

Abstract
Restrictive dermopathy is a lethal human genetic disorder characterized by very tight, thin, easily eroded skin, rocker bottom feet, and joint contractures. This disease was recently reported to be associated with a single heterozygous mutation in ZMPSTE24 and hypothesized to be a digenic disorder (Navarro et al., Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet 13:2493-2503, 2004). ZMPSTE24 encodes an enzyme necessary for the correct processing and maturation of lamin A, an intermediate filament component of the nuclear envelope. Here we present four unrelated patients with homozygous mutations in ZMPSTE24 and a fifth patient with compound heterozygous mutations in ZMPSTE24. Two of the three different mutations we found are novel, and all are single base insertions that result in mRNA frameshifts. As a consequence of the presumed lack of ZMPSTE24 activity, prelamin A, the unprocessed toxic form of lamin A, was detected in the nuclei of both cultured cells and tissue from restrictive dermopathy patients, but not in control nuclei. Abnormally aggregated lamin A/C was also observed. These results indicate that restrictive dermopathy is an autosomal recessive laminopathy caused by inactivating ZMPSTE24 mutations that result in defective processing and nuclear accumulation of prelamin A.
Keywords: STE24 protein, lamin A, nuclear envelope, FATP4 protein
Abbreviation: RD, restrictive dermopathy
Introduction
Restrictive dermopathy (RD), also known as lethal tight skin contracture syndrome (MIM 275210), is a rare genetic disorder that results in death, usually within several hours or days of birth. Babies with RD have tight, translucent, partially eroded skin, joint contractures, rocker bottom feet, and distinctive craniofacial abnormalities that typically include micrognathia, a facial expression with the mouth fixed in an “O” position, a small pinched nose, and low-set ears (Mau et al., 1997; Sillevis Smitt et al., 1998; Welsh et al., 1992). In most patients death results from respiratory distress due to severely restricted movements.
Of the few RD cases reported in the literature, many arose in children of consanguineous parents, and the disease recurs within families; its mode of inheritance had therefore been presumed to be autosomal recessive (Sillevis Smitt et al., 1998; Welsh et al., 1992). Recently, however, Navarro et al. (Navarro et al., 2004) used a candidate gene approach to identify a single inactivating heterozygous mutation in ZMPSTE24 (c.1085dupT) associated with RD in six unrelated, nonconsanguineous families of French and Dutch heritage. ZMPSTE24 encodes a zinc metalloproteinase required for lamin A maturation (Bergo et al., 2002; Pendas et al., 2002), suggesting that RD is a laminopathy that affects the structure of the nucleus. Because only one allele was found to be mutated in each affected individual, and the parents carrying that allele were normal, the authors hypothesized a digenic mode of inheritance for RD (Navarro et al., 2004). Contrary to this, we have identified four patients with three different homozygous mutations and one patient with compound heterozygous mutations in ZMPSTE24 that are associated with RD, providing strong support for a simple autosomal recessive mode of inheritance. We also show that in two patients with different homozygous mutations, there are nuclear abnormalities associated with accumulation of unprocessed prelamin A, thereby providing novel evidence that RD is a laminopathy.
Materials and Methods
Amplification of genomic DNA and sequencing.
The Washington University School of Medicine Human Studies Committee approved this study, which was conducted according to the Declaration of Helsinki principles. Informed parental consent was obtained in all cases, except for one that involved archived tissues; for this a waiver of consent was approved. Genomic DNA was extracted from the blood of six affected individuals from five different kindreds and from their parents and available unaffected siblings using QiaAMP DNA Blood Kit (Qiagen Inc., Valencia, CA). DNA was extracted from paraffin embedded tissues from a seventh affected individual as described (Coombs et al., 1999). Exons and intron-exon junctions from genomic DNA samples were amplified using KlentaqLA (BD Biosciences Clontech, Palo Alto, CA). Each 20 μl reaction contained 1x KLA buffer, 125 μM dNTPs, 2.5 mM MgCl2, 10 pmoles of each primer, 20 ng genomic DNA, and 1 U KlentaqLA. Cycling conditions were a 3 min initial denaturation at 95° C, followed by 35 cycles of 30 s at 94° C, 1 min at 57° C, and 3 min at 70° C. PCR products were gel-purified and recovered using the Wizard SV Gel Clean-up System (Promega, Madison, WI). The Washington University Protein and Nucleic Acid Chemistry Lab performed automated sequencing reactions using internal primers with Big-Dye Terminator 3.1 and an Applied Biosystems 3730 DNA sequencer (Applied Biosystems, Foster City, CA). All PCR and sequencing primer sequences are available upon request.
