

Abstract
Objective:
Pancreatic cancer is the most deadly of all gastrointestinal (GI) malignancies, yet relatively little is known regarding mechanisms of tumor development including the role of inflammation.
Summary Background Data:
Chronic pancreatitis (CP) increases the risk of developing cancer by 10- to 20-fold; mediators of the chronic inflammatory process and the surrounding fibrotic stroma likely support a transformation to malignancy, yet the exact mechanisms remain undefined. The purpose of our present study was to determine potential inflammatory components in epithelial and stromal cells that may contribute to both CP and pancreatic cancers.
Methods:
Specimens of normal pancreas, CP, and pancreatic cancer were examined using laser-capture microdissection (LCM), gene array, and immunohistochemistry.
Results:
Gene array analysis from LCM-dissected tissues demonstrated: (i) increased expression of interleukin-8 (IL-8), an activator of the inflammatory factor nuclear factor-κB (NF-κB), and (ii) decreased expression of IκB (an inhibitor of NF-κB) in CP ductal cells compared with normal ducts. Compared with CP, cancers demonstrated: (i) increased expression of tumor related genes including S100A4, cyclin E1, and epidermal growth factor (EGF) receptor, and (ii) expression of matrix metalloproteinase 2, a pro-invasive factor for tumor cells, which was not present in the CP stroma. Increased staining of both the p50 NF-κB subunit and IKKα kinase (a protein that allows activation of NF-κB) was noted in CP and cancers.
Conclusions:
Our results demonstrate that similar inflammatory components and downstream effectors are present in CP and pancreatic cancers. Importantly, these findings suggest that a common pathway for pancreatic cancer development may be through a chronic inflammatory process including stroma formation. These findings may lead to novel strategies for pancreatic cancer prophylaxis based on inhibition of inflammatory mediators.
Patients with chronic pancreatitis (CP) are at increased risk for developing pancreatic cancer. We demonstrate in this study that nuclear factor–κB (NF-κB) expression, present in both CP and pancreatic cancer, may provide a causative link between these diseases. Inhibition of inflammatory mediators, such as NF-κB, may provide an effective target for the early treatment of pancreatic cancer.
The causative link between chronic inflammation and cancer was described nearly 200 years ago by the French Surgeon Jean Nicholas Marjolin when he noted the development of squamous cell carcinoma at the site of a chronically inflamed open wound.1 Since that initial description, a variety of inflammatory diseases have been recognized as contributing to the development of cancer, including several cancers of the gastrointestinal (GI) tract. For example, there is increased risk of colorectal carcinoma in patients with inflammatory bowel disease involving the colon,2 and this risk appears to increase with the severity of the inflammation and a longer duration of illness.3–5 Further, antiinflammatory medications can decrease the risk of colorectal cancer,6,7 thus chemopreventive strategies may be uniquely effective against tumors that arise from chronic inflammation, especially for diseases that currently lack effective therapies. Pancreatic cancer is the fourth leading cause of cancer death in the United States;8 surgical resection offers the only possibility for cure, yet fewer than 15% of patients are candidates for tumor resection at the time of diagnosis.9,10 The lethality of this cancer is related to its rapid growth and propensity to invade adjacent organs and metastasize; novel strategies that can halt the progression of premalignant conditions will provide the most effective treatments to improve the prognosis of pancreatic cancer.
Various genetic alterations have been reported in pancreatic cancers;11–14 however, relatively few studies have assessed inflammatory components that may play a more critical role in pancreatic cancer development. Chronic pancreatitis (CP) significantly increases the risk of developing pancreatic cancer,15–17 which suggests chronic inflammation within the pancreas may be a predisposing factor to the development of cancer. Nuclear factor-κB (NF-κB) and interleukin-8 (IL-8) are key mediators of the inflammatory process in CP;18 both have been implicated in the development of other malignancies.19,20 The exact mechanisms and inflammatory mediators that link CP and pancreatic cancer remain undefined.
A dense fibrotic stroma that forms around the remaining acinar cells in CP contains inflammatory cells, proliferating fibroblasts, and cytokines. Similarly, pancreatic cancer induces a strong desmoplastic reaction that may provide a source of inflammatory mediators and growth factors to support tumor growth and metastases.21 This stroma is composed of the same cell types in both CP and pancreatic cancer, thus it may provide a source of cytokine expression and growth factors which facilitates the development and progression of pancreatic cancer from CP. The inflammatory mediators that lead to the development of cancer remain undefined.
We have previously shown that inhibition of NF-κB can sensitize pancreatic cancer cells to apoptosis,22 and activation of NF-κB is an important mediator of pancreatic inflammation.23 NF-κB activity has also been recognized as a key regulator of tumor cell growth and sensitivity to apoptosis.24 The purpose of our present study was to analyze normal pancreas, CP, and pancreatic cancer (cancer cells and surrounding stroma) to determine potential inflammatory components that may contribute to both CP and pancreatic cancers. We demonstrate here that components of the NF-κB pathway are highly expressed in both CP and pancreatic cancer, while markers of pancreatic adenocarcinoma were found only within the tumor tissue. Additionally, cells within the tumor stroma expressed growth factors and pro-invasive agents that could facilitate the aggressiveness typically seen with pancreatic cancer. Further study of inflammatory mechanisms of pancreatic cancer development may identify new targets for effective chemoprevention and improve the prognosis of pancreatic cancer.
