

Abstract
Background
Sphingomyelin (SM) is the major phospholipid in cell membranes and in lipoproteins. In human plasma, SM is mainly found in atherogenic lipoproteins; thus, high levels of SM may promote atherogenesis.
Methods
We investigated in a median follow up of 6.0 years the association of SM with the incidence of a combined endpoint (myocardial infarction and cardiovascular death) in stable and unstable patients, and its relation to other marker of atherosclerosis in 1,102 patients with angiographically documented CAD and 444 healthy controls.
Results and discussion
Logistic regression analysis showed that SM categorized by median was associated with an elevated risk for CAD (HR 3.2, 95%CI 2.5–4.0, p < 0.05). SM levels were correlated with apoB (r = 0.34) and triglyceride levels (r = 0.31). In patients with stable angina (n = 614), SM categorized by median was not related to incidence of a combined endpoint (cardiovascular death and myocardial infarction) (p = 0.844 by Log-rank test). However, in patients with acute coronary syndrome (n = 488), elevated SM was related to the combined endpoint (p < 0.05 by Log-rank test), also in a multivariate Cox regression analysis including potential confounders (HR 1.8, 95%CI 1.0–3.3, p < 0.05).
Conclusion
The results of our study reveal that 1) human plasma SM levels are a risk factor for CAD; 2) the pro-atherogenic property of plasma SM might be related to metabolism of apoB-containing or triglyceride-rich lipoproteins; and 3) plasma SM levels are a predictor for outcome of patients with acute coronary syndrome.
Introduction
Atherogenesis is initiated by the interaction of cholesterol-rich lipoproteins with the arterial wall [1]. Many processes have been implicated in early atherogenesis, including lipoprotein oxidation [2], lipoprotein retention and aggregation [1,3], endothelial alteration, macrophage chemotaxis and foam cell formation, and smooth muscle cell migration and alteration [4]. However, subendothelial retention and aggregation of atherogenic lipoproteins have emerged as the primary pathogenic processes [1].
Two sets of evidence indicate that aortic and plasma sphingomyelin (SM) levels are closely related to the development of atherosclerosis: First, it has long been known that SM accumulates in atheromas formed in humans and in animal models [5-7]. The low-density lipoprotein (LDL) extracted from human atherosclerotic lesions is much richer in SM than the LDL from plasma [8,9]. A substantial amount of the SM found in arteries and atherosclerotic lesions appears to arise from synthesis in arterial tissue [10]. The SM concentration is also significantly increased in macrophages treated with acetyl-LDL plus an acyl-CoA:cholesterol acyltransferase inhibitor [11]. However, even in atherosclerotic lesions, the apparent rate of SM formation is relatively low compared with the rate of total phospholipid synthesis [12], suggesting that other factors might also contribute to intimal SM accumulation. Secondly, plasma SM levels are increased in human familial hyperlipidemias, especially in familial hypercholesterolemia [13], and also in animal models of atherosclerosis [14-16]. The concentration of SM is an important determinant of the susceptibility of lipoprotein SM to sphingomyelinase (SMase) [9,16], which might in turn cause atherogenic lipoprotein aggregation and atherosclerosis [17]. These findings suggest that plasma SM levels may be a risk factor for atherosclerosis.
In order to further investigate the role of SM in atherosclerosis, we measured plasma SM levels in 1,102 patients with angiographically proven coronary artery disease (CAD) and 444 healthy controls and its relationship to other risk factors and clinical outcome.
Methods
Study population
Between November 1996 and July 2000, we recruited 1,102 patients suffering from symptoms of CAD (614 patients with stable angina [SAP]; 488 with acute coronary syndrome [ACS]) admitted to the second medical department of the Johannes Gutenberg-University Mainz or the Bundeswehrzentralkrankenhaus Koblenz for diagnostic coronary angiography. The sole inclusion criterion was the presence of a stenosis > 30% in at least one major coronary artery. The study is described in detail elsewhere [18]. Exclusion criteria were lack of CAD as defined above and evidence of significant concomitant disease, in particular severe valvular heart disease, known cardiomyopathy, malignancy, inflammatory diseases, or a febrile condition. Patients completed a questionnaire about smoking habits, history of diabetes mellitus, hypertension, hyperlipoproteinemia, current drug use, and family history of premature CAD (documented in one first-degree relative before age 65). Diabetes mellitus was diagnosed in patients who had previously undergone dietary treatment or received additional oral antidiabetic or insulin medication or who had a current fasting blood sugar level > 125 mg/dl; hypertension was diagnosed in patients who had received antihypertensive treatment or had been diagnosed as hypertensive (blood pressure > 160/90 mmHg); hyperlipoproteinemia was diagnosed in patients who had been given lipid-lowering medication or had a history of cholesterol levels > 240 mg/dl.
