

Abstract
When a parent virus replicates inside its host, it must first use its own genome as the template for replication. However, once progeny genomes are produced, the progeny can in turn act as templates. Depending on whether the progeny genomes become templates, the distribution of mutants produced by an infection varies greatly. While information on the distribution is important for many population genetic models, it is also useful for inferring the replication mode of a virus. We have analyzed the distribution of mutants emerging from single bursts in the RNA bacteriophage φ6 and find that the distribution closely matches a Poisson distribution. The match suggests that replication in this bacteriophage is effectively by a stamping machine model in which the parental genome is the main template used for replication. However, because the distribution deviates slightly from a Poisson distribution, the stamping machine is not perfect and some progeny genomes must replicate. By fitting our data to a replication model in which the progeny genomes become replicative at a given rate or probability per round of replication, we estimated the rate to be very low and on the on the order of 10−4. We discuss whether different replication modes may confer an adaptive advantage to viruses.
In recent years, viruses have been increasingly used in the laboratory to study the fitness effects of mutations (1, 4, 8, 10, 14, 15, 16, 25). The increase is likely due to two reasons. First, these fitness effects, which are generally quantified as the distribution of the magnitude, the sign (deleterious versus beneficial), and the interaction (additive versus nonadditive) of the mutations, are key to many evolutionary models, and viruses have become a major focus of evolutionary studies. Much of our predictive theories in evolution have come from population genetic models that make assumptions about the fitness effects of mutations. For example, depending on whether the interaction between deleterious mutations is log-additive, the models predict distinctly different advantages for the evolution of recombination (5, 15). Second, many issues addressed by such models have now also been raised in the context of viruses, especially RNA viruses, which have a much higher mutation rate than DNA viruses (11, 12). Evolutionary topics as wide ranging as the quasi-species concept, game theory, the origin of life, the constancy of the molecular clock, the divergence and convergence of isolated populations, and the evolution of recombination have all been recently considered in relation to viruses (1-3, 5-7, 15, 17, 18, 22, 24, 29, 31, 32).
As the more qualitative aspects of the distribution of mutational effects and interactions have become known, it has become desirable to extract more quantitative information. However, more quantitative analyses require also more exact information on another type of distribution, that of mutations and mutants in a population. The distinction between mutations and mutants is important and is made herein. A mutation is the event that changes a nucleotide sequence. The mutated sequence and any copies are mutants. Because mutations are generated randomly, the distribution of mutations across bursts (or across progeny for a virus that does not burst) is expected to be Poisson (27). However, the distribution of mutants across bursts may or may not be Poisson, depending on the mode of viral replication inside the host cell.
If the parental virus is the only template used for production of progeny, the distribution remains Poisson because mutants do not replicate. With only one template, the replication model conforms to a “stamping machine” model (28), and the resulting Poisson distribution has the characteristic of having a mean that equals the variance or a variance-to-mean ratio of 1 (27). On the other hand, if the progeny can also serve as templates for additional progeny, the distribution has a variance-to-mean ratio much greater than 1 because mutant progeny produce more mutant viruses. In the worst-case scenario, if a mutant progeny arises as the first progeny virus produced by the parental virus, half of the progeny will be mutant. If all progeny can act immediately as templates, the replication model is effectively exponential or geometric growth, and the resulting distribution conforms to the Luria-Delbruck distribution (20). If only a fraction of the progeny replicate, the replication model is a hybrid between geometric growth and stamping machine and deviates less from a Poisson distribution. Thus, knowing the mean and variance of the distribution of mutants across bursts is not only important for any detailed analysis of the distribution of mutational effects but also useful for inferring the mode of replication of a virus. Luria (19) concluded that the phage T2 replicated by geometric process because the distribution of mutants had a variance that was much greater than the mean. Denhardt and Silver (9) argued for a stamping machine in the phage φX174 because the distribution was Poisson.
In this study, we measured the distribution of mutants in the RNA bacteriophage φ6 (26, 30) by assaying the production of wild-type revertants from an amber mutant. φ6 has been used extensively for mutation accumulation experiments (1, 2, 4, 7). It has a double-stranded (ds) genome consisting of 13,383 bp (18). Its replication is known to occur by semiconservative, plus-strand (message-sense) displacement (18). After infecting and entering a host cell, the infecting or parental genome begins replication by synthesizing a new plus strand, which displaces the old plus strand of the parental genome. Although the parental genome is now ready for a second round of replication, the displaced strand is not. It must first be encapsidated and copied to produce the first ds progeny genome. Thus, because the progeny genome may or may not be ready to replicate when the parental genome initiates the second round of replication, the mode of replication may be either by a geometric growth or by a stamping machine model. We find that the distribution of mutants in φ6 deviates only slightly from a Poisson distribution. Thus, replication in φ6 is effectively a stamping machine in which the ds progeny genomes replicate with a very low probability.