Cell culture and immunofluorescence.
Skin fibroblasts from a RD patient and normal skin fibroblasts (Detroit 551 from the American Type Culture Collection, Manassas, VA) were cultured in Eagle’s Minimal Essential medium (EMEM) modified to contain 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 1.5 g/L sodium bicarbonate, and 10% fetal bovine serum. For immunostaining, cells were cultured for several days on glass Falcon BioCoat CultureSlides (BD Biosciences Discovery Labware, Bedford, MA). Cells were washed twice with PBS, fixed 10 min at 4° C in 2% fresh paraformaldehyde in PBS, washed three times in PBS, permeabilized for 10 min in cold 100% methanol, and allowed to air-dry. Liver tissue from an unrelated RD patient and kidney tissue from a control were frozen in OCT, sectioned on a cryostat, and air-dried for 30 minutes. Sections were fixed and permeabilized as described for cultured cells.
For immunofluorescence, samples were blocked in either 4% BSA in PBS or 1% heat-inactivated normal goat serum in PBS. Antibodies were as follows: anti-lamins A and C (MAB3538) and FITC-conjugated anti-goat IgG from Chemicon (Temecula, CA); anti-prelamin A (sc-6214) and anti-emerin (sc-15378) from Santa Cruz Biotechnology (Santa Cruz, CA); Alexa 488-conjugated anti-mouse IgG1 from Molecular Probes (Eugene, OR). All antibody dilutions were in PBS containing 1% BSA, and washes were in PBS. Following primary and secondary antibody incubations at room temperature for 1 h each and washing, slides were mounted in 1 mg/ml p-phenylenediamine/0.1× PBS/90% glycerol.
Confocal microscope images were obtained using a Zeiss compound microscope (Carl Zeiss International, Germany) with a BioRad MRC 1024 confocal adaptor (BioRad Laboratories, Inc., Hercules, CA). All other micrographs were obtained with a Spot 2 cooled color digital camera (Diagnostic Instruments, Sterling Heights, MI) attached to a Nikon Eclipse E800 compound microscope (Nikon USA, Melville, NY). Images were imported into Adobe Photoshop 5 and Adobe Illustrator 9 for processing and layout.
Results
We became interested in RD upon discovering a spontaneous, autosomal recessive mouse mutation that we named wrinkle free (wrfr). wrfr −/− mice have very tight, thick skin, and their mouths are fixed in an open position (Moulson et al., 2003). They die within 24 hours of birth from an inability to breathe properly or to suckle, due to severely restricted movements. The overall phenotype is thus reminiscent of RD. Positional cloning of wrfr revealed a retrotransposon insertion in Slc27a4, which encodes fatty acid transport protein 4 (Moulson et al., 2003).
In an effort to determine whether SLC27A4 plays a role in human RD, we obtained blood and/or tissues from members of six RD kindreds, three of which were known or suspected to involve consanguinity. DNA was prepared from individuals with RD, all of whom died within hours or days of birth and exhibited the classic features described by Witt (Witt et al., 1986). DNA was also prepared from parents and any unaffected full siblings when available. We sequenced SLC27A4 exons and intron-exon junctions in three unrelated patients but found no mutations. To formally rule out SLC27A4 as a candidate, and to identify potential candidate regions, we performed genome-wide single nucleotide polymorphism (SNP) genotyping using Affymetrix GeneChip Mapping arrays (Matsuzaki et al., 2004) on a small but potentially informative subset of these DNAs (data not shown). Using DNA-Chip (dCHIP) software (Lin et al., 2004) to display the results, and assuming an autosomal recessive mode of inheritance, we identified a 15 Mb locus on chromosome 1p32-35 as a major RD candidate region, while the SLC27A4 locus at 9q34 was ruled out. Consistent with the report of Navarro et al., 1p32-35 contains ZMPSTE24. Therefore, we sequenced ZMPSTE24 exons and flanking intronic sequences amplified by PCR from RD family members’ DNA to look for mutations and to determine their association with RD.
