1 sequences.
The infectious plasmid clone of CPV-d was mutagenized using uracilated ssDNA in an M13 vector (16), altering residues near the N terminus of VP1 as listed in Table 1. The mutated sequences were reintroduced into the CPV-d genome using BspEI and BsgI sites at nucleotides 2075 and 2773 in the genome and sequenced completely across the region replaced. To generate virus, 5-μg amounts of plasmids were transfected into NLFK cells by electroporation or with Lipofectamine (Gibco-BRL, Gaithersburg, Md.) in different experiments. Cells were then incubated for 3 days and then lysed with 0.5% NP-40 and frozen and thawed once. The lysates were clarified at 10,000 × g for 15 min, and then the capsids were pelleted at 100,000 × g for 3 h and the virus in the pellet was banded in 10 to 40% sucrose gradients. In other experiments the lysates were layered over preformed CsCl gradients (1.25 to 1.45 g/cm3) and centrifuged at 200,000 × g for 6 h at 12°C. Gradients were fractionated and capsids were detected by hemagglutination assay, and then the full virus fractions were pooled, pelleted at 100,000 × g for 2 h, and resuspended in PBS. Virus samples were tested for TCID50 in NLFK cells, while capsids were quantified by hemagglutination.
TABLE 1.
N-terminal VP1 sequences and infectivities of CPV mutants
| Mutant | Sequencea | Ratio of infectious titer (TCID50) and hemagglutinin titerb |
|---|---|---|
| CPV-d | 1-MAPPAKRARRGLV | 122 ± 61 |
| CPVΔVP1 | 1-PAPPAKRARRGLV | <1 |
| CPV-ARRR | 1-MAPPAARARRGLV | 61 ± 41 |
| CPV-KARR | 1-MAPPAKAARRGLV | 36 ± 22 |
| CPV-KRAR | 1-MAPPAKRAARGLV | 4.75 ± 5 |
| CPV-AAAR | 1-MAPPAAAAARGLV | <1 |
The sequence shown is the first 13 residues of VP1, showing the substitutions (boldface) prepared in the CPV genomes tested here.
Data are shown as ratio ± standard deviation from four separate experiments.
To compare the ssDNA production after transfection, NLFK cells seeded at a density of 2 × 104 per cm2 in 9-cm2 dishes were transfected with 5 μg of plasmids by using Lipofectamine, and the cells were lysed 3 days later using a modified Hirt procedure (34). The DNA was digested for 2 h with 3 U of DpnI per sample and electrophoresed on 1% agarose gels with 1 μg of ethidium bromide per ml. DNA was transferred to a nylon membrane by Southern blotting and detected with a 32P-labeled fragment of the CPV genome. The hybridized filter was exposed to X-ray film.
RESULTS
VP1 N-terminal sequences become exposed after treatments and in cells.
After treatment of capsids with urea or heat, the VP1 unique region could be readily detected by the anti-VP1-2-13 and by the anti-VP1 unique region antiserum (Fig. 1). Both antibodies detected the VP1 unique sequence at about the same urea concentration. Under the conditions of VP1 N-terminal release, the capsids were still detected with MAb 8, which recognizes intact capsids (51). DNA was released at a low level by heating full capsids to 55°C or by exposure to 3 M urea, and it was exposed at maximal levels by treatment at 75°C or with 6 M urea, when the capsids most likely disintegrated (Fig. 2).
FIG. 1.

Exposure of the VP1 unique region detected by antibodies after urea or temperature treatments of full or empty capsids. The VP1 sequence was detected with rabbit anti-VP1 or with anti-VP1-2-13 MAb, intact capsids were detected with MAb 8, and the total capsid proteins were detected with a rabbit anticapsid antibody. (A) Treatment of capsids by heating for 30 min at the indicated temperatures prior to applying to the membrane. (B) Treatment of capsids with various urea concentrations for 30 min prior to applying to the membrane. (C) Antibody binding to full or empty capsids after treatment with various concentrations of urea. Data represents the means from four separate experiments and are presented as percentages of the maximal binding seen for each antibody. Open symbols, empty capsids; closed symbols, full capsids; circles, MAb 8; squares, rabbit anticapsid; diamonds, anti-VP1-2-13 MAb; triangles, rabbit anti-VP1.
FIG. 2.