MATERIALS AND METHODS
Strains and culture conditions.
The bacteriophage used was strain φ6-sus297, an amber mutant of φ6 (26). The permissive host for sus297 is the bacterial strain S4, a suppressor mutant of Pseudomonas pseudoalcaligenes ERA-pLM2. The nonpermissive host of φ-sus297 is the suppressor-minus bacterial strain Pseudomonas syringae pv. phaseolicola HB10Y. All strains were kindly provided by L. Mindich (Public Health Research Institute, New York, N.Y.). Conditions for growth of phage and bacteria have been previously described (4, 7, 26).
Burst size estimates.
S4 was grown to a log-phase density of 5 × 108 cells ml−1 in LC medium (26) and then centrifuged and resuspended in half the volume of LC medium to yield a density of 109 cells ml−1. The adsorption of the phage to the host cells was initiated by mixing 10 μl of a 108 ml−1 lysate of sus297 and 1 ml of resuspended cells. At these densities of phage and bacteria, the multiplicity of infection is approximately 10−3. Host cells were at log phase and concentrated to ensure maximum phage adsorption. At the high cell density, 99.999% of the phage are adsorbed after of 30 min (28) on the basis of the adsorption rate coefficient estimated by Vidaver et al. (30). The adsorption mixture was shaken gently during the adsorption period. After the adsorption period, the mixture was serially diluted into three Erlenmeyer flasks, designated A, B, and C, which contained, respectively, 10, 9, and 10 ml of conditioned (aerated and adjusted to 25°C) LC medium. The dilutions were made by serially transferring 100 μl of the adsorption mixture to A, then transferring 1 ml from A to B, and finally transferring 100 μl from B to C. All three flasks were then shaken at 200 rpm for better aeration. By plating phage from B and C over a period of 240 min, the entire course of the burst was monitored.
Pseudo-single-burst experiments.
The pseudo-single-burst procedure was similar to the methods for the burst size estimates (above), but with the following modifications. To increase the number of phage, 100 μl of a 108 ml−1 lysate of sus297 was added to 1 ml of resuspended cells. At these densities, the multiplicity of infection is approximately 10−2 and still much less than 1. Flasks A, B, and C contained 10, 9, and 9 ml of conditioned LC medium. Serial dilutions were made by transferring 100 μl of the adsorption mixture to A, then transferring 1 ml from A to B, and finally transferring 1 ml from B to C. Immediately after dilution, 50 aliquots of 100 μl were removed from C and dispensed into 50 separate 13- by 100-mm test tubes, designated burst tubes, which were incubated in a 25°C water bath with gentle shaking for 150 min. The 30-min adsorption period (see above) and the 150-min incubation total 180 min. A period of 180 min had been determined from the burst size experiments (see Fig. 1) to be sufficient to complete a burst. The progeny of the bursts in each of the 50 tubes was then plated on the nonpermissive host HB10Y to assay for wild-type revertants. Care was taken to ensure that the entire progeny of in each burst tube was plated and one plate was used for each tube. To prevent any bacterial or phage replication after 180 min, all 50 tubes were placed on ice until plating. To obtain an accurate and precise estimate of the number of phage (or infective cells) that had been pooled in each of the 50 tubes, 200 μl from C was plated on the permissive host S4, with fivefold replication, immediately after the 50 tubes had been dispensed and well before the burst began.
FIG. 1.

Burst size of φ6 sus297 on permissive host S4. See the text for details.
RESULTS
Burst size.
After infecting and replicating in a host cell, φ6 releases its progeny by lysing the cell. The number of progeny released per infected host cell is the burst size. To determine the burst size of sus297, we infected the permissive host S4 with the phage at a low multiplicity of infection in order to avoid any interference between phages coinfecting the same host cell (29). Samples of the infection mixture were then used to infect the same host until a burst was detected. PFU obtained before the burst correspond to free phage and cells infected with phage. After the burst, the number of PFU is expected to rise, and the ratio of PFU before and after the burst provides an estimate of the burst size, herein designated B (Fig. 1). From three independent burst size experiments, an average value was estimated to be 76 ± 16 (mean ± standard error).
