

Abstract
A novel route to epoxysorbicillinol as well as dimers of sorbicillin is reported. The synthesis is—in principle—amenable to enantioselectivity. The key step is an oxidative dearomatization to produce a stable and highly malleable p-quinol intermediate, which undergoes a highly diastereoselective epoxidation.
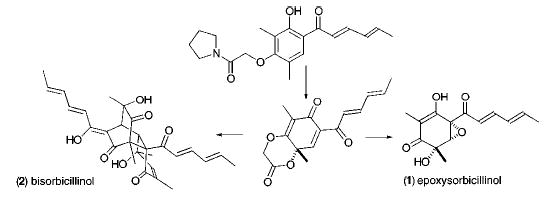
Cyclohexanones 1–3 (Figure 1) belong to a family of structurally diverse natural products isolated from both marine and terrestrial sources that encompass a wide range of biological activities.1 A highly functionalized tautomeric cyclohexadienone, 4,2 has been proposed as the common intermediate in the biosynthetic pathway.3b Indeed, several dimeric members of this natural product family, 2–3 along with trichodimerol, have yielded to synthesis by resolution of the racemic precursor 5, followed by its saponification and dimerization. These investigations3 testify to the difficulties that surround manipulating a ring system that contains both a dienic and dienophilic segment, predisposed toward dimerization.4
Figure 1.
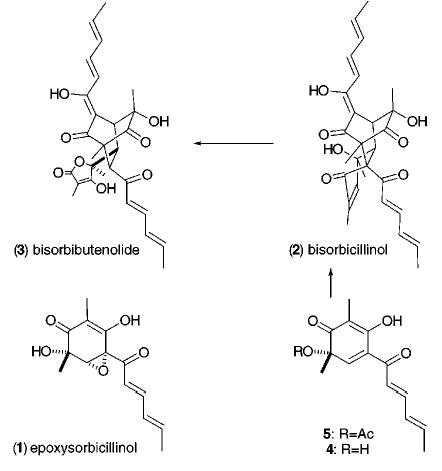
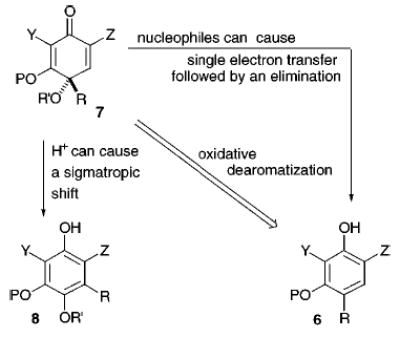
We were interested in developing a synthetic method that avoided resolution of 5 and furnished a sturdier ring system that was less prone toward dimerization. Such a process might lead to both monomeric and dimeric members of the sorbicillin family of natural products and prove useful in the synthesis of other complex molecules. Our plan entailed developing an asymmetric oxidative dearomatization procedure starting with the resorcinol derivative, 6,5 and affording the chiral quinoid 7, which we anticipated would be more manageable and therefore more synthetically useful than its counterpart, the cyclohexa-2,4-dienone 5. The ultimate synthetic value of 7, however, remained in doubt. The adequacy of the 3°-alcoholic stereocenter in 7 to control the formation of subsequent stereocenters was questionable and potentially obviated efficient application of 7 to 1. Another issue was the stability of 7. If the 3°-alcohol residue is protected, the addition of certain nucleophilic reagents is known to cause single electron transfer and elimination, reforming 6, rather than adding to the enone or ketone (Figure 2).6 In addition, in acidic media 7 is predisposed toward a sigmatropic shift resulting in 8.7 Perhaps, a structural feature could be incorporated in 7 that would promote stability, govern the formation of a particular diastereomer in subsequent reactions, and expedite the eventual development of an enantioselective method.
Figure 2.