A consanguineous Dutch family had one child affected with RD and five unaffected children. We identified in this family the same c.1085dupT insertion reported by Navarro et al. (Navarro et al., 2004) (Fig 1A and B). This duplication in exon 9 causes a frameshift at amino acid 362 (of 475 total) and premature termination, resulting in a truncated protein. In contrast to the findings of Navarro et al., here the mutation was homozygous in the affected patient, while both parents were heterozygous (Fig 1A and B). DNA was also available from four of the five unaffected siblings; three were heterozygous for the insertion, and one did not carry it. We found this same mutation in a nonconsanguineous American family of German ancestry (Fig 1C). The mutation was homozygous in one affected child, and both parents and one brother were heterozygous for the mutation; the other brother did not carry the mutation. No DNA from the second affected child was available.
Figure 1. Family pedigrees, ZMPSTE24 genotypes, and sequence chromatograms.
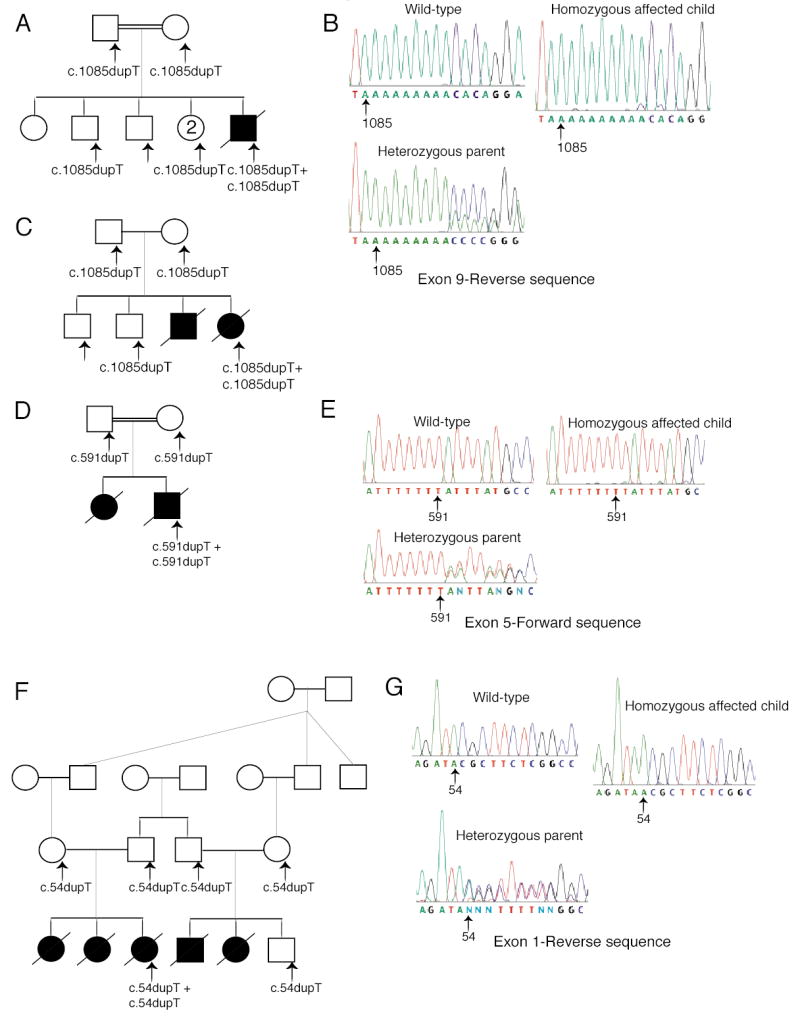
Arrows indicate individuals whose DNA was tested. ZMPSTE24 mutations are shown below the pedigree symbols. (A) Pedigree of a consanguineous Dutch family; the father’s maternal grandmother and the mother’s paternal grandmother were sisters. The affected child had a homozygous thymidine duplication in ZMPSTE24 exon 9. (B) Sequence chromatograms of exon 9 (reverse direction) show a duplicated thymidine in a stretch of nine. Overlap of the normal and mutated strand sequences occurred after the insertion in this and in all the other heterozygous samples described below. (C) Pedigree of a nonconsanguineous American family carrying the same mutation as the Dutch family. (D) Pedigree from a consanguineous Guatemalan family; the father’s paternal grandfather was a paternal uncle of the mother. (E) Sequence chromatograms of exon 5 (forward direction) showing the duplicated thymidine in a stretch of seven. (F) Five affected children were born to two pairs of related parents from a Mennonite kindred; the fathers were brothers of each other, and the mothers were first cousins of each other. (G) Sequence chromatograms of ZMPSTE24 exon 1 (reverse direction) show an extra thymidine homozygous in the affected child.