From Fig. 1, we also determined that 180 min was sufficient for the burst to be completed, and we used this value in our design of our pseudo-single-burst experiments (see Materials and Methods).
Distribution of mutants across single bursts. (i) Preliminary analysis.
The distribution of mutants can be measured across individual single bursts (a single-burst experiment) or across pools of single bursts (pseudo-single-burst experiment). In practice, a single-burst experiment is practical only if the expected number of mutations per burst is one or greater. Otherwise, most bursts will not contain a mutant and the number of bursts that have to be sampled becomes too large for a practical experiment. The expected number of mutations per burst is the product u(B − 1), where u is the mutation rate. The burst size, B, is reduced by one because the infecting template is assumed not to mutate (12), in which case B − 1 replication events must take place in order for B progeny to be produced. From preliminary results, we had determined that sus297 carried an amber mutation that reverted to wild type at a rate on the order of 10−6 (see below). With our estimate of B of 76 (above), u(B −1) is approximately 0.000075, in which case we were forced to take the pseudo-single-burst approach.
We infected the permissive host S4 with sus297 at a low multiplicity of infection, pooled the infected cells, and then assayed their joint progeny, after a single burst, for wild-type revertants by plating on the nonpermissive host HB10Y. Each pool was plated on a single plate. A total (N) of 1,650 plates (or pools) were scored, and the average number of infected cells per plate (K) was 793. The average number per plate was determined by sampling the infected cells, on S4, before the burst. Note that the number of infected cells per plate is equivalent to the number of bursts assayed per plate. Thus, a total (T, calculated as N × K) of 1,650 × 793, or 1,308,450, infected cells or bursts were sampled in our experiment.
The distribution of wild-type revertants across plates showed a high number of cases with a single wild-type revertant (Table 1). This outcome strongly suggested a Poisson distribution and thus a stamping machine model of replication for φ6, but the presence of some plates with a very large number of mutants indicated otherwise. However, because we had examined a large number of infected cells, a concern was that some of the cells were infected by wild-type revertants present in the original sus297 population used for our experiment. The fact that two of the plates had 69 and 79 mutants, which are very close to our estimate for B of 76, suggested that those plates may have indeed contained a cell that was infected with a wild-type revertant from the start. To access the likelihood of having wild-type revertants in the original sus297 population, we estimated umin and umax, the minimum and maximum mutation rates for reversion from amber to wild type in sus297, on the basis of the data in Table 1.
TABLE 1.
Distribution of wild-type revertants and mutations across plates
| No. of plates [g(i)]a | No. of mutants/plate (i) | Minimum no. of mutationsb | Maximum no. of mutationsb | g(i)i | g(i)i2 |
|---|---|---|---|---|---|
| 1,404 | 0 | 0 | 0 | 0 | 0 |
| 205 | 1 | 205 | 205 | 205 | 205 |
| 32 | 2 | 32 | 64 | 64 | 128 |
| 5 | 3 | 5 | 15 | 15 | 45 |
| 1 | 4 | 1 | 4 | 4 | 16 |
| 1 | 8 | 1 | 8 | 8 | 64 |
| 1 | 69 | 1 | 69 | ||
| 1 | 79 | 1 | 79 | ||
| 1,650 (total) | 246 | 444 | 296c | 458c |
The mutants contained in one plate correspond to those in the progeny produced by one pool of pseudo-single bursts. For the final analysis of the data, the plates containing 69 and 79 mutants were removed from the table, in which case the total decreased from 1,650 to 1,648.
Per the number of plates indicated in the first column. The minimum was calculated assuming that all mutants on a single plate were derived from one mutation occurring within a single infected cell. The maximum was calculated assuming that all mutants on a single plate were derived from unique mutations occurring possibly within the same or different infected cells.
The mean number of mutants per plate is E(i) = Σg(i)/1,648 = 0.1796. Variance is V(i) = Σg(i)i2/(1,648 − 1) − E(i)2 = 0.2458. The variance-to-mean ratio is 0.2458/0.1796 = 1.37.