Patients were followed up for a median 6.0 years. The majority of the patients presented at our clinic for follow-up; a small number of them were interviewed by telephone by trained medical staff. At follow-up, information was obtained about cardiovascular death or nonfatal myocardial infarction (n = 163). Information about the cause of death or clinical events was obtained from hospital or general practitioner charts.
Healthy control subjects (n = 444) were recruited either from general practitioners' offices in the course of routine check-up visits or by newspaper announcement, which briefly described the study design and invited healthy individuals aged ≥40 years to participate in this study as control subjects. Of the individuals who presented, we selected those without any clinical or anamnestic evidence of atherosclerosis and without evidence of any pathological ECG pattern. All individuals who presented received reports of any classical and treatable risk factors for personal use.
In general, study and control patients were of German nationality and were inhabitants of the Rhein-Main Area. The study was approved by the ethics committee of the University of Mainz. Participation was voluntary, and each study subject gave written informed consent.
Laboratory methods
Blood was drawn from all subjects under standardized conditions after an overnight fasting period and before coronary angiography was performed. Samples were placed on ice immediately and within 30 minutes were centrifuged at 4,000 rpm for 10 minutes, divided into aliquots, and frozen at -80°C until analysis.
Plasma SM levels were measured as described previously [19]. SM was analyzed concurrently in patients and controls and the laboratory personnel was unaware of the individual's study allocation.
Plasma lipid levels (total cholesterol, Roche Diagnostics, Germany; high-density lipoprotein cholesterol (HDL-C), Rolf Greiner Biochemica, Flacht, Germany; LDL-C, calculated according to the Friedewald formula; triglycerides, Roche Diagnostics, Germany) were determined immediately. Apolipoprotein A-I (apoA-I) and apoB concentrations were determined by immunoturbodimetric assays (Tina-quant, Roche Diagnostics, Germany). The lipoprotein(a) [Lp(a)] concentration was determined using an enzyme-linked immunosorbent assay-based method supplied by Immuno Ltd. (Dunton Green, Kent, UK). C-reactive protein (CRP) was determined by a highly sensitive, latex particle-enhanced immunoassay (detection range of 0 to 20 mg/l); the between-day imprecision of this assay (n = 21) was 2.14% and 1.44% at mean levels of 1.90 mg/l and 4.33 mg/l (Roche Diagnostics, Germany).
Statistical analysis
Demographic and clinical variables of cases and controls were compared by Chi square test for categorical and ANOVA test for continuous variables. Because of skewed distribution for triglyceride and Hs-CRP, median values were presented and the Mann-Whitney test applied for these variables. The distributions for plasma concentrations of SM were skewed and all analyses were calculated with log-transformed values; however, exponential values are presented. For correlation of SM with further variables, Pearson's correlation coefficients were calculated, using log-transformed values of SM, triglycerides and high-sensitive CRP (Hs-CRP) for each group individually. Case and control r-values were compared using Fisher's Z-transformation. We aimed to assess any evidence of association between SM and CAD in models, assuming both linear and nonlinear effects. We thus divided cases and controls into two groups stratified by median (48.1 mg/dl) on the basis of the overall study population and applied a logistic regression model.
Survival was analyzed by the Kaplan-Meyer method and Log-rank test. In all survival analyses, the endpoint was a combined endpoint of death of cardiovascular causes and nonfatal myocardial infarction. Data on patients who died of other causes were censored at the time of death. The association of SM with outcome was evaluated by Cox regression analyses adjusted for potential confounders. In these analyses, SM was considered in two groups stratified by median; other markers were considered either as continuous (log-transformed) or categorical variables. Hazard ratios (HRs) and 95 % confidence intervals (CIs) are reported with 2-tailed probability values. P < 0.05 was considered to be significant. All analyses were carried out using SPSS 11.5 software.
Results
Demographic and clinical measures
Baseline data regarding case and control subjects are outlined in Table 1. As expected, the prevalence of classical risk factors such as current smoking (27.3% versus 11.0%), history of diabetes mellitus (21.6% versus 3.7%), hypertension (71.9% versus 28.6%), and family history of premature CAD (38.2% versus 20.1%) was more frequent in cases than in controls. Furthermore, a significantly higher rate of ACE inhibitor (47.8% versus 8.3%) and statin (35.8% versus 6.3 %) prescriptions was noted in cases than in controls. The higher prescription of statins might well have caused the significantly lower levels of total cholesterol and LDL-C levels noted in the cases. HDL-C and apoA-I levels were lower in cases than in controls, as expected. Lp(a) and triglycerides but not apoB levels were higher in the patients. Phospholipid transfer protein (PLTP) and cholesterol ester transfer protein (CETP) activity and Hs-CRP concentrations were also elevated in patients.