Exposure of the 3" end of the viral DNA after temperature or heat treatment of full capsids. After treatments, capsids were washed and incubated with T7 DNA polymerase and 35S-dATP, and then the products were electrophoresed in agarose gels and detected on X-ray film. The labeled DNA band is the monomer-length double-stranded form produced from the viral genome.
To determine whether the VP1 N terminus became exposed on capsids within cells, full capsids were injected into cells which were then incubated for 1, 4, or 8 h. Antibody staining showed little detectable VP1 after 1 h, but by 4 h VP1 was detected around and inside the nucleus, while consistent nuclear staining was seen after 8 h (Fig. 3). Parallel studies with empty capsids showed similar staining patterns (results not shown). Anti-VP1-2-13 MAb showed background staining of control cells and could not be used in this study.
FIG. 3.

Exposure of the VP1 unique region on purified capsids injected into cells. Full virus capsids were injected into cells, which were then incubated for 1, 4, or 8 h at 37°C before fixation. The cells were incubated with rabbit anti-VP1 unique region followed by TxR-conjugated anti-rabbit IgG. Capsids were detected using MAb 8 and FITC-conjugated anti-mouse IgG. Confocal images show a 1-μm section from the center of each cell, while phase-contrast images of the same cells are also shown.
Anti-VP1 MAb or polyclonal antibodies block CPV infection.
Anti-VP1-2-13 MAb reduced the infectivity of CPV by over 1,000-fold when the virus was incubated for 1 h with 80 μg of IgG per ml, but no reduction in titer was seen when the antibody was at a concentration of 8 μg/ml or lower. No neutralization was seen by the rabbit anti-VP1 unique region antibody at a dilution of 1:40 or 1:400.
We have shown previously that anticapsid monoclonal antibodies injected into cells block CPV infection (47). Here we show that infection was also blocked by injection of antibodies against the N-terminal end of VP1 (Fig. 4). Cells injected with anti-VP1-2-13 MAb or rabbit anti-VP1 unique region antibodies prior to CPV inoculation showed only 0 to 0.3% infection as measured by NS1 expression. Experiments were repeated three times, and 30 to 42% of the noninjected or control IgG-injected cells became infected (Fig. 4).
FIG. 4.

Effect of injection with anti-VP1-2-13 or rabbit IgG against the complete VP1 unique region on infection of the cells by CPV. (A and B) Injection of anti-VP1-2-13 antibody (A) or control mouse IgG (B). The cells were costained for IgG (FITC, green) and for viral NS1 protein (red). (C) Percentage of cells that became infected after injection with anti-VP1-2-13, with the rabbit anti-VP1 unique region, or with control IgG or without injection in the same cultures. Error bars indicate standard deviations.
To determine whether the injected anti-VP1-2-13 MAb affected viral gene expression or replication, cells were injected with the MAb along with an infectious CPV plasmid clone. NS1 expression was seen in between 22 and 35% of the injected cells (Fig. 5A), and in those cells the IgG formed cytoplasmic and nuclear aggregations indicating VP1 production. The use of polyclonal anticapsid antibodies confirmed that the capsids were present in the aggregates (Fig. 5B).
FIG. 5.
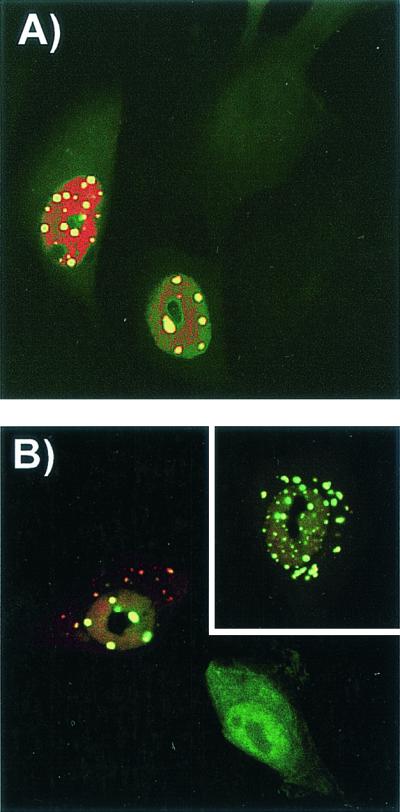
Detection of CPV replication and of capsid protein localization in cells injected simultaneously with anti-VP1-2-13 and the CPV infectious plasmid and then incubated for 24 h at 37°C. (A) Expression of NS1 in cells coinjected with anti-VP1-2-13 and a CPV infectious plasmid. The anti-VP1-2-13 was detected with an FITC-labeled anti-mouse IgG (green), and NS1 was detected with a TxR-labeled anti-NS1 IgG (red). (B) Localization of CPV capsid proteins in cells coinjected with anti-VP1-2-13 and a CPV infectious plasmid. The injected MAb was detected with FITC-labeled anti-mouse IgG (green), and capsid proteins were detected with a specific rabbit anticapsid IgG followed by TxR-labeled anti-rabbit IgG (red). The inset shows another cell subjected to the same treatment.