It is usually difficult to estimate the mutation rate from the observed frequency of mutants because a single mutant may or may not be the product of one mutation. The observed frequency can be corrected to estimate the mutation rate, but that requires knowing the exact mode of replication, which creates a circularity because that was partly the objective of the present study. However, as discussed earlier, the distribution of mutations is expected to be Poisson. As a result, the observed frequency of bursts with no mutants, and hence also no mutations, can be used to estimate the mutation rate by using the Poisson formula. If u(B − 1) is the expected number of mutations per burst, f(0), the expected frequency of bursts with no mutants, is the Poisson null class
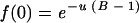 |
(1) |
Solving equation 1 for u gives
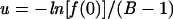 |
(2) |
Using equation 2, umin was estimated by assuming that all mutants on a plate (Table 1) were derived from a single mutation. In other words, on a plate with eight mutants, all eight were produced by one mutation occurring within a single infected cell. In effect, umin assumes a geometric growth model in which the mutant progeny becomes a replicative template. Obversely, umax was estimated by assuming that all mutants on a plate were produced by unique mutations or, in effect, a stamping machine model. By a stamping machine the unique mutations may or may not occur within the same infected cell.
If all mutants on a plate are assumed to be derived from a single mutation, the minimal number of mutations that occurred during our experiment was 246 (Table 1), and the proportion of plates with no mutants [p(0)] is (T − 246)/T. Thus, with a B value of 76 and equation 2, umin is 2.5 × 10−6. If all mutants on a plate are derived from unique mutations, p(0) is (T − 444)/T (Table 1) and umax is 4.5 × 10−6.
The likelihood that the plates with 69 and 79 mutants are explained by having wild-type revertants in the original sus297 population can be determined by calculating the expected number of revertants in T number of phage. The minimum expectation is given by umin × T as 3.27, and the maximum is given by umax × T as 5.9. Thus, having two revertants among the 1,308,450 phage sampled is very likely, and the two plates with 69 and 79 mutants are best explained by the presence by two cells each initially infected with a revertant. After 69 and 79, the next largest size class was 8 mutants per plate (Table 1). However, because 8 is so much smaller than our estimated burst size of 76, plates with 8 or fewer mutants could not be attributed to bursts caused by revertants present in the original sus297 population and were therefore not removed from the data set. With the 69- and 79-mutant plates removed, the corrected data set was subjected to a final analysis.
Final analysis.
Although we estimated umin and umax by examining the proportion of infected cells or bursts not producing any mutants, such an analysis can give only the maximum and minimum bounds of u. To obtain a more exact estimate of u, we reanalyzed the data by retaining the experiment's true data structure, which scored the number of mutants per plate. With the data structured by plates, u is estimated instead by p(0). Following the logic used to derive equation 1, the expected value of p(0) is again the null class of the Poisson, except that the expected number of mutations per plate is u(B − 1)K, where K (the number of infected cells or bursts per plate) is 793. Thus,
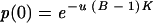 |
(3) |
Because the two plates with 69 and 79 mutants were discarded, the observed value of p(0) was 1,404/N, where N is 1,650 − 2, or 1,648 (Table 1). With these observed values of p(0), B, N, and K, the solution of equation 3 yields
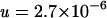 |
(4) |
As indicated above, the fact that a large number of plates contained only one mutant was suggestive of a stamping machine model of replication for φ6, but the presence of plates with many mutants indicated the possibility of some replication by the progeny genomes. Even after the plates with 69 and 79 mutants were removed, the distribution still appeared to be overrepresented for plates with many mutants. For example, if the replication mode were a stamping machine, the distribution should have been Poisson with variance-to-mean ratio of 1. Instead, even after the plates with 69 and 79 mutants were subtracted, the variance and mean of the distribution were 0.2457 and 0.1796 (Table 1), which yielded a value of
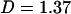 |
(5) |
where D is the ratio of the variance to the mean.