Table 1.
Demographic and clinical measures in cases and controls. For cases and controls categorical variables are presented as percentage of patients; chi-square test was used for group differences; and continuous variables are presented as mean values (standard deviation) or *median values (25/75th interquartiles) because of skewed distribution. The p values were obtained by *Mann-Whitney test. #: data for 913 patients for ejection fraction, data for 200 controls/1026 patients for apoA-I and apoB, for 200 controls/905 patients for Hs-CRP, and for 200 controls/1060 patients for Lp(a).
| Variable | Cases n = 1102 | Controls n = 444 | P- value |
| Age, years | 61.1 (10.1) | 59.7 (7.4) | <0.001 |
| BMI, kg/m2 | 27.2 (3.7) | 26.7 (4.3) | 0.02 |
| Sex, male % | 74.7 | 72.7 | 0.43 |
| Current smoking, % | 27.3 | 11.0 | < 0.001 |
| Diabetes mellitus, % | 21.6 | 3.7 | <0.001 |
| Hypertension, % | 71.9 | 28.6 | <0.001 |
| Family history of CAD, % | 38.2 | 20.1 | <0.001 |
| Statins, % | 35.8 | 6.3 | < 0.001 |
| ACE inhibitors, % | 47.8 | 8.3 | <0.001 |
| Angiotensin receptor blockers, % | 5.0 | 3.8 | 0.327 |
| Ejection fraction <40%#, % | 7.6 | - | - |
| Hs-CRP*, mg/l# | 4.4 (1.9/12.4) | 1.5 (0.9/3.8) | <0.001 |
| PLTP, U/l | 25.5 (9.8) | 22.4 (7.4) | <0.001 |
| CETP, U/l | 49.9 (20.9) | 41.4 (17.4) | <0.001 |
| Total cholesterol, mg/dl | 219.7 (45.7) | 238.4 (41.8) | <0.001 |
| LDL cholesterol, mg/dl | 141.0 (40.1) | 155.5 (35.1) | <0.001 |
| HDL cholesterol, mg/dl | 48.8 (14.7) | 59.4 (15.9) | <0.001 |
| Triglyceride*, mg/dl | 141 (102/193) | 120 (87/167) | <0.001 |
| Apolipoprotein A-I, g/l# | 1.31 (0.24) | 1.62 (0.28) | <0.001 |
| Apolipoprotein B, g/l# | 1.20 (0.28) | 1.19 (0.23) | 0.3 |
| Lipoprotein(a), mg/dl# | 36.3 (38.9) | 24.2 (30.4) | <0.001 |
Relationship between SM and CAD
Figure 1 shows that patients with CAD had higher plasma SM levels than healthy controls (mean 51.8 mg/dl versus mean 44.9 mg/dl; p < 0.001 by ANOVA).
Figure 1.
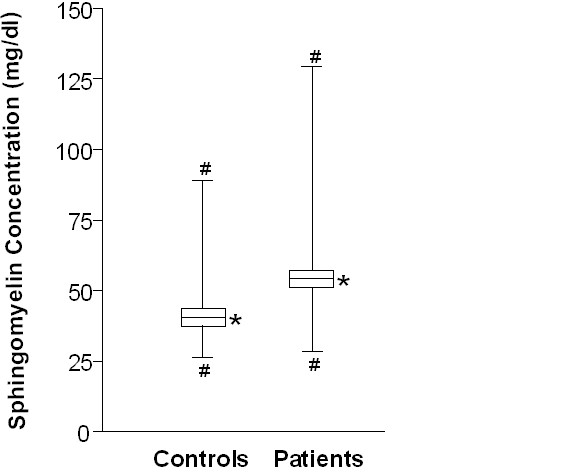
Plasma SM concentrations in patients and controls (p < 0.001 by ANOVA). *: mean with 95% confidence intervals, #: minimum/maximum.
Logistic regression analysis showed that plasma SM categorized by median was associated with an elevated risk for CAD (HR 3.2, 95%CI 2.5–4.0, for subjects with SM ≥48.1 mg/dl compared to subjects with SM < 48.1 mg/dl).
Correlation with other risk factors
To investigate the possibility that plasma SM levels could act as a marker for triglyceride-rich lipoproteins or apoB-containing lipoprotein metabolism, we performed correlation analyses as outlined in Table 2. In all subjects correlations were found between plasma SM and apoB (r = 0.34) and between plasma SM and triglycerides (r = 0.31). Moreover, these relationships were significantly stronger in patients than in controls (for apoB, r = 0.36 for patients and r = 0.25 for controls, respectively, p = 0.03; for triglyceride, r = 0.32 for patients and r = 0.11 for controls, respectively, p < 0.001, see Table 2).
Table 2.