To test the effect of the anti-VP1 unique region antibodies on capsid nuclear transport, we injected those antibodies into cells along with capsids. Thirty to 50% of the cells injected with purified capsids showed high levels of accumulation in the nucleus by 6 h after injection (Fig. 6). However, when the capsids were coinjected with anti-VP1-2-13 MAb or rabbit anti-VP1, both full and empty capsids were retained in the cytoplasm of most of the cells (Fig. 6). Thirty-nine percent of the cells coinjected with full capsids and control mouse IgG showed strong nuclear localization.
FIG. 6.

Nuclear localization of full or empty CPV capsids after injection into the cytoplasm of cells in the presence or absence of anti-VP1-2-13, rabbit (Rab.) anti-VP1, or control mouse IgG. The cells were incubated for 6 h after injection and then fixed and stained for capsids with a directly conjugated anticapsid antibody. (A) Percentage of cells injected with capsids showing substantial nuclear localization after 6 h of incubation. The data for each experiment are shown for each capsid and antibody combination. (B and C) Representative fields of cells showing the localization of capsids within cells 6 h after injection of virus and antibody (B) or virus alone (C) into the cells.
Mutations of some VP1 N-terminal residues affect infectious virus production.
Site-directed mutant genomes with changes in the VP1 N-terminal sequences had two effects on the viral infection. After transfection of the plasmids into NLFK cells by either electroporation or Lipofectamine treatment, significant hemagglutinin titers of all viruses were recovered. All plasmids appeared to generate similar proportions of ssDNA, suggesting that they were packaging the genome and producing full capsids (Fig. 7). Mutants CPV-ARRR and CPV-KARR both replicated to give high-titer stocks that had infectivities similar to that of the wild-type CPV (Table 1). In contrast, only very low titers or no infectious viruses were recovered from the mutants CPVΔVP1, CPV-KRAR, and CPV-AAAR, even when the full capsids were purified by sucrose or CsCl gradient centrifugation (Table 1).
FIG. 7.

Production of replicative-form DNA (dsRF DNA) and genomic ssDNA after transfection of cells with the wild-type (CPV-wt) or mutant (Table 1) infectious plasmids. The DNA recovered by a modified Hirt lysis procedure was digested with DpnI prior to electrophoresis in a 1% agarose gel with 1 μg of ethidium bromide per ml. The viral DNAs were detected by Southern blotting.
DISCUSSION
Infection of cells by parvoviruses is a complex process which takes the capsid into the cell, through the endosomal pathways, and into the cytoplasm, and then the viral DNA or a DNA-protein complex is transported into the nucleus, where the DNA replicates and is packaged. Several specific regions of the surface of the capsid can affect cell infection and viral host range, most likely by affecting specific receptor binding (5, 33, 34). However, less is known about the later steps in the infection pathway or the viral structural changes that can influence those steps.
Here we show that the VP1 unique region has a specific role in the cell infection process of CPV. The N-terminal sequence found in CPV was similar to the sequence of the same region of the VP1s of related rodent or porcine viruses, and some similarity was also present in bovine parvovirus (Fig. 8). However, more distantly related parvoviruses (Aleutian disease virus, adeno-associated virus, and the human parvovirus B19) show no significant homology. The VP1 unique sequence has been shown to be required for infection of cells by MVM but to be normally buried within the MVM capsid and not readily detected by antibodies (8, 44). Those results for MVM were confirmed here, and we also showed that both the VP1 N terminus and the 3′ end of the viral DNA were released by treatment with urea or heat under conditions where many of the capsids were still largely intact (Fig. 1 and 2). Although the VP1 of the full capsids was exposed by heating at between 65 and 75°C or by treatment with 4 to 5 M urea, and the 3′ end of the genome was exposed under similar conditions, our data do not show whether those events were due to the same structural changes in the capsids. In studies of MVM the release of the VP1 unique region and the viral DNA appeared to be closely linked and likely resulting from the same effects on the capsids (8). The sequence examined here is in a region which is apparently not required for the PLA2 activity of the VP1 unique region (57), and our data suggest that an effect of this very N-terminal sequence is to control the nuclear transport of the capsids after they enter the cytoplasm during infection.