For these reasons the [P] and [R′] residues of 7 were to be combined in a δ-lactone, 9, a structure we speculated could be formed by electrophilic cyclization of an amide, which was tethered ortho to a cationic site generated by exposure of 6 [P = –CH2C(O)NR2] or a similar resorcinol, such as 12, to an oxidant [cf. Figure 3]. Scrutiny of the chemical literature for effective conditions for this intramolecular oxidative cyclization revealed, however, that electrophilic amide cyclizations generally yielded γ-lactones,8 while oxidative dearomatization usually involved an ipso closure.9 Thus, precedent argued against the success of our strategy for 1–3. Nevertheless, the oxidation was investigated to test these notions in a racemic sense.
Figure 3.
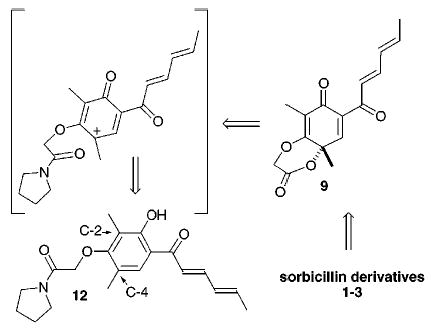
Coupling of sorbicillin 102b with the α-hydroxy amide 1110 using Mitsunobu’s conditions produces 12 (90%, Scheme 1). Oxidation of this resorcinol nucleus with 1 equiv of PhI-(OCOCF3)2 followed by aqueous workup cleanly affords 9 (45%) along with epoxide 13 (5%). No other regioisomer or derivative is apparent in 1H NMR spectra obtained for the crude mixture. However, the mass transfer is inexplicably low. The stability of 9 was remarkable, but not entirely unexpected. The 3° [OR′] substituent disposed equatorially in 9 is improperly aligned to eliminate, and being an electron-deficient component of a lactone, a poor facilitator of a sigmatopic shift. Although the regiospecificity of the cyclization was predictable,11 the formation of epoxide 13 in small quantities was quite surprising. We had anticipated that formation of 13 would require a Julia epoxidation or some similar type of procedure. However, treatment of 12 with 2.2 equiv of PhI(OCOCF3)2 exclusively affords the epoxide 13 as the sole diastereomer. Alternatively, treatment of 9 with 1.1 equiv of either PhI(OCOCF3)2 or treatment with a similar amount of PhIO affords 13 in almost quantitative yield. Thus, we assume that 9 precedes 13 along the synthetic pathway and further speculate that PhI(OCOCF3)2 in CH2-Cl2 exists in equilibrium with PhIO. Epoxidation of 9 most likely occurs by conjugate addition of PhIO to the activated enone and cleavage of the [PhI–Osubstrate] bond by the resulting enolate.12 The initial addition of PhIO occurs anti to the angular methyl residue, which shields one side of the enone and vinylogous ester from reaction. The relative stereochemistry in 13 was assigned using NOE and comparison with 16, a structure that was rigorously established using X-ray cystallography (Figure 4).13
Scheme 1.
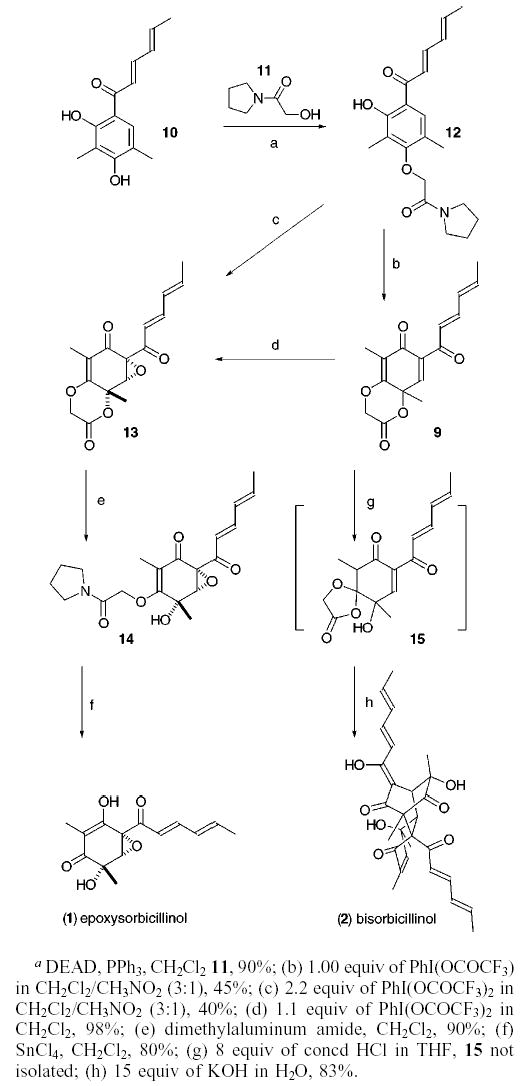
Figure 4.