Pearson coefficients of correlation between SM and other continuous variables (age, further lipid variables, CETP, PLTP, and Hs-CRP), stratified by CAD. Comparison of r-values of cases and controls by Fisher's Z-transformation (p- and Z-values presented)
| Sphingomyelin‡ | ||||
| All | Cases | Controls | p-value (Z-value) | |
| Age, years | - 0.03 | 0.06 | -0.03 | 0.11 (1.6) |
| BMI, kg/m2 | 0.08 | 0.09 | 0.06 | 0.59 (-0.54) |
| Total cholesterol, mg/dl | 0.17 | 0.25 | 0.14 | 0.04 (2.03) |
| LDL cholesterol, mg/dl | 0.13 | 0.20 | 0.13 | 0.2 (1.28) |
| HDL cholesterol, mg/dl | - 0.14 | -0.08 | -0.01 | 0.12 (-0.24) |
| Triglyceride, mg/dl‡ | 0.31 | 0.32 | 0.11 | <0.001 (3.92) |
| Apolipoprotein A-I, g/l | -0.05 | 0.05 | -0.07 | 0.03 (2.13) |
| Apolipoprotein B, g/l | 0.34 | 0.36 | 0.25 | 0.03 (2.15) |
| Lipoprotein(a), mg/dl | 0.03 | 0.01 | -0.04 | 0.37 (0.89) |
| CETP, U/l | -0.11 | -0.03 | -0.19 | < 0.001 (2.88) |
| PLTP, U/l | 0.01 | -0.06 | 0.08 | 0.01 (-2.94) |
| Hs-CRP, mg/l‡ | 0.05 | 0.05 | 0.12 | 0.21 (1.25) |
‡: log-transformed variables
Weak associations were found between plasma SM levels and total cholesterol (r = 0.17), LDL-C (r = 0.13), HDL-C (r = -0.14) and CETP (r = -0.11) in all subjects. The correlations between SM and total cholesterol, LDL-C, and HDL-C were stronger in patients; however, the correlation between SM and CETP was stronger in controls.
There were no or only very weak associations between plasma SM levels and other risk factors, including age, BMI, apoA-I, PLTP, and Lp(a). It is of interest that SM concentrations were not related to Hs-CRP levels (r = 0.05).
For the possible relationship of plasma SM to categorical risk factors, ANOVA tests were performed separately for patients and controls. Statin intake seemed to influence plasma concentrations in patients, but a significant trend for lower plasma SM concentrations was not observed in patients receiving statin treatment (p = 0.068). Sex, diabetes (in patients), and left ventricular ejection fraction (dichotomized for <40%) did not affect the concentration of plasma SM (see Table 3). The small number of controls with diabetes might explain the significantly elevated SM plasma concentrations in this subgroup. We found a trend for higher plasma SM concentrations in smokers. However, SM was significantly decreased in patients receiving ACE inhibitors (51.3 mg/dl versus 49.7 mg/dl, p = 0.015) but not in controls taking this medication (44.3 mg/dl versus 42.8 mg/dl, p = 0.309) (Table 3).
Table 3.
Mean of plasma SM levels according to cardiovascular risk factors.
| Controls | Patients | |||||
| n | SM in mg/dl | p-value | n | SM in mg/dl | p-value | |
| Sex | ||||||
| female | 121 | 44.7 ± 1.2 | 0.398 | 279 | 51.3 ± 1.25 | 0.210 |
| male | 323 | 43.9 ± 1.21 | 823 | 50.3 ± 1.24 | ||
| Diabetes mell. | ||||||
| - | 428 | 43.9 ± 1.21 | <0.001 | 863 | 50.7 ± 1.24 | 0.362 |
| + | 16 | 52.8 ± 1.26 | 239 | 49.9 ± 1.26 | ||
| Hypertension | ||||||
| - | 317 | 44.0 ± 2.29 | 0.628 | 309 | 51.5 ± 1.25 | 0.077 |
| + | 127 | 44.4 ± 1.24 | 793 | 50.2 ± 1.25 | ||
| Smoking | ||||||
| - | 395 | 43.9 ± 1.21 | 0.048 | 801 | 50.2 ± 1.24 | 0.057 |
| + | 49 | 46.4 ± 1.21 | 301 | 51.6 ± 1.26 | ||
| Statins | ||||||
| - | 411 | 44.0 ± 1.21 | 0.46 | 690 | 51.0 ± 1.24 | 0.068 |
| + | 28 | 45.3 ± 1.19 | 395 | 49.7 ± 1.26 | ||
| ACE inhibitors | ||||||
| - | 407 | 44.3 ± 1.21 | 0.309 | 575 | 51.3 ± 1.26 | 0.015 |
| + | 37 | 42.8 ± 1.26 | 527 | 49.7 ± 1.23 | ||
| Ejection fraction | ||||||
| <40% | - | - | - | 69 | 50.4 ± 1.24 | 0.325 |
| >40% | - | - | 844 | 51.4 ± 1.21 | ||
Cardiovascular risk in patients
In patients with stable angina, SM was not related to the combined endpoint (p = 0.844 by log-rank test, see Figure 1a). However, in a group of patients with acute coronary syndrome and plasma SM concentrations above the median (48.1 mg/dl), a higher proportion of patients with myocardial infarction and cardiovascular death was found (p < 0.05 by Log-rank test, see Figure 1b). For further statistical evaluation of these data, univariate (model 1, Table 4) and multivariate (model 2, model 3, Table 4) Cox regression analyses were performed. The multivariate model including age, sex, body -mass index, smoking, diabetes mellitus, arterial hypertension, hyperlipoproteinemia, family history of CHD, statins, log-transformed triglycerides, and apoB showed that patients with ACS and SM ≥48.1 mg/dl were at an elevated risk for further cardiovascular events (HR 1.8, 95%CI 1.0–3.3, p = 0.048, Table 4).