MATERIALS AND METHODS
Materials
The PixCell II LCM system (Arcturus Engineering, Mountain View, CA) was used for laser capture microdissection. RNaeasy and RLT lysis buffer were purchased from Qiagen (Valencia, CA). DNase I was purchased from Roche (Basel, Switzerland). The AmpoLabeling-LPR kit and the Human Cancer Pathway Finder Gene Array were purchased from Superarray Bioscience (Frederick, MD). Primary antibodies recognizing the NF-κB subunit p50, IKK kinase, epidermal growth factor (EGF), or EGF receptor (EGFR) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The DAKO EnVision+System, Peroxidase(DAB)Kit was purchased from DAKO Corporation (Carpinteria, CA).
Tissue Specimens
Pancreatic tissue was obtained under an IRB-approved protocol from patients undergoing pancreatic resection. Two specimens of histologically normal pancreas were obtained from resections performed for a diagnosis (ie, serous cystadenoma) other than CP or pancreatic cancer. CP tissue was taken from resections performed due to extensive disease within the head of the pancreas; no malignancy was identified in any of these 9 specimens. Pancreatic cancer specimens (n = 11) were all confirmed to be moderate or poorly differentiated adenocarcinomas by a pathologist. Specimens were either fixed within 24 hours and embedded in paraffin (for immunohistochemistry) or snap frozen in liquid nitrogen immediately after resection (for gene array analysis and immunohistochemistry) and stored at −80°C.
Laser Capture Microdissection (LCM)
Specimens of CP or pancreatic cancer tissue were cut into sections (10-μm thickness) and mounted on uncoated glass slides. Before LCM, sections were fixed in 70% ethanol for 1 minute at a room temperature, followed by stepwise dehydration with 95% ethanol twice and 100% ethanol twice for 1 minute, and incubation in xylene for 1 minute at a room temperature. After the sections were air-dried, cancer cells or ductal cells from CP tissue and stromal cells were selectively microdissected by the PixCell II LCM system as we have done previously.25,26
Isolation of RNA and Gene Array Analysis
Laser-captured cell nests or normal pancreatic tissue was resuspended with RLT lysis buffer, and total RNA was extracted using the RNeasy Kit according to the manufacturer’s protocol. During this step, DNase I was added to remove any contaminating genomic DNA. Total RNA was amplified and used to synthesize cDNA probes for gene array analysis using the AmpoLabeling-LPR kit. Microarray analysis was performed using the Human Cancer Pathway Finder Gene Array according to the manufacturer’s protocol. Briefly, biotinylated cDNA probes were hybridized to membranes that contain complementary sequences from 96 genes related to cancer growth, cell cycle regulation, and metastases. Membranes were washed after overnight hybridization, and signals were detected using a nonradioactive chemiluminescent detection system. Images were obtained using a digital camera and quantitated using GE array analyzer software. Comparisons of expression were performed after background levels were subtracted and values were normalized to housekeeping gene expression (GAPDH and β-actin) on each array.
Immunohistochemistry
Paraffin-embedded tissue or frozen tissue embedded in OCT compound was sectioned and mounted on glass slides. After protein denaturation, slides were incubated with primary antibodies to the NF-κB subunit p50, IKK kinase, EGF, or EGFR. Immunostaining was detected using the DAKO EnVision+System, Peroxidase(DAB)Kit according to the manufacturer’s protocol.
RESULTS
Expression of Pro-Inflammatory Cytokine IL-8 Is Increased in CP
To determine the expression of inflammatory factors in CP, compared with normal pancreatic tissue, gene array analysis was performed using RNA from LCM-dissected tissues. Expression of IL-8 was increased more than 9-fold in the cells isolated from CP tissue when compared with normal pancreatic tissue (Fig. 1). IL-8 is an activator of the inflammatory factor NF-κB,27 which suggests NF-κB activity is high in CP tissues. Additionally, gene array analysis demonstrated decreased expression of IκB (an inhibitor of NF-κB) in CP ductal cells compared with normal ducts, further supporting the possibility that NF-κB activity increased with CP.
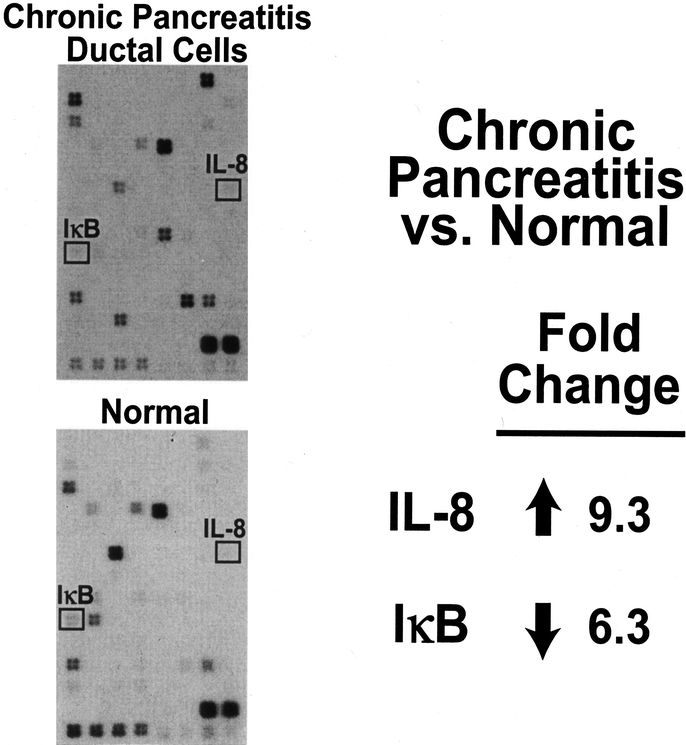
FIGURE 1. IL-8 expression is increased, and IκB expression is decreased in ductal cells from CP compared with normal pancreatic tissue, based on gene array analyses.