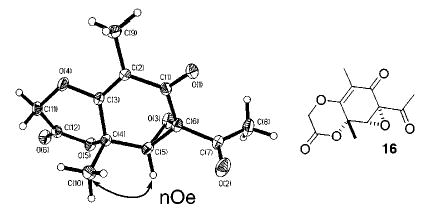
Vinyl ether 13 proved quite resistant to hydrolysis conditions, which are usually employed with vinylogous esters of this type.14 Therefore, the lactone was submitted to a Weinreib procedure, which results in amide 14 (90%).15 This material succumbs to SnCl4-mediated dealkylation of the vinylogous ester and affords epoxysorbicillinol (1) (80%) after acid cleavage of the tin enolate with 2 N HCl.
Access to the dimeric sorbicillinoids is achieved by cleaving the tether to reveal the underlying cyclohexa-2,4-dienone core. This is accomplished by treatment of 9 with concentrated HCl, which contracts the lactone and produces what we suppose is the mixed ketal 15.16 Subsequent addition of aqueous KOH to the reaction mixture followed by acidic workup produces bisorbicillinol (2) in 83% by the [4 + 2] cycloaddition expected for these conditions.3b Synthesis of 2 also constitutes a formal synthesis of bisorbibutenolide (3).
The intramolecular cyclization leading to 9 should simplify the development of an asymmetric oxidative dearomatization procedure. During the conversion of 12 to 9, the oxygen atom of the amide tether is restricted to two possible topologies of closure, a compact transition state and a noncompact transition state (cf. 18a‡ and 18b‡ for examples). In principle, asymmetry can be induced during the cyclization by controlling both the reactive face of the amide carbonyl and the topology of the cyclization. To this end, a stereocenter installed in 12 at either one of two sites, between the carbonyl and oxygen atoms in the tether moiety or adjacent to the nitrogen atom in the pyrrolidine ring, may lead to optically enriched products.
Although we have not yet completed satisfactory asymmetric syntheses of 1, 2, and 3, similar treatment of 17, a compound derived by Mitsunobu inversion of S-lactic acid (Scheme 2), affords (+)-2 in 51% ee when the diastereomeric mixture of analogues that corresponds to methylated derivatives of 9 is carried forward without separation.17 We suspect that A-1,3 strain between the chiral methyl residue and the pyrrolidine ring restricts the cyclization to transition states 18a‡ and 18b‡, where the methyl group is disposed in a pseudo-axial orientation. These two transition states lead to opposite enantiomers of 2. Because (+)-2 is obtained as the major product, the pathway proceeding through the dipole-stabilized intermediary 18b‡ appears to be the energetically favored reaction coordinate for this transformation.18
Scheme 2.