Table 4.
Cox regression analyses of SM stratified by median (48.1 mg/dl) in patients with acute coronary syndrome in univariate (model 1) and multivariate models (model 2 included age, sex, BMI, smoking, diabetes mellitus, arterial hypertension, and family history of CHD; model 3 further adjusted for statins, apoB, and log-transformed triglyceride)
| SM <48.1 mg/dl | SM ≥48.1 mg/dl | P-value | |
| OR Model 1 | --- | 0.6 | 0.049 |
| 95%CI | 0.3–0.9 | ||
| OR Model 2 | --- | 1.9 | 0.019 |
| 95%CI | 1.1–3.4 | ||
| OR Model 3 | --- | 1.8 | 0.048 |
| 95%CI | 1.0–3.3 | ||
Discussion
In the present study we confirmed our previous observation that plasma SM levels are a risk factor for CAD [19]. Furthermore, we revealed that plasma SM levels showed the strongest correlation with plasma apoB and triglycerides levels, suggesting that human plasma SM levels could be a marker for triglyceride-rich or apoB-containing lipoprotein remnant accumulation found in human atherosclerosis as we suggested previously [20]. Moreover, we found that plasma SM levels are a predictor for event-free survival of patients with acute coronary syndrome, suggesting a possible link between plasma SM levels and the instability of coronary plaques. We also found a not significant trend for lower plasma SM concentrations in patients under statin treatment. Diabetes mellitus or low ejection fraction did not affect plasma SM concentrations.
Why is SM related to the atherosclerotic process? SM might be a marker for an inflammatory effect, and inflammatory markers such as CRP have been shown to be important risk factors for atherosclerosis [21]. However, plasma SM levels did not correlate with Hs-CRP in our study (Table 2). Thus, it is unlikely that SM acts as a surrogate inflammatory marker.
Plasma SM levels are related to atherogenic lipoprotein aggregation, which is the initial step in the development of atherosclerosis [17]. It is well known that SM content is much higher in apoB-containing or triglyceride-rich lipoproteins than in HDL [22]. This may indicate that apoB-containing or triglyceride-rich lipoproteins are atherogenic but that HDL is not. It is also well known that aggregated lipoproteins are prominent in atherosclerotic lesions [3,23-26]. In addition, processes that promote lipoprotein aggregation before or during retention dramatically increase the amount of lipoprotein retained [27,28], which, in turn, might amplify the atherogenic responses to this retention [1,28]. The mechanisms responsible for increased retention as a result of aggregation include increased binding of the aggregates to the subendothelial matrix [27,28] and decreased efflux of the aggregates from the arterial wall due to their increased size [30,31]. Atherogenic lipoprotein aggregation after hydrolysis by sphingomyelinase is the most physiologically plausible mechanism [32,9,34]. Hydrolysis of approx. 25% of the SM of LDL to ceramide in vitro by bacterial SMase leads to the formation of aggregated and fused particles that are excellent inducers of macrophage foam cell formation [28,32,34]. There is evidence to show that a mammalian arterial-wall SMase may carry out a similar reaction in atherosclerotic lesions in vivo [33]. Schissel et al. [35,36] discovered a novel form of SMase called secretory SMase, or S-SMase. Although S-Smase is secreted by macrophages, it is secreted even more abundantly by endothelial cells [37]. Thus, S-SMase could be present in arteries before lesions develop and therefore might explain very early lipoprotein aggregation [3]. Further evidence implicating a role for S-SMase in lipoprotein aggregation in atherosclerotic lesions includes its ability to hydrolyze and aggregate atherogenic lipoprotein [9], its increased secretion from endothelial cells treated with cytokines that have been implicated in atherogenesis [39], and its presence in atherosclerotic lesions as demosntrated by immunohistochemistry [38].