Activators of NF-κB Are Expressed in Both CP and Pancreatic Cancer
Gene array analysis demonstrated increased IL-8 expression in CP, which can activate NF-κB. NF-κB activation requires the expression of its subunits as well as the kinases that permit its translocation to the nucleus where it induces transcription of pro-inflammatory and oncogenic factors.28 To determine whether the key components of the NF-κB pathway are expressed in normal pancreatic tissue, CP, or pancreatic cancer, immunohistochemical staining was performed on all specimens collected (Fig. 2). Increased staining of both the p50 NF-κB subunit and IKKα kinase (a protein that allows activation of NF-κB) was noted in CP and cancers; in contrast, minimal to no staining was identified in normal pancreas. The staining of p50 and IKK was seen predominately in the acinar and ductal cells in CP or tumor cell cancers; there was minimal staining in the fibrotic stroma or tumor stroma. Taken together with the results of the gene array, we demonstrate that increased NF-κB activity differentiates normal pancreatic tissue from CP and pancreatic cancer.
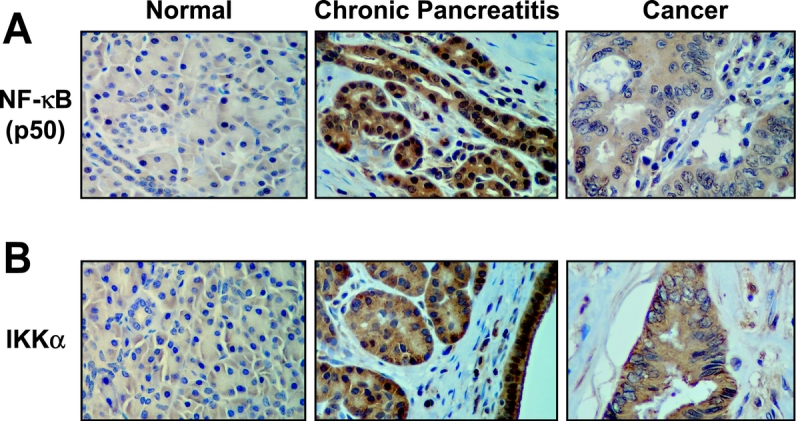
FIGURE 2. Expression of the NF-κB subunit p50 and activating kinase IKK are increased in CP and pancreatic cancer as shown by the brown immunohistochemical staining.
Expression of Tumor-Related Genes Is Increased in Pancreatic Cancer
We hypothesize that inflammatory mediators are expressed in early stages of malignant progression, which then give rise to expression of specific oncogenes that stimulate pancreatic cancer growth and metastases. To determine whether these oncogenic factors are expressed in tumor cells but absent in benign tissues, we again used gene array analysis to simultaneously measure the expression of multiple genes. Compared with CP or normal tissue, cancers demonstrated an increased expression of tumor-related genes including S100A4, cyclin E1, and EGFR (Fig. 3), which activates NF-κB.29 The calcium-binding protein S100A4 (also known as mts-1) is expressed in most pancreatic cancer cell lines and invasive pancreatic carcinomas, but not in normal tissue or early intraductal neoplasia;30 this correlates well with our finding of higher expression only in the tumor cells. Cyclin E1 expression was highest in tumor cells, but it was also elevated in ductal cells from CP tissue compared with normal pancreatic cells. Additionally, expression of the cyclin-dependent kinase (cdk) inhibitor p27kip1 was highest in CP cells and nearly absent in normal and tumor cells, which suggests the chronic inflammatory state seen in CP stimulates cell growth. This stimulation, as indicated by cyclin E1 expression, is also present in tumor cells, but regulation by inhibitors of the cell cycle (eg, p27kip1) may be lost in tumor cells, allowing them to proliferate rapidly. Finally, the expression of EGFR was higher in tumor cells compared with ductal cells in CP. When stimulated by its ligand (ie, EGF), EGFR activates signaling pathways that are important for pancreatic cancer growth.31 Taken together, our gene array results demonstrate that factors known to stimulate growth and metastases in pancreatic cancer are more highly expressed in tumor cells compared with normal ductal cells or ductal cells from CP.
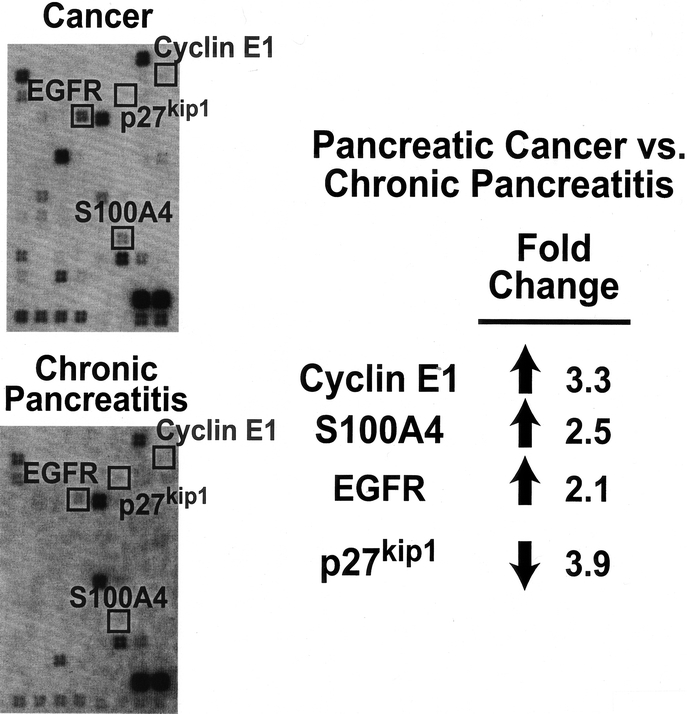
FIGURE 3. Mediators of tumor growth and invasion are more highly expressed in pancreatic cancer compared with CP based on gene array analyses.