FIG. 8.

Alignment of the VP1 N-terminal sequences from viruses related to CPV. More distantly related parvoviruses showed no significant homology in this region of the protein.
The VP1 unique region of the capsid became exposed in capsids injected into the cytoplasm of cells. Although little VP1-specific staining was seen 1 h after injection, it increased particularly in the nucleus by 4 or 8 h of incubation of the injected cells at 37°C. It is not clear whether this was a specific release of the N terminus or a partial disintegration of the CPV capsids, but the capsids within the cells still reacted strongly with MAb 8, indicating that most remained intact. The lumen of the nuclear pore is about 27 nm in diameter and therefore may allow the intact or partially disassembled capsid to enter the nucleus (10). Destabilization of the capsids within the cell to release the VP1 unique region may result from virus interactions with the receptor or with cytosolic factors, subcellular organelles, or the nuclear membrane. Complete or partial particle disassembly during viral entry has been reported for other nuclear-replicating viruses (18, 22, 23, 29).
Anti-VP1-2-13 MAb neutralized the virus outside the cell but did so only at the high concentration of 80 μg/ml and not at 8 μg/ml. Other MAbs recognizing the CPV capsid surface neutralize efficiently at concentrations of between 0.12 and 1.2 μg/ml (56). The low efficiency of neutralization by the anti-VP1 antibodies may be due to the low concentration or transient exposure of the VP1 on capsids or possibly to a requirement for the capsids and the antibodies to be taken into cells together for neutralization to occur. As both rabbit polyclonal and mouse monoclonal anti-VP1 antibodies inhibited viral infection after cytoplasmic injection (Fig. 4), the VP1 unique region must be exposed on the capsid within the cell during the infection process. The injected IgG and the capsid proteins became aggregated in the cells that were coinjected with anti-VP1-2-13 MAb and infectious plasmid (Fig. 5), and that effect was not seen in cells injected with nonspecific IgG that became infected (e.g., in Fig. 4B). Whether the NS1 and capsid proteins seen in the plasmid-injected cells are due to viral replication or to expression from the plasmid is not known. However, only a proportion of the cells that contained injected IgG showed protein expression, although all of those cells should have received significant amounts of plasmid, and so much of the expression seen is likely to result from viral genome replication. Coinjection of antibodies and capsids showed that the process of full or empty capsid transport into the nucleus was markedly decreased by both anti-VP1 antibodies (Fig. 6), indicating that binding of the VP1 N terminus by the antibodies can interfere with capsid nuclear transport.
The exposure of these VP1 unique sequences which are normally internal to the capsid may resemble results seen for picornaviruses and nodaviruses, where an apparent “breathing” of the capsid allows sequences internal in the structure revealed by X-ray crystallography to be exposed or released to the exterior (11, 19, 20, 43, 53). The need for a high concentration of IgG for neutralization by anti-VP1-2-13 MAb most likely explains the lack of neutralization seen with the rabbit anti-VP1 unique region antisera seen here or in previous studies of MVM, as those antibodies would have a lower concentration of the VP1-specific IgG (8).
Human B19 virus capsids assembled from VP1 and VP2 by baculovirus expression have the VP1 unique region exposed to the outside, and B19 virus infectivity for erythroid cells can be neutralized by anti-VP1 antibodies (2, 15, 37, 38). Baculovirus expression of CPV can lead to some incorrectly assembled aggregates of the capsid proteins (55), but the results obtained here suggest similarities between the capsids of erythroviruses and the other parvoviruses in the exposure of VP1.
In earlier studies we characterized the nuclear transport of BSA conjugated with peptides mimicking the N terminus of VP1, and we showed that changing basic amino acids at positions 6, 7, and 9 in the VP1 sequence to Gly affected their ability to mediate nuclear import of BSA (45). Here we showed that the same N-terminal sequence also contains information required for viral infectivity. The site-directed mutations in CPV-KRAR or CPV-AAAR diminished infectivity of the virus, indicating the importance of this sequence for the viral life cycle. Interestingly, changing of Lys 6 or Arg 7 to Ala did not significantly affect viral infection, although when those residues were changed to Gly in the peptide studies they prevented nuclear import of BSA (45). The CPVΔVP1 virions showed virtually no detectable infectivity, as has been reported previously for MVM (44), and those capsids would lack both the putative NLS sequences and the PLA2 activity of the virus (57).