Analysis of ZMPSTE24 in a consanguineous Guatemalan family with two affected children revealed a novel single base duplication in exon 5 (c.591dupT) (Fig 1D and E). This mutation was homozygous in one affected child and heterozygous in both parents (Fig 1D and E); DNA from the second child was not available. This mutation causes a frameshift at amino acid 198 and premature termination 19 codons downstream, resulting in a severely truncated protein.
A novel single base duplication was present in ZMPSTE24 exon 1 (c.54dupT) in a Mennonite kindred with five affected children out of six born to two families (Fig 1F and G). DNA was only available from one affected child; the mutation was homozygous in her and heterozygous in both her parents. The other parents and their unaffected son were heterozygous for the same mutation. This duplication causes a frameshift at amino acid 19 and premature termination 27 codons downstream, resulting in a severely truncated protein. Interestingly, RD has been reported in a Mennonite kindred before (Lowry et al., 1985), suggesting that this unique mutation is segregating in the Mennonite population.
We also assayed DNA extracted from archived paraffin embedded tissues taken from a Dutch RD patient. Two different heterozygous mutations were detected: the exon 5 mutation found in the Guatemalan family (c.591dupT) and the previously identified exon 9 insertion (c.1085dupT) (Navarro et al., 2004). We conclude that this patient is compound heterozygous, though we can not formally prove it because parental DNAs are not available.
The only known substrate for ZMPSTE24 is lamin A, which is encoded by the LMNA gene. This gene also encodes lamin C, a shorter form that is translated from an alternatively spliced mRNA. Lamin A maturation requires ZMPSTE24 activity, but lamin C maturation does not. Lamin A/C distribution is abnormal in RD patients reported to have a heterozygous c.1085dupT mutation in ZMPSTE24 (Navarro et al., 2004). Because the c.1085dupT mutation in the American patient from whom we obtained fibroblasts was homozygous, we examined whether there was aberrant distribution of lamin A or C using an antibody which recognizes both (Fig 2A, B). Lamin A/C appeared to be distributed in clusters in the nucleus, as opposed to the homogeneous, smooth, uniform distribution in nuclei from normal fibroblast cultures.
Figure 2. Immunofluorescent localization of lamins A and C and prelamin A in control and RD patient fibroblasts.