MMP-2 and EGF Are Expressed in Tumor Stroma
The dense fibrosis found in CP closely resembles tumor stroma seen with pancreatic carcinoma histologically, but the interrelationship between these mesenchymal cells and pancreatic cancer cells is poorly understood. To determine patterns of gene expression within the fibrosis found in CP and tumor stroma, we performed gene array analysis using RNA isolated by LCM from the stromal cells surrounding the CP and pancreatic cancers. These expression patterns were also compared with our previous gene array results comparing normal ductal cells and pancreatic cancer cells to determine whether the expression patterns differ between epithelial cells and mesenchymal cells. The tumor stroma cells had increased expression of matrix metalloproteinase 2 (MMP-2), a pro-invasive factor for tumor cells,32 when compared with stromal cells found in CP (Fig. 4A). NF-κB can increase MMP-2 expression,33 thus further supporting the hypothesis that inflammation may facilitate the development and progression of pancreatic cancer. Additionally, expression of EGF, which can activate NF-κB,29 was increased in the tumor stroma when compared with either tumor cells or the mesenchymal cells in CP. Conversely, expression of the EGFR was higher in tumor cells than in the stroma or ductal cells in CP tissue (data not shown). This pattern of expression suggests a mitogenic relationship between tumor stroma and the adjacent cancer, as EGF is produced within the tumor stroma and activates the EGFR on the surface of tumor cells.
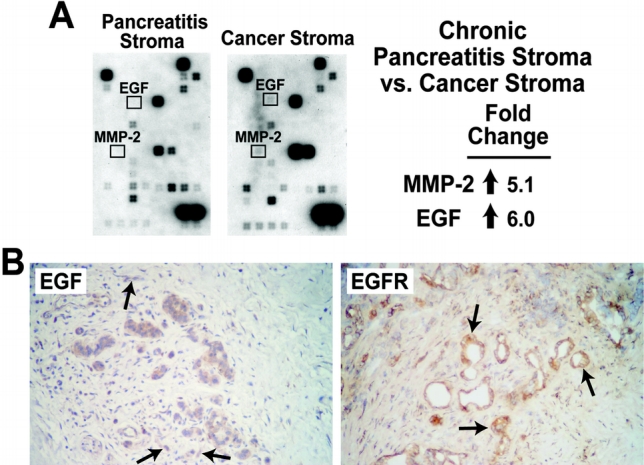
FIGURE 4. Tumor stroma contains high expression of EGF and MMP-2 compared with pancreatitis stroma based on gene array analyses (A); immunohistochemistry staining for EGF or EGFR (shown in brown) demonstrated EGF expression predominantly in tumor stroma and EGFR expression predominantly in tumor cells. Only genes with greater than 2-fold change were considered significant. (B) Frozen tissue from pancreatic cancer specimens were sectioned, mounted on slides, and immunostained for epidermal growth factor (EGF), or EGF receptor (EGFR). Staining was detected using a horseradish peroxidase technique as described in Methods. Positive expression of either EGF or EGFR is shown in brown (arrows).
To further confirm the expression of EGF and EGFR in pancreatic cancer and the surrounding stroma, we performed immunohistochemistry on pancreatic cancer tissue. Staining of EGFR was seen predominately in tumor cells and not in the surrounding stroma, which is consistent with our gene array results (Fig. 4B). Staining of EGF was seen in both stroma and cancer cells, although its activity is dependent on the presence of the EGF receptor, which was found almost exclusively on tumor cells. Together, these findings suggest that the fibrotic stroma that results from chronic inflammation may provide a source of growth factors and proteases that facilitate pancreatic cancer growth and invasion.
DISCUSSION
The link between inflammation and the development of cancer has been recognized for a number of years. In 1863, Rudolf Virchow noted leukocytes in neoplastic tissue and suggested that this reflected the origin of cancer at sites of chronic inflammation.34 In pancreatic cancer, inflammatory mediators, including NF-κB, cyclo-oxygenase-2, and TNFα are known to facilitate tumor cell growth and metastasis;19 however, the importance of inflammation as the origin of pancreatic cancer development has not been described.
We demonstrate in this study that inflammatory mediators, most notably NF-κB, are expressed in pancreatic cancer cells and in CP, which is a strong risk factor for the development of pancreatic cancer.15,16 Up-regulated or constitutive expression of NF-κB has been identified in many forms of cancer including other GI malignancies such as colorectal cancer and hepatocellular carcinoma.20 Activated RelA, a subunit of the active NF-κB complex, is constitutively activated in human pancreatic cancer specimens when compared with normal pancreatic tissue.35 Activation of NF-κB inhibits induction of apoptosis in pancreatic cancer cells36 and can activate signaling pathways that stimulate pancreatic cancer cell growth,29 thus suggesting an important role for NF-κB in the progression of pancreatic cancer.
In CP, this activation of NF-κB is likely due to cytokines (ie, IL-8) and other components of the chronic inflammatory response, most notably reactive oxygen species.37 The chronic inflammatory process leads to activation of stellate cells and the development of pancreatic fibrosis, which consists of an inflammatory infiltrate and proliferating mesenchymal cells. The combination of NF-κB expression in ductal cells and the presence of this inflammatory stroma create a cellular milieu rich in growth factors that favor the proliferation of hyperplastic cells which may have been mutated from exposure to reactive oxygen species and cytokines. Consistent with this hypothesis, previous investigators have shown that NF-κB activates cyclin D1 expression, a regulatory protein that promotes cell cycle activity.38 In our present study, we found that expression of another activator of cell cycle progression, cyclin E1, is increased in CP ductal cells when compared with normal cells. The increased expression of cyclin E may induce cell proliferation, depending on the expression of the cell cycle inhibitors such as p27kip1, which is also increased in CP. Coexpression of p27kip1 and cyclin E1 may prevent cyclin E1-induced proliferation in CP ductal cells, however the expression of cyclin E1 remains high in tumor cells and expression of p27kip1 is reduced, which favors cell proliferation.