As we suggested previously [20], SM might be a surrogate marker for the metabolism of triglyceride-rich and apoB-containing lipoproteins. In the present study, we found the strongest correlations between plasma SM and apoB or triglycerides (Table 2). Humans spend most of their lives in the postprandial state. The long-term effect of repeated, prolonged, and exaggerated bouts of alimentary lipemia may be disturbances in the metabolism of endogenous lipoproteins that are rich in SM and atherogenic [39]. It is conceivable that postprandial SM remnants in apoB-containing or triglyceride-rich lipoproteins may not only serve as a clearance marker of these particles, but also have their own impact on atherosclerosis. If somehow the clearance of postprandial apoB-containing- or triglyceride-rich lipoproteins was blocked, the SM-rich particles could likely be aggregated in the arterial wall after encountering sphingomylinase there, and development or instability of atheroscerotic plaques could be the consequence. However, epidemiological studies of postprandial SM levels and atherosclerosis are still lacking.
Moreover, atherosclerotic LDLs exhibit an increase in SM [9], which is believed to stimulate hydrolysis of LDL by secreted Smase. In addition, the ceramide content of lipoproteins found in atherosclerotic lesions is increased (up to a 50-fold enrichment as compared with plasma LDL) [36]. These two mechanisms may be further involved in apoptosis of affected cells such as smooth muscle cells or macrophages via the SM/ceramide pathway, resulting in destabilization of atherosclerotic plaques.
Conclusion
Overall, plasma SM is risk factor for CAD and a risk factor for further cardiovascular events in patients with acute coronary syndrome. Its relationship to apoB-containing or triglycerides-rich lipoproteins as shown in our data could partly explain its proatherogenic properties.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
All authors have made substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data. All authors have been involved in drafting the manuscript or revising it critically for important intellectual content, and have given final approval of the version to be published.
Figure 2.
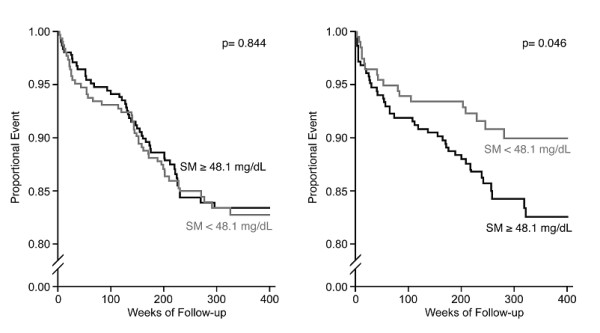
Kaplan-Meyer survival plots for cardiovascular mortality according to SM plasma concentrations in patients with stable (Figure 2a) and unstable angina (Figure 2b).
Acknowledgments
Acknowledgements
This study was supported by National Institute of Health grants HL-64735 and HL-69817. Axel Schlitt was supported by Deutsche Herzstiftung.
Contributor Information
Axel Schlitt, Email: axel.schlitt@medizin.uni-halle.de.
Stefan Blankenberg, Email: stefan.blankenberg@gmx.de.
Daoguang Yan, Email: yandaoguang@hotmail.com.
Hans von Gizycki, Email: Han.VonGizycki@downstate.edu.
Michael Buerke, Email: michael.buerke@medizin.uni-halle.de.
Karl Werdan, Email: karl.werdan@medizin.uni-halle.de.
Christoph Bickel, Email: cbickel@t-online.de.
Karl J Lackner, Email: lackner@zentrallabor.klinik.uni-mainz.de.
Juergen Meyer, Email: meyer@2-med.klinik.uni-mainz.de.
Hans J Rupprecht, Email: hj.rupprecht@gp-ruesselsheim.de.
Xian-Cheng Jiang, Email: xjiang@downstate.edu.