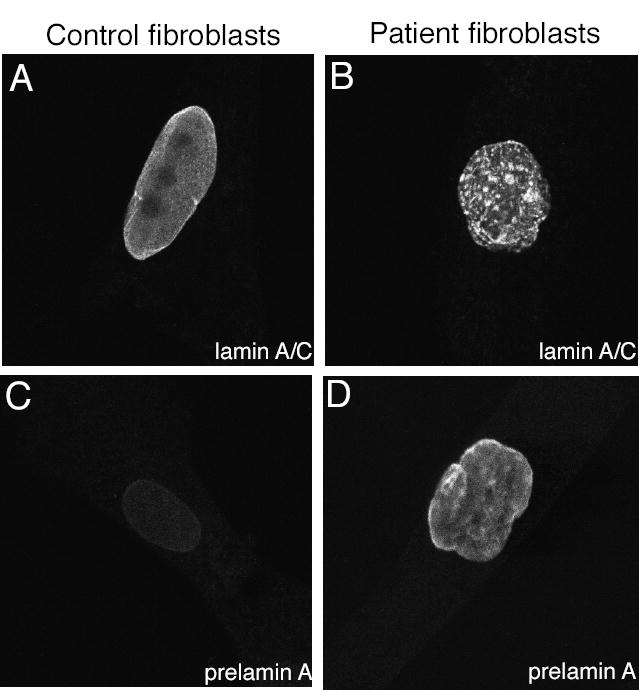
(A) Confocal micrographs show localization of lamins A and C in nuclei of a control fibroblast and (B) a fibroblast from a RD patient. (C) Confocal micrographs show staining for prelamin A in a control fibroblast and (D) in a fibroblast from a RD patient.
Accumulation of unprocessed prelamin A, which is toxic (Fong et al., 2004), has been reported in Zmpste24 knockout mice (Bergo et al., 2002; Pendas et al., 2002). To examine whether there was accumulation of prelamin A in RD patients as well, cultured fibroblasts were stained with a prelamin A antibody. Clear prelamin A staining was observed in RD fibroblast nuclei, while no prelamin A was detected in control fibroblasts (Fig 2C, D); these results were confirmed by western blotting (data not shown). The presence of prelamin A in RD nuclei was also observed in frozen liver sections from the Guatemalan patient (Fig 3B), but no prelamin A was detectable in kidney nuclei from a control (Fig 3A). In contrast, emerin, another component of the nuclear envelope, was present in both control and RD nuclei (Fig 3C-D). This demonstrates a specific defect in prelamin A processing.
Figure 3. Immunofluorescent localization of prelamin A and emerin in control tissue and tissue from a patient with RD.
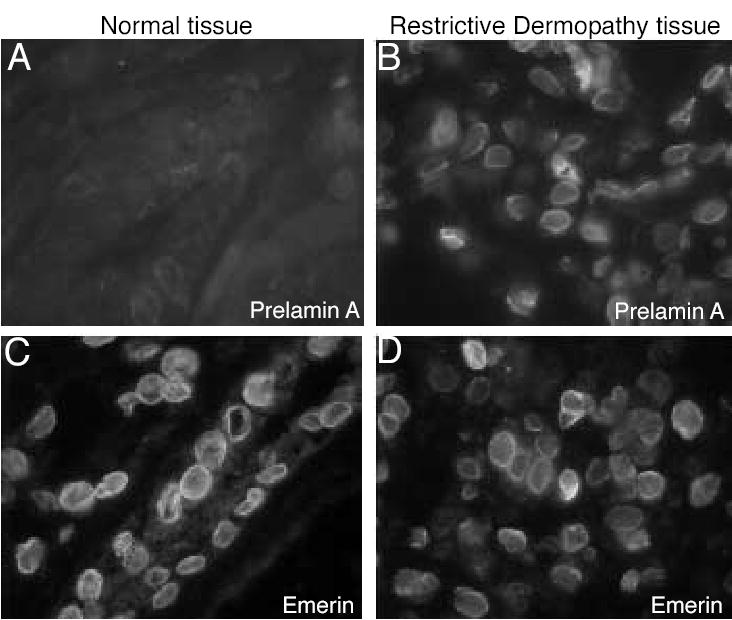
(A) Prelamin A was undetectable in a kidney section from a normal patient but clearly present in nuclei in a liver section from a RD patient (B). (C) Nuclei in control cells and (D) in the RD patient’s cells are labeled with anti-emerin to show the presence of nuclear envelopes.