The most likely explanation for why our observed data deviated from Poisson is that the replication mode of φ6 is primarily a stamping machine in which the progeny become replicative templates with a small probability. In fact, it should be possible to estimate such a probability by constructing a model and determining what value the probability must take to best explain our observation of a D value of 1.37. Thus, we simulated our data set by constructing a four-state computer model to represent replication of φ6 within a single infected cell. The four states corresponded to replicate template genomes and nonreplicative genomes, which in turn could be either mutated or nonmutated. The number of replicative genomes is denoted by R(0) and R(1) and the number of nonreplicative genomes is denoted by NR(0) and NR(1), where the indexes 0 and 1 represent, respectively, mutated and nonmutated states. In the context of the present experiment, 0 is the amber mutation in sus297 and 1 is the wild-type revertant. Because all of our bursts were due to cells infected with a single parental sus297 phage, R(0) is 1 and R(1), NR(0), and NR(1) are 0 at the start of the infection. For each round of replication after infection, all replicative genomes were each assumed to produce one progeny nonreplicative genome. Mutated replicative genomes produced nonmutated nonreplicative genomes with a probability equal to the mutation rate (u), and they produced mutated nonreplicative genomes with a probability of 1 − u. Nonmutated replicative genomes produced only nonmutated nonreplicative genomes. Including the mutational transition from nonmutated replicative genomes to mutated nonreplicative genomes would not have changed the analysis by much, because most mutated nonreplicative genomes will be produced from mutated replicative genomes, which will always be more abundant than nonmutated replicative genomes for the conditions examined by the model. After each round of replication, nonreplicative genomes were converted to replicative genomes with a probability m. To generate a burst, replication rounds were continued until R(0) + R(1) + NR(0) + NR(1) = B. The total number of mutants produced by a burst is therefore R(1) + NR(1). Both m and u are probabilities per round of genome replication.
Using our model as the basis of replication of φ6, we simulated our observed data set (Table 1) with a computer program in BASIC. The core of the model was the randomization of replication during rounds leading up to each burst. During each round, mutations were generated according to random numbers drawn from a uniform distribution between 0 and 1. If the random number was less than u, a mutated replicative template was recorded to have produced a nonmutated nonreplicative progeny. If the number was greater than u, the production of an mutated nonreplicative progeny was recorded. u was set at the value estimated in equation 4. In a similar manner, the conversion from nonreplicative to replicative genomes was generated according to random numbers compared to the value of m, which was explored for a range of values from 10−1 to 10−7. For each value of m, the computer model generated 400 randomized data sets. For each randomized set, 793 bursts were first simulated to represent one plate. The number of mutants [R(1) + NR(1)] was summed for all 793 bursts to provide the number of mutants for the plate. The process was repeated until the number of mutants per plate was generated for 1,648 plates to complete one randomized data set. The mean and variance of the number of mutants per plate and the ratio (D) were then determined (Table 1) for each randomized set. Thus, for each value of m, 400 randomized values of D were generated.
To determine which value of m best explained our observed D value of 1.37, the expected 95% confidence limits of D were determined from the 400 randomized values. After the values were ranked, the limits were taken as the boundaries for the top and bottom 2.5% percentiles. These confidence limits describe the variation that is expected in D by chance alone for a value of m, given the size of our observed data set with K being 793 bursts per plate and N being 1,648 plates. Thus, m values with confidence limits that contained a D value of 1.37 could account for our observed data. It was clear from the results that a very narrow range of D was appropriate (Fig. 2). With an m value of 1, replication was effectively a geometric model and D was much greater, as expected from a Luria-Delbruck distribution. With an m value of <10−5, the conversion of genomes from nonreplicative to replicative was so rare that replication was effectively stamping machine and the limits were tightly dispersed around a D value of 1, as expected by from a Poisson distribution. Only an m value of 10−4 generated limits that contained 1.37.
FIG. 2.

Variance-to-mean ratios for the number of mutants expected for different values of m, where m is the probability per round of genome replication that a progeny virus becomes a replicative template. Bars represent the 95% confidence limits of the variance-to-mean ratios generated by the model (see the text for details). The dashed line denotes a ratio of 1.37, which was the value observed in our experiment. Only an m value of 10−4 generated 95% limits that contained the observed value of 1.37.
Our data and analysis suggest therefore that replication in the bacteriophage φ6 is primarily by a stamping machine process with a small probability, on the order of 10−4 per round of replication, that a newly synthesized genome becomes a replicative template.
DISCUSSION
We used the distribution of mutants across bursts to investigate whether the bacteriophage φ6 replicated by a stamping machine or geometric growth model. We found that our observed distribution had a variance-to-mean ratio of 1.37. With a pure stamping machine model, the distribution is expected to be Poisson, with a variance-to-mean ratio of 1 (27). By replicating with a computer model the data structure of our experiment, we determined that our observed ratio was best explained by a stamping machine model of replication in which newly synthesized progeny templates have a probability (m) of 10−4 per replication round of becoming replicative templates themselves. Thus, replication in φ6 is not a pure stamping machine model. Because progeny templates can replicate, there is an element of geometric growth. However, because the burst size of φ6 is about 100 (see above) (30), the proportion of bursts in which progeny templates become replicative is on the order of 100 × 10−4, or 10−2. Thus, replication in φ6 is effectively by a stamping machine model because approximately 99% of infections are pure stamping machine.