References
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 1991;88:1785–1792. doi: 10.1172/JCI115499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nievelstein PF, Fogelman AM, Mottino G, Frank JS. Lipid accumulation in rabbit aortic intima 2 hours after bolus infusion of low density lipoprotein. Arterioscler Thromb. 1991;11:1795–1805. doi: 10.1161/01.atv.11.6.1795. [DOI] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–808. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Smith EB. Intimal and medial lipids in human aorta. Lancet. 1960;1:799–803. doi: 10.1016/S0140-6736(60)90680-2. [DOI] [PubMed] [Google Scholar]
- Bottcher CJF. In: Phospholipids of atherosclerotic lesions in the human aorta. In Evolution of the Atherosclerotic Plaque. Jones RJ, editor. Chicago University Press, Chicago; 1963. pp. 109–116. [Google Scholar]
- Portman OW, Illingworth RD. Arterial metabolism in primates. Primates Med. 1976;9:145–223. [PubMed] [Google Scholar]
- Guyton JR, Klemp KF. Development of the lipid-rich core in human atherosclerosis. J Lipid Res. 1996;16:4–11. doi: 10.1161/01.atv.16.1.4. [DOI] [PubMed] [Google Scholar]
- Schissel SL, Jiang X, Tweedie-Hardman J, Jeong T, Camejo EH, Najib J, Rapp JH, Williams KJ, Tabas I. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH. J Biol Chem. 1998;273:2738–2746. doi: 10.1074/jbc.273.5.2738. [DOI] [PubMed] [Google Scholar]
- Zilversmit DB, McCandless EL, Jordan PH, Henly WS, Ackerman RF. The synthesis of phospholipids in human atheromatous lesions. Circulation. 1961;23:370–375. doi: 10.1161/01.cir.23.3.370. [DOI] [PubMed] [Google Scholar]
- Okwu AK, Xu XX, Shiratori Y, Tabas I. Regulation of the threshold for lipoprotein-induced acyl-CoA:cholesterol acyltransferase stimulation in macrophages by cellular sphingomyelin content. J Lipid Res. 1994;35:644–655. [PubMed] [Google Scholar]
- Eisenberg S, Stein Y, Stein O. Phospholipases in arterial tissue. J Clin Invest. 1969;48:2320–2329. doi: 10.1172/JCI106198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel C, Marcel YL, Darignon J. Plasma phospholipids in the different types of primary hyperlipoproteinemia. J Lab Clin Med. 1972;79:611–612. [PubMed] [Google Scholar]
- Rodriguez JL, Ghiselli GC, Torreggiani D, Sirtori CR. Very low density lipoproteins in normal and cholesterol-fed rabbits: lipid and protein composition and metabolism. Atherosclerosis. 1976;23:73–83. doi: 10.1016/0021-9150(76)90119-2. [DOI] [PubMed] [Google Scholar]
- Portman OW. Atherosclerosis in nonhuman primates: Sequences and possible mechanisms of changes in phospholipid composition and metabolism. Ann NY Acad Sci. 1969;162:120–136. doi: 10.1111/j.1749-6632.1969.tb56356.x. [DOI] [PubMed] [Google Scholar]
- Jeong TS, Schissel SL, Tabas I, Pownall HJ, Tall AR, Jiang X. Increased sphingomyelin content of plasma lipoproteins in apolipoprotein E knockout mice reflects combined production and catabolic defects and enhances reactivity with mammalian sphingomyelinase. J Clin Invest. 1998;101:905–912. doi: 10.1172/JCI870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. Nonoxidative modifications of lipoprotein in atherosclerosis. Annu Rev Nutr. 1999;19:123–139. doi: 10.1146/annurev.nutr.19.1.123. [DOI] [PubMed] [Google Scholar]
- Barbaux SC, Blankenberg S, Rupprecht HJ, Francomme C, Bickel C, Hafner G, Nicaud V, Meyer J, Cambien F, Tiret L. Association between P-selectin gene polymorphisms and soluble P-selectin levels and their relation to coronary artery disease. Arterioscler Thromb Vasc Biol. 2001;21:1668–1673. doi: 10.1161/hq1001.097022. [DOI] [PubMed] [Google Scholar]
- Jiang XC, Paultre F, Pearson TA, Reed RG, Francis CK, Lin M, Berglund L, Tall AR. Plasma sphingomyelin level as a risk factor for coronary artery disease. Arterioscler Thromb Vasc Biol. 2000;20:2614–2618. doi: 10.1161/01.atv.20.12.2614. [DOI] [PubMed] [Google Scholar]
- Schlitt A, Hojjati MR, von Gizycki H, Lackner KJ, Blankenberg S, Schwaab B, Meyer J, Rupprecht HJ, Jiang XC. Serum sphingomyelin levels are related to the clearance of postprandial remnant-like particles. J Lipid Res. 2005;46:196–200. doi: 10.1194/jlr.C400011-JLR200. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–979. doi: 10.1056/NEJM199704033361401. [DOI] [PubMed] [Google Scholar]