Discussion
In this study we identified two novel mutations and a third previously described mutation in ZMPSTE24 that are associated with restrictive dermopathy. Importantly, we also show that RD is a simple autosomal recessive disorder. Though ZMPSTE24 had already been implicated in RD, only one heterozygous mutation was identified, and it was also present in unaffected family members, leading to the conclusion that RD was a digenic disorder (Navarro et al., 2004). Rather than there being a digenic mode of inheritance, we favor the possibility that there exist ZMPSTE24 mutations that are difficult to detect by conventional PCR-based methods. This is perhaps the most straightforward explanation for how there could be an absence of mature lamin A even with an apparently wild-type copy of ZMPSTE24. Such mutations could lie in the gene’s promoter, or could involve segmental deletions that are complemented--only as far as PCR is concerned--by normal regions of the other allele. Indeed, we could not find any mutations in a pair of affected fraternal twins from Finland, though SNP genotyping showed that the twins share genotypes on a segment of chromosome 1 containing the ZMPSTE24 locus (data not shown).
All mutations identified thus far to cause RD are single base duplications that result in mRNA frameshifts, which likely prevent production of a functional protein. However, one missense ZMPSTE24 mutation (c.1018T>C; p.Trp340Arg) has been found, in a Belgian patient with severe mandibuloacral dysplasia with type B lipodystrophy (MADB—MIM 608612) (Agarwal et al., 2003). This patient was actually compound heterozygous; the other allele bore the same c.1085dupT inactivating mutation that Navarro et al. 2004 and we have subsequently found to be associated with RD. The mutant protein carrying the p.Trp340Arg substitution is partially active (Agarwal et al., 2003), and this easily explains the fact that MADB is a less severe disease than RD.
Accumulation of prelamin A has been reported in Zmpste24 knockout mice (Bergo et al., 2002; Pendas et al., 2002) and in cultured cells from human patients with Hutchinson-Gilford progeria syndrome (HGPS—MIM 176670) (Goldman et al., 2004); its presence has also been hypothesized but not proven in other human laminopathies (Agarwal et al., 2003; Navarro et al., 2004). Ours is the first demonstration of accumulation of prelamin A in affected human tissue in situ (Fig. 4A), suggesting that it is not merely a consequence of cell culture. Accumulation of prelamin A has recently been shown to be toxic; disease phenotypes in Zmpste24 −/− mice are “rescued” by only a 50% reduction in the amount of prelamin A, via knockout of one Lmna allele (Fong et al., 2004). The presence of large amounts of prelamin A may also account for the increased severity of RD as compared to laminopathies with either lamin A mutations or missense mutation in ZMPSTE24. Given the lack of detectable prelamin A in normal cells, it is possible that a simple immunofluorescence assay for prelamin A in fetal cell nuclei could serve as a prenatal test for RD and perhaps other laminopathies.
We found lamins A and C clustered and distributed in aggregates in the nuclei of RD patient fibroblasts, whereas normal fibroblast nuclei showed a homogeneous and smooth, even distribution, concentrated at the nuclear periphery. Abnormal distribution of lamins A and C in foci, aggregations, and honeycombs has been reported in a number of human patients carrying lamin A/C gene mutations (Capanni et al., 2003; Caux et al., 2003; Muchir et al., 2004; Novelli et al., 2002) as well as in cells transfected with lamin A/C mutants (Holt et al., 2003; Ostlund et al., 2001).
The mechanism whereby mutations in LMNA and ZMPSTE24 that result in aberrant lamin A processing cause disease has not been determined. Several lines of evidence suggest that it is likely to be a combination of impaired nuclear stability and the resulting nuclear deformations (Lammerding et al., 2004) causing secondary alterations in heterochromatin localization and significant changes in gene expression (Nikolova et al., 2004). (For a review of these hypotheses, see reference (Worman and Courvalin, 2004).) In addition, lamin A binds directly or indirectly to a number of nuclear proteins, including emerin (Lee et al., 2001; Sakaki et al., 2001) and nesprin (Mislow et al., 2002), and attach them to the nuclear envelope. Lamin A also binds to transcription factors such as MOK2 (Dreuillet et al., 2002), retinoblastoma (RB) (Mancini et al., 1994), and SREBP1 (Lloyd et al., 2002). (See (Zastrow et al., 2004) for a review of lamin-binding proteins.) Mislocalization of lamin A, as we and others have observed, is likely indicative that its binding partners are also mislocalized and not bound to the nuclear envelope, which may have profound secondary effects on cells.