We have shown that the VP1 N terminus of CPV becomes exposed after treatments with urea and heat, that it is also exposed within the cell, and that the N-terminal basic sequence is required for successful cell infection. It will be interesting to further characterize the viral nuclear import process to reveal more about the mechanisms involved and to determine whether the N-terminal sequence serves as a unique NLS on the capsid or whether it is one of a family of sequences needed for viral import into the nucleus.
Acknowledgments
Gail Sullivan provided excellent technical assistance.
This work was supported by grants NR 64951 from the Academy of Finland and AI28385 and AI33468 to C.R.P. from the National Institutes of Health.
Footnotes
‡ Present address: Department of Molecular Biology, Princeton University, Princeton, NJ 08544.
REFERENCES
- 1.Agbandje, M., R. McKenna, M. G. Rossmann, M. L. Strassheim, and C. R. Parrish. 1993. Structure determination of feline panleukopenia virus empty particles. Proteins 16:155-171. [DOI] [PubMed] [Google Scholar]
- 2.Anderson, S., M. Momoeda, M. Kawase, S. Kajigaya, and N. S. Young. 1995. Peptides derived from the unique region of B19 parvovirus minor capsid protein elicit neutralizing antibodies in rabbits. Virology 206:626-632. [DOI] [PubMed] [Google Scholar]
- 3.Bui, M., G. Whittaker, and A. Helenius. 1996. Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J. Virol. 70:8391-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casal, J. I., J. P. Langeveld, E. Cortes, W. W. Schaaper, E. van Dijk, C. Vela, S. Kamstrup, and R. H. Meloen. 1995. Peptide vaccine against canine parvovirus: identification of two neutralization subsites in the N terminus of VP2 and optimization of the amino acid sequence. J. Virol. 69:7274-7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang, S. F., J. Y. Sgro, and C. R. Parrish. 1992. Multiple amino acids in the capsid structure of canine parvovirus coordinately determine the canine host range and specific antigenic and hemagglutination properties. J. Virol. 66:6858-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole, C. N., and C. M. Hammell. 1998. Nucleocytoplasmic transport: driving and directing transport. Curr. Biol. 8:R368-R372. [DOI] [PubMed] [Google Scholar]
- 7.Corbett, A. H., and P. A. Silver. 1997. Nucleocytoplasmic transport of macromolecules. Microbiol. Mol. Biol. Rev. 61:193-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotmore, S. F., M. D'Abramo, A., C. M. Ticknor, and P. Tattersall. 1999. Controlled conformational transitions in the MVM virion expose the VP1 N-terminus and viral genome without particle disassembly. Virology 254:169-181. [DOI] [PubMed] [Google Scholar]
- 9.Cotmore, S. F., and P. Tattersall. 1989. A genome-linked copy of the NS-1 polypeptide is located on the outside of infectious parvovirus particles. J. Virol. 63:3902-3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dworetzky, S. I., R. E. Landford, and C. M. Feldherr. 1988. The effects of variations in the number and sequence of targeting signals on nuclear uptake. J. Cell Biol. 107:1279-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fricks, C. E., and J. M. Hogle. 1990. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64:1934-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glotzer, J. B., A. I. Michou, A. Baker, M. Saltik, and M. Cotten. 2001. Microtubule-independent motility and nuclear targeting of adenoviruses with fluorescently labeled genomes. J. Virol. 75:2421-2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorlich, D., and U. Kutay. 1999. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15:607-660. [DOI] [PubMed] [Google Scholar]
- 14.Görlich, D., and I. W. Mattaj. 1996. Nucleocytoplasmic transport. Science 271:1513-1518. [DOI] [PubMed] [Google Scholar]
- 15.Kawase, M., M. Momoeda, N. S. Young, and S. Kajigaya. 1995. Most of the VP1 unique region of B19 parvovirus is on the capsid surface. Virology 211:359-366. [DOI] [PubMed] [Google Scholar]
- 16.Kunkel, T. A. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA 82:488-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langeveld, J. P., J. I. Casal, E. Cortes, G. van de Wetering, R. S. Boshuizen, W. M. Schaaper, K. Dalsgaard, and R. H. Meloen. 1994. Effective induction of neutralizing antibodies with the amino terminus of VP2 of canine parvovirus as a synthetic peptide. Vaccine 12:1473-1480. [DOI] [PubMed] [Google Scholar]