The value we estimated for m was much lower than expected because indirect evidence had suggested that progeny φ6 genomes could replicate at a much higher rate. If reoviruses provide an example by analogy, the incorporation pattern of labeled uridine has shown that progeny genomes replicate (21, 23). Reoviruses are ds RNA viruses that replicate by a conservative plus-strand displacement mode that is otherwise similar to φ6. In φ6, procapsids synthesized from cloned cDNA are capable in vitro of taking up plus-strand RNA, completing minus-strand synthesis to form a ds progeny, and producing new plus strands (18). The fact we find very little replication by the progeny genomes argues that some of the steps observed in vitro are delayed by regulation in vivo.
The demonstration that a virus such as φ6 replicates primarily by a stamping machine is useful for the purpose of estimating mutation rates. Because the distribution will be much more Poisson than Luria-Delbruck, mutation rates can be estimated without much error as the frequency of mutants in one large pool of pseudo-single bursts, instead of applying equation 3 to a very large collection of many pools. For example, by using a collection of many pools (N = 1,648 plates), we obtained our best estimate of u of 2.7 × 10−6 (equation 4) in the final analysis. However, had we pooled all of our pseudo-single bursts and then selected for mutants on one plate, we would have obtained a single plate with 444 mutants (Table 1). These mutants would have been among the progeny phage produced by T number of bursts or among T × (B − 1), or 98,133,750, phage. Thus, the frequency of mutants in the hypothetical single pool would have been 444/98,133,750, or 4.5 × 10−6. This estimate of u differs less than twofold from our more accurate estimate of 2.7 × 10−6. Thus, u in φ6 or any other virus that replicates by an equivalent stamping machine could be estimated within a factor of two by simply screening for mutants in a single pool of pseudo-single bursts. Note also that much of the twofold error is due to the inclusion of the plates with 69 and 79 mutants, which were removed from our estimate of u of 2.7 × 10−6 (see Results). If T is reduced so that the probability of having a revertant in the original sus297 population is also reduced, the accuracy of an estimate of u based on a single pool is increased greatly.
Because RNA viruses are thought to have generally a high mutation rate, our estimate of u of 2.7 × 10−6 for φ6 was lower than expected. Drake and Holland (12) estimated the genomic mutation rate (U) to be between 1 and 0.1 for most RNA viruses, where U is G × u, G is the genome size in nucleotides, and u is the per-nucleotide mutation rate. For φ6, G is approximately 104 (18), in which case we had anticipated that u is approximately 10−4 to 10−5. If the amber mutation in sus297 can be reverted by only one nucleotide change, then our u value of 2.7 × 10−6 is a per-nucleotide estimate. If there is more than one way to revert, then our value is an overestimate of the per-nucleotide rate. In either case, our estimate was over 1 order of magnitude lower than expected from Drake and Holland's estimates. Perhaps φ6 is an exception and RNA viruses are more variable than previously suspected. However, because our estimate was obtained for a single amber mutation, it is not appropriate to generalize. Additionally, there may be a simpler explanation. The sus297 mutant of φ6 was originally chosen for its stability as a marker for genetic crosses, in which case we could have selected for an amber mutation that naturally reverted at a low rate.
The existence of different modes of replication in viruses raises the question of whether one or another provides an adaptive advantage. Geometric growth clearly provides the power of replication speed. If B is 100, a stamping machine requires 100 − 1, or 99, rounds of replication. Because 99 ≈ 26.6, geometric growth requires only 6.6 rounds of replication to achieve an equivalent burst size. However, a stamping machine could be advantageous if the rate of deleterious mutations is high. As indicated previously, viruses have become commonly used to measure the rate of deleterious mutations, and high values have generally been reported for RNA viruses. To illustrate the effect of a high rate of deleterious mutations, let g(0) be the proportion of genomes synthesized without any mutations during the course of one infection and burst. If replication is by a stamping machine, the proportion is given by the Poisson null class, or
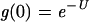 |
(6) |
However, if replication is by geometric growth, g(0) will depend on the number of doublings that occur during one infection and burst cycle. After the first doubling, the proportion of mutation-free genomes is given by equation 6. However, after the second doubling, the proportion is reduced by a factor of e−U. Following the argument,
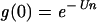 |
(7) |
after n doublings.