Our findings also suggest potential mechanisms for a role of the fibrotic stroma in pancreatic cancer progression. In pancreatic cancer specimens, EGF expression was higher in the tumor stroma than in either cancer cells or fibrotic stroma from CP. Further, expression of EGFR was predominately in tumor cells, where activation of signaling pathways downstream from EGFR can stimulate pancreatic cancer cell growth and invasion. Thus the tumor stroma may provide a source of critical growth factors, such as EGF, to facilitate the aggressive biologic behavior of pancreatic cancer. The importance of EGF in regulating pancreatic cancer cell growth is well established. Treatment with inhibitors to EGFR result in sensitization to other apoptosis-inducing drugs39 and the concomitant presence of EGFR and its ligand EGF is associated with enhanced tumor aggressiveness and shorter survival periods after tumor resection.40 Additionally, activation of EGF signaling in transgenic mouse pancreata produces pancreatic tumors that closely resemble human pancreatic adenocarcinoma in terms of histology, aggressiveness, and lethality.41 In this study, we have identified the inflammatory tumor stroma as a possible source of EGF, further suggesting the importance of inflammation in pancreatic cancer development.
Expression of MMP-2 was increased in tumor stroma when compared with CP, which further links NF-κB activation and pancreatic cancer. MMP-2 is a key regulator of pancreatic cancer cell invasion and formation of metastases.42 In vitro studies have also shown that NF-κB can induce MMP-2 expression after stimulation with reactive oxygen species.33,43 We have shown previously that inhibition of MMP-2 expression in pancreatic cancer cells is associated with reduced invasiveness.44 Also expression of MMP-2 correlates with the amount of desmoplastic reaction within pancreatic tumors,45 which provides a potential mechanism for NF-κB–induced stroma formation. The close interaction between MMP-2 and NF-κB and the importance of both in regulating pancreatic cancer invasiveness further support a significant role for inflammation in the development of pancreatic cancer.
Results of our study as well as others46,47 suggest a progression of pancreatic cancer that begins with the establishment of chronic inflammation. Pancreatic inflammation is associated with reactive oxygen species production, cytokine release, and up-regulation of pro-inflammatory transcription factors (ie, NF-κB). Mediators of the inflammatory response can induce genetic damage, cell proliferation, and inhibition of apoptosis in the pancreas. The inflammation also induces stroma formation that can facilitate the growth of transformed cells through secretion of EGF and production of proinvasive factors (eg, MMP-2).
In conclusion, we demonstrate that inflammatory mediators are expressed in CP and pancreatic cancer, providing a potential explanation for the higher rate of pancreatic cancer found in patients with CP. Further studies will more clearly elucidate other important inflammatory mediators that activate key components of malignant transformation in the development of pancreatic cancer. Aspirin, an inhibitor of many inflammatory mediators, has already been shown to decrease the incidence of pancreatic cancer.48 We expect that as the link between chronic inflammation and pancreatic cancer becomes more established, other antiinflammatory agents can be tested to determine their effectiveness in halting the progression of pancreatic cancer. Early intervention is likely the best strategy to improve the prognosis of this devastating disease.
ACKNOWLEDGMENTS
We thank Eileen Figueroa and Karen Martin for manuscript preparation. Buckminster Farrow is a recipient of a Jeane B. Kempner award.
Discussions
Dr. Henry A. Pitt (Milwaukee, Wisconsin): This paper by Drs. Farrow, Evers, and colleagues from Galveston clearly establishes at the molecular genetic level, a link between chronic inflammation and pancreatic cancers. The authors have shown that NF-κB plays a key role in this process, and they have confirmed that epidermal growth factor and matrix metalloproteinase 2 also are important in tumor growth and invasion. These observations are likely to lead to new strategies for cancer prevention, one of the ultimate goals in medicine.
My first question relates to the patients that you have studied. You analyzed tissue from only 2 patients with a normal pancreas. Is this number sufficient for to you be comfortable that your findings are generalizable, or do you need more patients in this category?
In comparison, you had 9 patients with CP and 11 with pancreatic cancer. Were these patients similar with respect to age, gender, diabetes, and other factors that may have influenced your results?
In addition, a subset of patients with CP has recently been described who have lymphoproliferative sclerosing pancreatitis. These patients have a more active inflammatory response than the average patient with CP. Do you know whether any of your CP patients had this variety of the disease?
One of the interesting negative findings from your analysis is that TNFα was not upregulated in either your CP or your pancreatic cancer patients. TNFα is known to facilitate pancreatic tumor cell growth and metastases; therefore, were you surprised that you did not find alterations in TNFα? Or do you have any technical explanation for the negative observations?
Both CP and pancreatic cancer involve intra- and peripancreatic nerves, with pain being a common symptom for both diseases. I know that you focused in the latter part of your presentation on stroma rather than on the tumor cells. Would you speculate, however, as to whether any of your findings provide any insight regarding the interaction among inflammation, pancreatic nerves, and pancreatic cancer?
Finally, I agree with your observations that they are likely to lead to new strategies for prevention rather than for treatment. Would you speculate, however, as to whether COX-2 inhibitors, amino-salicylates, anti-growth factor drugs, or anti-metalloproteinase medications are most likely to be successful in preventing cancer?