- 18.Lanzrein, M., A. Schlegel, and C. Kempf. 1994. Entry and uncoating of enveloped viruses. Biochem. J. 302:313-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis, J. K., B. Bothner, T. J. Smith, and G. Siuzdak. 1998. Antiviral agent blocks breathing of the common cold virus. Proc. Natl. Acad. Sci. USA 95:6774-6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li, Q., A. G. Yafal, Y. M. Lee, J. Hogle, and M. Chow. 1994. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J. Virol. 68:3965-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lombardo, E., J. C. Ram&ıacute;rez, M. Agbandje-McKenna, and J. M. Almendral. 2000. A beta-stranded motif drives capsid protein oligomers of the parvovirus minute virus of mice into the nucleus for viral assembly. J. Virol. 74:3804-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marsh, M., and A. Helenius. 1989. Virus entry into animal cells. Adv. Virus Res. 36:107-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marsh, M., and A. Pelchen-Matthews. 1993. Entry of animal viruses into cells. Rev. Med. Virol. 3:173-185. [Google Scholar]
- 24.Martin, K., and A. Helenius. 1991. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 65:232-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore, M. S. 1998. Ran and nuclear transport. J. Biol. Chem. 273:22857-22860. [DOI] [PubMed] [Google Scholar]
- 26.Moroianu, J. 1998. Distinct nuclear import and export pathways mediated by members of the karyopherin beta family. J. Cell. Biochem. 70:231-239. [PubMed] [Google Scholar]
- 27.Nakielny, S., and G. Dreyfuss. 1997. Nuclear export of proteins and RNAs. Curr. Opin. Cell Biol. 9:420-429. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura, T., N. Kawai, and I. Ichihara. 1991. Interaction of endocytotic vacuoles with the inner nuclear membrane in simian virus 40 entry into CV-1 cell nucleus. Cell Struct. Funct. 16:441-445. [DOI] [PubMed] [Google Scholar]
- 29.Ojala, P. M., B. Sodeik, M. W. Ebersold, U. Kutay, and A. Helenius. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 20:4922-4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Neill, R. E., R. Jaskunas, G. Blobel, P. Palese, and J. Moroianu. 1995. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J. Biol. Chem. 270:22701-22704. [DOI] [PubMed] [Google Scholar]
- 31.Paradiso, P. R. 1981. Infectious process of parvovirus H-1: correlation of protein content, particle density, and viral infectivity. J. Virol. 39:800-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parker, J. S., and C. R. Parrish. 2000. Cellular uptake and infection by canine parvovirus involves rapid dynamin-regulated clathrin-mediated endocytosis, followed by slower intracellular trafficking. J. Virol. 74:1919-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker, J. S. L., W. J. Murphy, D. Wang, S. J. O'Brien, and C. R. Parrish. 2001. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 75:3896-3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker, J. S. L., and C. R. Parrish. 1997. Canine parvovirus host range is determined by the specific conformation of an additional region of the capsid. J. Virol. 71:9214-9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parrish, C. R. 1991. Mapping specific functions in the capsid structure of canine parvovirus and feline panleukopenia virus using infectious plasmid clones. Virology 183:195-205. [DOI] [PubMed] [Google Scholar]
- 36.Pelkmans, L., J. Kartenbeck, and A. Helenius. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3:473-483. [DOI] [PubMed] [Google Scholar]
- 37.Rosenfeld, S. J., K. Yoshimoto, S. Kajigaya, S. Anderson, N. S. Young, A. Field, P. Warrener, G. Bansal, and M. S. Collett. 1992. Unique region of the minor capsid protein of human parvovirus B19 is exposed on the virion surface. J. Clin. Investig. 89:2023-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saikawa, T., S. Anderson, M. Momoeda, S. Kajigaya, and N. S. Young. 1993. Neutralizing linear epitopes of B19 parvovirus cluster in the VP1 unique and VP1-VP2 junction regions. J. Virol. 67:3004-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simpson, A. A., V. Chandrasekar, B. Hebert, G. M. Sullivan, M. G. Rossmann, and C. R. Parrish. 2000. Host range and variability of calcium binding by surface loops in the capsids of canine and feline parvoviruses. J. Mol. Biol. 300:597-610. [DOI] [PubMed] [Google Scholar]