Depending on the values of n and U and assuming that most mutations are deleterious, the difference between equations 6 and 7 can be large. For example, U is 0.002 for most DNA viruses, whereas U is 1 for most RNA viruses. Thus, if DNA viruses replicate by a stamping machine, g(0) is e−0.002, or 0.998. By geometric growth, using DNA phages as an example, B is 100 and n is 6.6 (see above). Thus, g(0) is 0.9986.6, or 0.987, and the gain of replicating by a stamping machine over geometric growth for avoiding deleterious mutations is on the order of only 1%. However, for an RNA virus with the same value of n, the difference is significant. By a stamping machine, g(0) is e−1, or 0.37. By geometric growth, g(0) is 0.376.6, or 0.0014, and the relative advantage of a stamping machine is over 26,000%.
The above analysis suggests that RNA viruses have much more to gain by evolving a stamping machine mode of replication. The fact that we find support for a stamping machine in φ6 supports such a viewpoint. DNA viruses, on the other hand, have little to gain by evolving a stamping machine. Thus, they would have more to gain by evolving a geometric growth mode of replication and capitalizing on the resulting speed. However, the data for DNA phages are not supportive. As indicated previously, whereas the DNA phage T2 replicates by geometric growth, the DNA phage φX174 follows a stamping machine. Both T2 and φX174 have approximately the same genomic mutation rate (U), 0.003 (9, 13).
Determining whether the replication mode of viruses is itself an adaptive trait or whether it is simply a by-product of other constraints in the viral developmental system will require information on many more viruses. Additional information would help by providing larger sample sizes to determine whether any patterns or correlations are robust. For example, is φX174 the example that rejects the hypothesis that DNA viruses should replicate by a geometric growth, or is it the exception that proves the rule? We hope that our study encourages the collection of similar data sets and also demonstrates the utility of distributional data from pseudo-single-burst experiments. The value of pseudo-single-burst data is more than just complementing alternative approaches, such as using label incorporation during replication. Because pseudo-single-burst data track replication by mutated progeny genomes, the sensitivity of the analysis is increased and it is much easier to estimate m.
Acknowledgments
We thank Olivier Tennaillon and members of our laboratory for discussions and comments and Lenny Mindich for generously providing viral and bacterial strains.
We thank the National Institutes of Health (National Institute of General Medical Sciences award GM60916) for support.
REFERENCES
- 1.Burch, C. B., and L. Chao. 1999. Evolution by small steps and rugged landscapes in the RNA virus φ6. Genetics 151:921-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burch, L. C., and L. Chao. 2000. Evolvability of an RNA virus determined by its mutational neighborhood. Nature 406:625-628. [DOI] [PubMed] [Google Scholar]
- 3.Chao, L. 1991. Levels of selection, evolution of sex in RNA viruses, and the origin of life. J. Theor. Biol. 153:229-246. [DOI] [PubMed] [Google Scholar]
- 4.Chao, L. 1990. Fitness of RNA virus decreased by Muller's ratchet. Nature 348:454-455. [DOI] [PubMed] [Google Scholar]
- 5.Chao, L. 1992. Evolution of sex in RNA viruses. Trends Ecol. Evol. 7:147-151. [DOI] [PubMed] [Google Scholar]
- 6.Chao, L. 1997. Evolution of sex and the molecular clock in RNA viruses. Gene 205:301-308. [DOI] [PubMed] [Google Scholar]
- 7.Chao, L., T. T. Trang, and T. T. Trang. 1997. The advantage of sex in the RNA phage φ6. Genetics 147:953-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de la Pena, M., S. F. Elena, and A. Moya. 2000. Effect of deleterious mutation-accumulation on the fitness of RNA bacteriophage MS2. Evolution 54:686-691. [DOI] [PubMed] [Google Scholar]
- 9.Denhardt, D., and R. B. Silver. 1966. An analysis of the clone size distribution of φX174 mutants and recombinants. Virology 30:10-19. [DOI] [PubMed] [Google Scholar]
- 10.Domingo, E. 2000. Viruses at the edge of adaptation. Virology 270:251-253. [DOI] [PubMed] [Google Scholar]
- 11.Domingo, E., and J. J. Holland. 1994. Mutation rates and rapid evolution of RNA viruses, p. 161-184. In S. S. Morse (ed.), The evolutionary biology of viruses. Raven Press, Ltd., New York, N.Y.