Dr. David J. Cole (Charleston, South Carolina): I appreciate the opportunity to discuss this interesting paper. I would like to congratulate the authors on what is an excellent and I believe timely study addressing molecular pathogenesis, or at least initial insights into that topic, related to pancreatic cancer. It is clear, given the static and currently dismal outcomes related to the treatment of pancreatic cancer, that novel insights, and therefore novel treatments, are going to be required before we can make substantive gains in the treatment of this disease.
Currently there is an increasing array of molecular techniques, hopefully insights, as a result of the completion of human genome project, which provides tools for addressing molecular pathogenesis. The strength of this study is that these investigators have started to apply these tools as a first step from which subsequent studies can hopefully have impact on the treatment of this disease. I have several questions for the authors.
Similar to our first discussant, 2 normal specimens is a fairly small sample size. My question is, is this enough for a comparative analysis? Especially when the differential expression is not a hundred-fold difference, but 6- to 9-fold. Similarly, were you able to confirm, given the vagaries of quantitation of microarray analysis, the actual RNA by different techniques; for example, PCR confirmation, Western blot, and so forth?
Secondly, did you look at other antiinflammatory or inflammatory mediators? One example might be cyclooxygenase 2. I did not see any commentary on that topic. Clearly you did a full microarray analysis, but you focused on specific mediators. Were there clearly any other genes that were elevated or suppressed in your analysis?
Finally, do you have any actual evidence that NF-κB has a causative effect with respect to the pathogenesis of pancreatic cancer as opposed to relational presence in the presence of inflammation?
Dr. David W. Mercer (Houston, Texas): This interesting paper examines the link between CP and the development of pancreatic cancer. I would like to compliment the authors on an excellent paper presentation, for providing me a copy of the manuscript, and for their novel observations that may possibly identify therapeutic targets for the treatment of patients at risk for developing pancreatic cancer. I do have several questions, and some of these are shared by the other discussants.
My first question relates to patient demographics. There really weren’t any demographics presented in the manuscript. I wonder, especially since a lot of these patients have pain and are on salicylates or nonsteroidals, which have been shown to decrease the incidence of pancreatic cancer, whether any of these patients were on these medications as part of their treatment?
Secondly, as I looked at the immunohistochemical stainings for the NF-κB, it looked to me as though the P-50 subunits were in the cytosol and not in the nucleus. In order to have transcriptional activity, these subunits need to be located in the nucleus. I wonder if you would comment on that.
Along those lines, transcriptional activity requires heterodimers. P-50 or P-65 homodimers usually don’t have transcriptional activity. Have you shown that heterodimers exist, or are these just P-50 homodimers?
Thirdly, as the other discussant inquired, were COX-2 and TNFα included on your chip array; if so, did they change?
Lastly, with regards to the MMPs, I think that is a very intriguing observation. However, there are other things that go along with gelatinases. One, there are other gelatinases involved, and were these measured?
Secondly, did you correlate that with gelatin zymography or in situ zymography to see if these changes in transcription correlated with changes in activity of these gelatinases?
Lastlly, when assessing MMP activity, you also have to look at the activity of endogenous tissue inhibitors of MMPs or TIMPs, to see if they change. If both MMP and TIMP activity increase simultaneously, there probably isn’t any significant change in net activity. Therefore I think it is important that you look at both.
Dr. J. Patrick O’Leary (New Orleans, Louisiana): I wonder which is the chicken and which is the egg? Are you proposing that these are causative or simply the result of the destructive process that occurs in the pancreas?
With regard to the MMPs, they also can be elevated in the diffuse inflammatory process. They can also be elevated in the presence of overgrowth of bacteria in the gut. I wonder if this might be a part of the process. So my basic question is: Is it causative or just the result of the tumor growth?
Dr. Buckminster Farrow (Galveston, Texas): Thank you to all the discussants for their insightful questions and comments.
Both Dr. Pitt and Dr. Cole asked about our use of only 2 normal specimens for comparison in the study. We used fairly strict criteria as to what we consider normal. We used pieces of pancreatic tissue that had no other malignant diagnoses in them, including no IPMNs and no forms of pancreatitis, and we found very few specimens that fit that stringent criteria. However, in all the comparisons that we did, the analysis remained the same. So we feel that those are sufficient for now, and we certainly continue to build our database of tissue to try to further enhance our data set.
Secondly, both Dr. Pitt and Dr. Mercer asked about our patient populations. In general, our patient populations with CP and pancreatic cancer in Galveston are from the same mold. We don’t see a lot of things outside the typical age and sex demographics for both those diseases. So we feel that these results would be somewhat applicable to the population as a whole.
Third, Dr. Pitt, you asked about the lymphoproliferative variant of CP. We saw none of that in the samples that we used for this analysis.
Fourth, the question about TNFα. That was included in our array, and we did not see any significant differences in the specimens between the CP and pancreatic cancer nor in the stroma specimens in terms of the TNFα expression. This may be because there really is no difference, or it may be due to our microdissection technique. We very carefully selected only epithelial and stroma cells and did not dissect any lymphoid cells that could be producing TNF and that could be confounding the true situation in the biological setting.
The next question was about nerve cells and their role in this. We actually have research ongoing looking at the importance of perineural invasion, the role of inflammation, and whether or not that can facilitate perineural invasion, but we have not necessarily looked specifically at nerve cell gene expression in this study. I will say that I think that it is definitely consistent with our hypothesis that ongoing inflammation in both CP and pancreatic cancer could be causing pain in both these patients, and it may be inflammatory mediators causing pain in both the clinical situations.
Finally, the question about prophylaxis with antiinflammable medications. As you pointed out, the use of salicylates to inhibit COX-2 has already been described for other tumors. We feel that this may be helpful in preventing pancreatic cancer, and some preliminary population-based studies have shown that.