- 40.Sodeik, B., M. W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strassheim, L. S., A. Gruenberg, P. Veijalainen, J.-Y. Sgro, and C. R. Parrish. 1994. Two dominant neutralizing antigenic determinants of canine parvovirus are found on the threefold spike of the virus capsid. Virology 198:175-184. [DOI] [PubMed] [Google Scholar]
- 42.Suomalainen, M., M. Y. Nakano, S. Keller, K. Boucke, R. P. Stidwill, and U. F. Greber. 1999. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144:657-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsang, S. K., P. Danthi, M. Chow, and J. M. Hogle. 2000. Stabilization of poliovirus by capsid-binding antiviral drugs is due to entropic effects. J. Mol. Biol. 296:335-340. [DOI] [PubMed] [Google Scholar]
- 44.Tullis, G. E., L. R. Burger, and D. J. Pintel. 1993. The minor capsid protein VP1 of the autonomous parvovirus minute virus of mice is dispensable for encapsidation of progeny single stranded DNA but is required for infectivity. J. Virol. 67:131-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vihinen-Ranta, M., L. Kakkola, A. Kalela, P. Vilja, and M. Vuento. 1997. Characterization of a nuclear localization signal of canine parvovirus capsid proteins. Eur. J. Biochem. 250:389-394. [DOI] [PubMed] [Google Scholar]
- 46.Vihinen-Ranta, M., A. Kalela, P. Makinen, L. Kakkola, V. Marjomaki, and M. Vuento. 1998. Intracellular route of canine parvovirus entry. J. Virol. 72:802-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vihinen-Ranta, M., W. Yuan, and C. R. Parrish. 2000. Cytoplasmic trafficking of the canine parvovirus capsid and its role in infection and nuclear transport. J. Virol. 74:4853-4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang, D., and C. R. Parrish. 1999. A heterogenous nuclear ribonucleoprotein A/B-related protein binds to single-stranded DNA near the 5′end or within the genome of feline parvovirus and can modify virus replication. J. Virol. 73:7761-7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weichert, W. S., J. S. Parker, A. T. M. Wahid, S. F. Chang, E. Meier, and C. R. Parrish. 1998. Assaying for structural variation in the parvovirus capsid and its role in infection. Virology 250:106-117. [DOI] [PubMed] [Google Scholar]
- 50.Whittaker, G. R., and A. Helenius. 1998. Nuclear import and export of viruses and virus genomes. Virology 246:1-23. [DOI] [PubMed] [Google Scholar]
- 51.Wikoff, W. R., G. Wang, C. R. Parrish, R. H. Cheng, M. L. Strassheim, T. S. Baker, and M. G. Rossmann. 1994. The structure of a neutralized virus: canine parvovirus complexed with neutralizing antibody fragment. Structure 2:595-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie, Q., and M. S. Chapman. 1996. Canine parvovirus capsid structure, analyzed at 2.9 Å resolution. J. Mol. Biol. 64:497-520. [DOI] [PubMed] [Google Scholar]
- 53.Yafal, A. G., G. Kaplan, V. R. Racaniello, and J. M. Hogle. 1993. Characterization of poliovirus conformational alteration mediated by soluble cell receptors. Virology 197:501-505. [DOI] [PubMed] [Google Scholar]
- 54.Yeung, D. E., G. W. Brown, P. Tam, R. H. Russnak, G. Wilson, I. Clark-Lewis, and C. R. Astell. 1991. Monoclonal antibodies to major nonstructural nuclear protein of minute virus of mice. Virology 181:35-45. [DOI] [PubMed] [Google Scholar]
- 55.Yuan, W., and C. R. Parrish. 2001. Canine parvovirus capsid assembly and differences in mammalian and insect cells. Virology 279:546-557. [DOI] [PubMed] [Google Scholar]
- 56.Yuan, W., and C. R. Parrish. 2000. Comparison of two single-chain antibodies that neutralize canine parvovirus: analysis of an antibody-combining site and mechanisms of neutralization. Virology 269:471-480. [DOI] [PubMed] [Google Scholar]
- 57.Zadori, Z., J. Szelei, M.-C. Lacoste, P. Raymond, M. Allaire, I. R. Nabi, and P. Tijssen. 2001. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 1:291-302. [DOI] [PubMed] [Google Scholar]