- Chapman MJ, Comparative analysis of mammalian plasma lipoproteins Meth Enzymol. 1986;128:70–143. doi: 10.1016/0076-6879(86)28063-5. [DOI] [PubMed] [Google Scholar]
- Aviram M, Maor I, Keidar S, Hayek T, Oiknine J, Bar-El Y, Adler Z, Kertzman V, Milo S. Lesioned low density lipoprotein in atherosclerotic apolipoprotein E-deficient transgenic mice and in humans is oxidized and aggregated. Biochem Biophys Res Commun. 1995;216:501–513. doi: 10.1006/bbrc.1995.2651. [DOI] [PubMed] [Google Scholar]
- Guyton JR, Klemp KF. Development of the lipid-rich core in human atherosclerosis. Arterioscler Thromb Vasc Biol. 1996;16:4–11. doi: 10.1161/01.atv.16.1.4. [DOI] [PubMed] [Google Scholar]
- Hoff HF, Morton RE. Lipoproteins containing apo B extracted from human aortas: structure and function. Ann NY Acad Sci. 1985;454:183–94. doi: 10.1111/j.1749-6632.1985.tb11857.x. [DOI] [PubMed] [Google Scholar]
- Steinbrecher UP, Lougheed M. Scavenger receptor-independent stimulation of cholesterol esterification in macrophages by low density lipoprotein extracted from human aortic intima. Arterioscler Thromb. 1992;12:608–25. doi: 10.1161/01.atv.12.5.608. [DOI] [PubMed] [Google Scholar]
- Oorni K, Hakala JK, Annila A, Ala-Korpela M, Kovanen PT. Sphingomyelinase induces aggregation and fusion, but phospholipase A2 only aggregation, of low density lipoprotein particles. Two distinct mechanisms leading to increased binding strength of LDL to human aortic proteoglycans. J Biol Chem. 1998;273:29127–29134. doi: 10.1074/jbc.273.44.29127. [DOI] [PubMed] [Google Scholar]
- Tabas I, Li Y, Brocia RW, Xu SW, Swenson TL, Williams KJ. Lipoprotein lipase and sphingomyelinase synergistically enhance the association of atherogenic lipoproteins with smooth muscle cells and extracellular matrix. A possible mechanism for low density lipoprotein and lipoprotein(a) retention and macrophage foam cell formation. J Biol Chem. 1993;268:20419–20432. [PubMed] [Google Scholar]
- Williams KJ, Tabas I. The response-to-retention hypothesis of atherogenesis, reinforced. Curr Opin Lipidol. 1998;9:471–474. doi: 10.1097/00041433-199810000-00012. [DOI] [PubMed] [Google Scholar]
- Nordestgaard BG, Wootton R, Lewis B. Selective retention of VLDL, IDL, and LDL in the arterial intima of genetically hyperlipidemic rabbits in vivo. Molecular size as a determinant of fractional loss from the intima-inner media. Arterioscler Thromb Vasc Biol. 1995;15:534–542. doi: 10.1161/01.atv.15.4.534. [DOI] [PubMed] [Google Scholar]
- Tozer EC, Carew TE. Residence time of low-density lipoprotein in the normal and atherosclerotic rabbit aorta. Circ Res. 1997;80:208–218. doi: 10.1161/01.res.80.2.208. [DOI] [PubMed] [Google Scholar]
- Pentikainen MO, Lehtonen EMP, Kovanen PT. Aggregation and fusion of modified low density lipoprotein. J Lipid Res. 1996;37:2638–2649. [PubMed] [Google Scholar]
- Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J Clin Invest. 1996;98:1455–1464. doi: 10.1172/JCI118934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Tabas I. Sphingomyelinase enhances low density lipoprotein uptake and ability to induce cholesteryl ester accumulation in macrophages. J Biol Chem. 1991;266:24849–24858. [PubMed] [Google Scholar]
- Suits AG, Chait A, Aviram M, Heinecke JW. Phagocytosis of aggregated lipoprotein by macrophages: low density lipoprotein receptor-dependent foam-cell formation. Proc Natl Acad Sci USA. 1989;86:2713–2717. doi: 10.1073/pnas.86.8.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. 1996;271:18431–18436. doi: 10.1074/jbc.271.31.18431. [DOI] [PubMed] [Google Scholar]
- Marathe S, Schissel SL, Yellin MJ, Beatini N, Mintzer R, Williams KJ, Tabas I. Human vascular endothelial cells are a rich and regulatable source of secretory sphingomyelinase. Implications for early atherogenesis and ceramide-mediated cell signaling. J Biol Chem. 1998;273:4081–4088. doi: 10.1074/jbc.273.7.4081. [DOI] [PubMed] [Google Scholar]
- Marathe S, Kuriakose G, Williams KJ, Tabas I. Sphingomyelinase, an enzyme implicated in atherogenesis, is present in atherosclerotic lesions and binds to specific components of the subendothelial extracellular matrix. Arterioscler Thromb Vasc Biol. 1999;19:2648–2658. doi: 10.1161/01.atv.19.11.2648. [DOI] [PubMed] [Google Scholar]
- Krauss RM. Atherogenicity of triglyceride-rich lipoproteins. Am J Cardiol. 1998;81:13B–17B. doi: 10.1016/S0002-9149(98)00032-0. [DOI] [PubMed] [Google Scholar]