However, there are also a lot of other targets, including NF-κB, that we feel are very important, and a lot of new inhibitors coming out that could be very effective through inhibition of NF-κB. In addition, we have done some work with PPAR-γ, which is an antiinflammatory transcription factor. Not only does it modulate tumor growth, but there is also a lot of description in the literature about the way PPAR-? activation can inhibit both pancreatic and hepatic fibrosis. It would be interesting to speculate whether inhibiting that activity may inhibit the development of the stroma seen in CP and in pancreatic cancer, and provide another way to prevent both these diseases.
Dr. Cole, you asked about studies in addition to our gene array data. We are still working on perfecting our technique of LCM to try to get suitable levels of protein to be able to do further studies such as Western blotting. As we perfect that technique, we certainly hope to have more information.
In regards to other inflammatory mediators. I have already mentioned that TNFα expression was unchanged. COX-2 is also included on this gene array, and we did not see any significant changes between CP and pancreatic cancer.
In regards to Dr. Cole’s last question, that I think Dr. O’Leary was alluding to in his question about NF-κB and whether there is really a causative effect here, I think that is obviously the key question. And what we hope to do is take this study further and develop models of pancreatic cancer based on NF-κB overexpression to truly test whether or not we can drive the formation of malignancy through sufficient expression and inflammatory mediators.
Dr. Mercer mentioned about the immunohistochemistry and the NF-κB subunit P-50. We did see, as he mentions, expression in the cytoplasm and not necessarily nuclear translocation. We are still looking at that more specifically and actually trying to develop better techniques for determining whether or not NF-κB activity is truly active at the transcriptional level in these tissues. Of note, we did also look at P-65 expression, which we did find in most samples, but there is not a significant difference between the tissue types, which is why we haven’t reported that specifically in the presentation.
In regards to MMPs, there was only 1 other MMP listed on this array, which we didn’t see a lot of change with. As I mentioned before, as we perfect our protein extraction techniques through LCM, we hope to be able to do other things like gelatin zymography to further enhance the results of our study.
Footnotes
Supported by grants from the National Institutes of Health (RO1 DK48498, R37AG10885, PO1 DK35608, and T32 DK07639).
Reprints: B. Mark Evers, MD, Department of Surgery, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555-0536. E-mail: mevers@utmb.edu.
REFERENCES
- 1.Marjolin J-N. Ulcer̈e. Dictionnaire de Medecine. Vol 21. Pratique, 1828.
- 2.Adelstein P, Baldwin JA, Fedrich J. Cancers of the large bowel: associated disorders in individuals. Cancer. 1979;43:2553–2557. [DOI] [PubMed] [Google Scholar]
- 3.Konig H, Hermanek P, Rosch W. Clinical conference: Unusual course of chronic colitis. Acta Hepatogastroenterol (Stuttg). 1976;23:227–231. [PubMed] [Google Scholar]
- 4.Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–1233. [DOI] [PubMed] [Google Scholar]
- 5.Munkholm P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment Pharmacol Ther 2003;18(Suppl 2):1–5. [DOI] [PubMed] [Google Scholar]
- 6.Eaden J. Review article: the data supporting a role for aminosalicylates in the chemoprevention of colorectal cancer in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2003;18(Suppl 2):15–21. [DOI] [PubMed] [Google Scholar]
- 7.Ryan BM, Russel MG, Langholz E, et al. Aminosalicylates and colorectal cancer in IBD: a not-so bitter pill to swallow. Am J Gastroenterol. 2003;98:1682–1687. [DOI] [PubMed] [Google Scholar]
- 8.Jemal A, Murray T, Samuels A, et al. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5–26. [DOI] [PubMed] [Google Scholar]
- 9.Yeo CJ, Cameron JL. Exocrine pancreas. In: Townsend CM Jr, Beauchamp RD, Evers BM, et al, eds. Sabiston Textbook of Surgery. The Biological Basis of Modern Surgical Practice. Philadelphia: W. B. Saunders Company; 2001:1112–1143. [Google Scholar]
- 10.Lillemoe KD, Yeo CJ, Cameron JL. Pancreatic cancer: state-of-the-art care. CA Cancer J Clin. 2000;50:241–268. [DOI] [PubMed] [Google Scholar]
- 11.Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156:1821–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansel DE, Kern SE, Hruban RH. Molecular pathogenesis of pancreatic cancer. Annu Rev Genomics Hum Genet. 2003;4:237–256. [DOI] [PubMed] [Google Scholar]
- 13.House MG, Herman JG, Guo MZ, et al. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg. 2003;238:423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimenez RE, Warshaw AL, Z’Graggen K, et al. Sequential accumulation of K-ras mutations and p53 overexpression in the progression of pancreatic mucinous cystic neoplasms to malignancy. Ann Surg. 1999;230:501–509; discussion 509–511. [DOI] [PMC free article] [PubMed]
- 15.Howes N, Neoptolemos JP. Risk of pancreatic ductal adenocarcinoma in chronic pancreatitis. Gut. 2002;51:765–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–1437. [DOI] [PubMed] [Google Scholar]