Interestingly, the neonatally lethal wrinkle free phenotype of Slc27a4 −/− mice that we and others have reported (Herrmann et al., 2003; Moulson et al., 2003) appears to be much more similar to the human RD phenotype than that exhibited by Zmpste24 −/− mice, which survive for several months. This may be coincidence, but the possibility exists that there is some special mechanistic relationship in humans between nuclear architecture and expression of genes involved in fatty acid homeostasis. Indeed, one of the features of the diverse laminopathies that are less severe than RD is lipodystrophy.
Genetic and allelic heterogeneity appear to explain in part the phenotypic differences among the related laminopathies Hutchinson-Gilford Progeria Syndrome (HGPS-MIM 176670), MAD, and most recently, RD. Navarro et al. included in their study two patients several months old with a skin disease less severe than typical RD, and they found one novel and one previously reported mutation in the LMNA gene. These are both likely to be sporadic, heterozygous, dominant negative mutations. The latter mutation had been found previously in several patients with HGPS. There is clearly a marked difference in disease severity between ZMPSTE24-based neonatally lethal RD and LMNA-based progeria. To avoid confusion, we propose that the descriptor “restrictive dermopathy” be reserved for individuals with the severe, neonatally lethal disease described by Witt (Witt et al., 1986). Children with some RD-like features who survive well past the neonatal period are more likely to manifest features of progeria or MAD and should be described as such. The ability to screen for ZMPSTE24 and LMNA mutations should make this distinction relatively straightforward.
Acknowledgments
We are extremely grateful to the families who participated in this study and to the referring physicians and genetic counselors for their invaluable contributions. We thank Edward Fox and the Dana-Farber Cancer Institute Microarray Core for performing SNP genotyping; Martin Pollak and Alan Beggs for advice; Phillip Stahl and Didier Hodzic for use of the confocal microscope; and Paul Goodfellow for helpful suggestions and comments on the manuscript. This work was funded by National Institutes of Health grant R01AR049269 to JHM.
References
- Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- Bergo MO, Gavino B, Ross J, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A. 2002;99:13049–13054. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capanni C, Cenni V, Mattioli E, et al. Failure of lamin A/C to functionally assemble in R482L mutated familial partial lipodystrophy fibroblasts: altered intermolecular interaction with emerin and implications for gene transcription. Exp Cell Res. 2003;291:122–134. doi: 10.1016/s0014-4827(03)00395-1. [DOI] [PubMed] [Google Scholar]
- Caux F, Dubosclard E, Lascols O, et al. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J Clin Endocrinol Metab. 2003;88:1006–1013. doi: 10.1210/jc.2002-021506. [DOI] [PubMed] [Google Scholar]
- Coombs NJ, Gough AC, Primrose JN. Optimisation of DNA and RNA extraction from archival formalin-fixed tissue. Nucleic Acids Res. 1999;27:e12. doi: 10.1093/nar/27.16.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreuillet C, Tillit J, Kress M, Ernoult-Lange M. In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nucleic Acids Res. 2002;30:4634–4642. doi: 10.1093/nar/gkf587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong LG, Ng JK, Meta M, et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci U S A. 2004;101:18111–18116. doi: 10.1073/pnas.0408558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann T, van der Hoeven F, Grone HJ, et al. Mice with targeted disruption of the fatty acid transport protein 4 (Fatp 4, Slc27a4) gene show features of lethal restrictive dermopathy. J Cell Biol. 2003;161:1105–1115. doi: 10.1083/jcb.200207080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt I, Ostlund C, Stewart CL, Man N, Worman HJ, Morris GE. Effect of pathogenic missense mutations in lamin A on its interaction with emerin in vivo. J Cell Sci. 2003;116:3027–3035. doi: 10.1242/jcs.00599. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Schulze PC, Takahashi T, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Haraguchi T, Lee RS, Koujin T, Hiraoka Y, Wilson KL. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci. 2001;114:4567–4573. doi: 10.1242/jcs.114.24.4567. [DOI] [PubMed] [Google Scholar]
- Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20:1233–1240. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet. 2002;11:769–777. doi: 10.1093/hmg/11.7.769. [DOI] [PubMed] [Google Scholar]
- Lowry RB, Machin GA, Morgan K, Mayock D, Marx L. Congenital contractures, edema, hyperkeratosis, and intrauterine growth retardation: a fatal syndrome in Hutterite and Mennonite kindreds. Am J Med Genet. 1985;22:531–543. doi: 10.1002/ajmg.1320220311. [DOI] [PubMed] [Google Scholar]
- Mancini MA, Shan B, Nickerson JA, Penman S, Lee WH. The retinoblastoma gene product is a cell cycle-dependent, nuclear matrix-associated protein. Proc Natl Acad Sci U S A. 1994;91:418–422. doi: 10.1073/pnas.91.1.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki H, Loi H, Dong S, et al. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 2004;14:414–425. doi: 10.1101/gr.2014904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mau U, Kendziorra H, Kaiser P, Enders H. Restrictive dermopathy: report and review. Am J Med Genet. 1997;71:179–185. doi: 10.1002/(sici)1096-8628(19970808)71:2<179::aid-ajmg11>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Mislow JM, Holaska JM, Kim MS, Lee KK, Segura-Totten M, Wilson KL, McNally EM. Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 2002;525:135–140. doi: 10.1016/s0014-5793(02)03105-8. [DOI] [PubMed] [Google Scholar]
- Moulson CL, Martin DR, Lugus JJ, Schaffer JE, Lind AC, Miner JH. Cloning of wrinkle-free, a previously uncharacterized mouse mutation, reveals crucial roles for fatty acid transport protein 4 in skin and hair development. Proc Natl Acad Sci U S A. 2003;100:5274–5279. doi: 10.1073/pnas.0431186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchir A, Medioni J, Laluc M, et al. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy, and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve. 2004;30:444–450. doi: 10.1002/mus.20122. [DOI] [PubMed] [Google Scholar]
- Navarro CL, De Sandre-Giovannoli A, Bernard R, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- Nikolova V, Leimena C, McMahon AC, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113:357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71:426–431. doi: 10.1086/341908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostlund C, Bonne G, Schwartz K, Worman HJ. Properties of lamin A mutants found in Emery-Dreifuss muscular dystrophy, cardiomyopathy and Dunnigan-type partial lipodystrophy. J Cell Sci. 2001;114:4435–4445. doi: 10.1242/jcs.114.24.4435. [DOI] [PubMed] [Google Scholar]
- Pendas AM, Zhou Z, Cadinanos J, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- Sakaki M, Koike H, Takahashi N, Sasagawa N, Tomioka S, Arahata K, Ishiura S. Interaction between emerin and nuclear lamins. J Biochem (Tokyo) 2001;129:321–327. doi: 10.1093/oxfordjournals.jbchem.a002860. [DOI] [PubMed] [Google Scholar]
- Sillevis Smitt JH, van Asperen CJ, Niessen CM, et al. Restrictive dermopathy. Report of 12 cases Dutch Task Force on Genodermatology. Arch Dermatol. 1998;134:577–579. doi: 10.1001/archderm.134.5.577. [DOI] [PubMed] [Google Scholar]
- Welsh KM, Smoller BR, Holbrook KA, Johnston K. Restrictive dermopathy. Report of two affected siblings and a review of the literature. Arch Dermatol. 1992;128:228–231. doi: 10.1001/archderm.128.2.228. [DOI] [PubMed] [Google Scholar]
- Witt DR, Hayden MR, Holbrook KA, Dale BA, Baldwin VJ, Taylor GP. Restrictive dermopathy: a newly recognized autosomal recessive skin dysplasia. Am J Med Genet. 1986;24:631–648. doi: 10.1002/ajmg.1320240408. [DOI] [PubMed] [Google Scholar]
- Worman HJ, Courvalin JC. How do mutations in lamins A and C cause disease? J Clin Invest. 2004;113:349–351. doi: 10.1172/JCI20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zastrow MS, Vlcek S, Wilson KL. Proteins that bind A-type lamins: integrating isolated clues. J Cell Sci. 2004;117:979–987. doi: 10.1242/jcs.01102. [DOI] [PubMed] [Google Scholar]