- 12.Drake, J. W., and J. Holland. 1999. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 96:13910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drake, J. W., B. Charlesworth, D. Charlesworth, and J. F. Crow. 1998. Rates of spontaneous mutation. Genetics 148:1667-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duarte, E., D. Clarke, A. Moya, E. Domingo, and J. Holland. 1992. Rapid fitness losses in mammalian RNA virus clones due to Muller's ratchet. Proc. Natl. Acad. Sci. USA 89:6015-6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elena, S. F. 1999. Little evidence for synergism among deleterious mutations in a nonsegmented RNA virus. J. Mol. Evol. 49:703-707. [DOI] [PubMed] [Google Scholar]
- 16.Elena, S. F., and A. Moya. 1999. Rate of deleterious mutation and the distribution of its effects on fitness in vesicular stomatitis virus. J. Evol. Biol. 12:1078-1088. [Google Scholar]
- 17.Fraile, A., J. L. Alonso-Prados, M. A. Aranda, J. J. Bernal, J. M. Malpica, and F. Garcia-Arenal. 1997. Genetic exchange by recombination or reassortment is infrequent in natural populations of a tripartite RNA plant virus. J. Virol. 71:934-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frilander, M., P. Gottlieb, J. Strassman, D. H. Bamford, and L. Mindich. 1992. Dependence of minus-strand synthesis on complete genomic packaging in the double-stranded RNA bacteriophage φ6. J. Virol. 66:5013-5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luria, S. 1951. The frequency distribution of spontaneous bacteriophage mutants as evidence for the exponential rate of phage production. Cold Spring Harbor Symp. Quant. Biol. 16:463-470. [DOI] [PubMed] [Google Scholar]
- 20.Luria, S. E., and M. Delbruck. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28:491-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan, E. M., and H. J. Zweerink. 1977. Characterization of the double-stranded RNA in replicase particles in reovirus-infected cells. Virology 77:421-423. [DOI] [PubMed] [Google Scholar]
- 22.Ruiz-Jarabo, C. M., A. Arias, E. Baranowski, C. Escarmis, and E. Domingo. 2000. Memory in viral quasispecies. J. Virol. 74:3543-3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiff, L. A., and B. N. Fields. 1991. Reoviruses and their replication, p. 587-618. In B. N. Fields and D. M. Knipe (ed.), Fundamental virology, 2nd ed. Raven Press, Ltd., New York, N.Y.
- 24.Schneider, W. L., and M. J. Roossinck. 2000. Evolutionarily related Sindbis-like plant viruses maintain different levels of population diversity in a common host. J. Virol. 74:3130-3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sierra, S., M. Davila, P. R. Lowenstein, and E. Domingo. 2000. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J. Virol. 74:8316-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinclair, J. F., J. Cohen, and L. Mindich. 1976. Isolation of suppressible nonsense mutations of bacteriophage φ6. Virology 75:198-208. [PubMed] [Google Scholar]
- 27.Sokal, R. R., and F. J. Rohlf. 1981. Biometry. Freeman, San Francisco, Calif.
- 28.Stent, G. S. 1963. Molecular biology of bacterial viruses. W. H. Freeman and Company, San Francisco, Calif.
- 29.Turner, P. E., and L. Chao. 1999. Prisoner's dilemma in an RNA virus. Nature 398:441-443. [DOI] [PubMed] [Google Scholar]
- 30.Vidaver, A. K., R. K. Koski, and J. L. Van Etten. 1973. Bacteriophage φ6: a lipid-containing virus of Pseudomonas phaseolicola. J. Virol. 11:799-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wichman, H. A., M. R. Badgett, L. A. Scott, C. M. Boulianne, and J. J. Bull. 1999. Different trajectories of parallel evolution during viral adaptation. Science 285:422-425. [DOI] [PubMed] [Google Scholar]
- 32.Wichman, H. A., L. A. Scott, C. D. Yarber, and J. J. Bull. 2000. Experimental evolution recapitulates natural evolution. Philos. Trans. R. Soc. Lond. B 355:1677-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]