- 17.Otsuki M. Chronic pancreatitis in Japan: epidemiology, prognosis, diagnostic criteria, and future problems. J Gastroenterol. 2003;38:315–326. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu K, Shiratori K, Hayashi N, et al. Thiazolidinedione derivatives as novel therapeutic agents to prevent the development of chronic pancreatitis. Pancreas. 2002;24:184–190. [DOI] [PubMed] [Google Scholar]
- 19.Farrow B, Evers BM. Inflammation and the development of pancreatic cancer. Surg Oncol. 2002;10:153–169. [DOI] [PubMed] [Google Scholar]
- 20.Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938–6947. [DOI] [PubMed] [Google Scholar]
- 21.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. [DOI] [PubMed] [Google Scholar]
- 22.Thomas RP, Farrow BJ, Kim S, et al. Selective targeting of the nuclear factor-kappaB pathway enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated pancreatic cancer cell death. Surgery. 2002;132:127–134. [DOI] [PubMed] [Google Scholar]
- 23.Ethridge RT, Hashimoto K, Chung DH, et al. Selective inhibition of NF-κB attenuates the severity of cerulein-induced acute pancreatitis. J Am Coll Surg. 2002;195:497–505. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz SA, Hernandez A, Mark Evers B. The role of NF-κB/INF-κB proteins in cancer: implications for novel treatment strategies. Surg Oncol. 1999;8:143–153. [DOI] [PubMed] [Google Scholar]
- 25.Sugiyama Y, Shingo D, Yoshida Y, et al. A large-scale gene expression comparison of microdissected, small-sized endometrial cancers with or without hyperplasia matched to same-patient normal tissue. Clin Cancer Res. 2003; In press. [PubMed]
- 26.Sugiyama Y, Sugiyama K, Hirai Y, et al. Microdissection is essential for gene expression profiling of clinically resected cancer tissues. Am J Clin Pathol. 2002;117:109–116. [DOI] [PubMed] [Google Scholar]
- 27.Shimada T, Terano A. Chemokine expression in Helicobacter pylori-infected gastric mucosa. J Gastroenterol. 1998;33:613–617. [DOI] [PubMed] [Google Scholar]
- 28.Baldwin AS Jr. Series introduction: the transcription factor NF-κB and human disease. J Clin Invest. 2001;107:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liptay S, Weber CK, Ludwig L, et al. Mitogenic and antiapoptotic role of constitutive NF-κB/Rel activity in pancreatic cancer. Int J Cancer. 2003;105:735–746. [DOI] [PubMed] [Google Scholar]
- 30.Rosty C, Ueki T, Argani P, et al. Overexpression of S100A4 in pancreatic ductal adenocarcinomas is associated with poor differentiation and DNA hypomethylation. Am J Pathol. 2002;160:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuda K, Idezawa T, You XJ, et al. Multiple mitogenic pathways in pancreatic cancer cells are blocked by a truncated epidermal growth factor receptor. Cancer Res. 2002;62:5611–5617. [PubMed] [Google Scholar]
- 32.Koshiba T, Hosotani R, Wada M, et al. Detection of matrix metalloproteinase activity in human pancreatic cancer. Surg Today. 1997;27:302–304. [DOI] [PubMed] [Google Scholar]
- 33.Philip S, Bulbule A, Kundu GC. Osteopontin stimulates tumor growth and activation of promatrix metalloproteinase-2 through nuclear factor-κB-mediated induction of membrane type 1 matrix metalloproteinase in murine melanoma cells. J Biol Chem. 2001;276:44926–44935. [DOI] [PubMed] [Google Scholar]
- 34.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Abbruzzese JL, Evans DB, et al. The nuclear factor-κB RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- 36.McDade TP, Perugini RA, Vittimberga FJ Jr, et al. Salicylates inhibit NF-κB activation and enhance TNF-α-induced apoptosis in human pancreatic cancer cells. J Surg Res. 1999;83:56–61. [DOI] [PubMed] [Google Scholar]
- 37.Kim H, Seo JY, Kim KH. NF-κB and cytokines in pancreatic acinar cells. J Korean Med Sci. 2000;15(Suppl):S53–S54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang ZQ, Buchsbaum DJ, Raisch KP, et al. Differential responses by pancreatic carcinoma cell lines to prolonged exposure to Erbitux (IMC-C225) anti-EGFR antibody. J Surg Res. 2003;111:274–283. [DOI] [PubMed] [Google Scholar]
- 40.Ozawa F, Friess H, Tempia-Caliera A, et al. Growth factors and their receptors in pancreatic cancer. Teratog Carcinog Mutagen. 2001;21:27–44. [DOI] [PubMed] [Google Scholar]
- 41.Greten FR, Wagner M, Weber CK, et al. TGFα transgenic mice. A model of pancreatic cancer development. Pancreatology. 2001;1:363–368. [DOI] [PubMed] [Google Scholar]
- 42.Matsuyama Y, Takao S, Aikou T. Comparison of matrix metalloproteinase expression between primary tumors with or without liver metastasis in pancreatic and colorectal carcinomas. J Surg Oncol. 2002;80:105–110. [DOI] [PubMed] [Google Scholar]
- 43.Yoon SO, Park SJ, Yoon SY, et al. Sustained production of H(2)O(2) activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-κB pathway. J Biol Chem. 2002;277:30271–30282. [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto K, Ethridge RT, Evers BM. Peroxisome proliferator-activated receptor gamma ligand inhibits cell growth and invasion of human pancreatic cancer cells. Int J Gastrointest Cancer. 2002;32:7–22. [DOI] [PubMed] [Google Scholar]
- 45.Ellenrieder V, Alber B, Lacher U, et al. Role of MT-MMPs and MMP-2 in pancreatic cancer progression. Int J Cancer. 2000;85:14–20. [DOI] [PubMed] [Google Scholar]
- 46.Hedin KE. Chemokines: new, key players in the pathobiology of pancreatic cancer. Int J Gastrointest Cancer. 2002;31:23–29. [DOI] [PubMed] [Google Scholar]
- 47.Maisonneuve P, Lowenfels AB. Chronic pancreatitis and pancreatic cancer. Dig Dis. 2002;20:32–37. [DOI] [PubMed] [Google Scholar]
- 48.Hitt E. Aspirin may lower risk of pancreatic cancer. Lancet Oncol. 2002;3:518. [DOI] [PubMed] [Google Scholar]