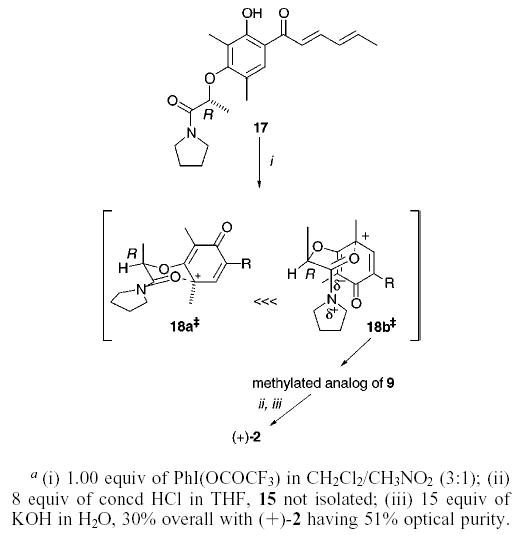
Studies directed at optimizing the yield19 and enhancing the enantioselectivity of the oxidative dearomatization are underway. Applications to other natural products exhibiting a highly oxidized core ring system such as manumycin, scyphostatin, rishirilide B, and diazaphilonic acid are envisioned.
Acknowledgments
Support from the University of California Cancer Research Coordinating Committee is greatly appreciated.
Footnotes
Supporting Information Available: Spectral characterization for 9, 12, 13, 14, 16, 17, and 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For isolation and medicinal value of sorbicillinoids, see: (a) Andrade R, Ayer WA, Mebe PP.Can J Chem 1992702526–2535. [Google Scholar]; b Andrade R, Ayer WA, Trifonov LS. Can J Chem. 1996;74:371–379. [Google Scholar]; c Andrade R, Ayer WA, Trifonov LS. Aust J Chem. 1997;50:255–257. [Google Scholar]; d Sperry S, Samuels GJ, Crews P. J Org Chem. 1998;63:10011–10014. [Google Scholar]; e Abe N, Yamamoto K, Murata T, Hirota A. Tennen Yuki Kagobutsu Toronkai Koen Yoshishu. 1999;41:553–558. [Google Scholar]; f Abe N, Murata T, Hirota A. Biosci, Biotechnol, Biochem. 1998;62:661–666. doi: 10.1271/bbb.62.661. [DOI] [PubMed] [Google Scholar]; g Shirota O, Pathak V, Hossain CF, Sekita S, Takatori K, Satake M. J Chem Soc, Perkin Trans 1. 1997;20:2961–2964. [Google Scholar]; h Mazzucco CE, Warr G. J Leukocyte Biol. 1996;60:271–277. doi: 10.1002/jlb.60.2.271. [DOI] [PubMed] [Google Scholar]; i Warr GA, Veitch JA, Walsh AW, Hesler GA, Pirnik DM, Leet JE, Lin PFM, Medina IA, McBrien KD. J Antibiot. 1996;49:234–240. doi: 10.7164/antibiotics.49.234. [DOI] [PubMed] [Google Scholar]; j Gao Q, Leet JE, Thomas ST, Matson JA, Bancroft DP. J Nat Prod. 1995;58:1817–1821. [Google Scholar]
- 2.Abe N, Sugimoto O, Tanji KI, Hirota A. J Am Chem Soc. 2000;122:12606–12607. [Google Scholar]
- 3.a Barnes-Seeman D, Corey EJ. Org Lett. 1999;1:1503–1504. doi: 10.1021/ol991070h. [DOI] [PubMed] [Google Scholar]; b Nicolaou KC, Vassilikogiannakis G, Simonsen KB, Baran PS, Zhong YL, Vidali VP, Pitsinos EN, Couladouros EA. J Am Chem Soc. 2000;122:3071–3079. doi: 10.1002/(sici)1521-3773(19991203)38:23<3555::aid-anie3555>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; c Nicolaou KC, Simonsen KB, Vassilikogiannakis G, Baran PS, Vidali VP, Pitsinos EN, Couladouros EA. Angew Chem, Int Ed. 1999;38:3555–3559. doi: 10.1002/(sici)1521-3773(19991203)38:23<3555::aid-anie3555>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; d Nicolaou KC, Jautelat R, Vassilikogiannakis G, Baran PS, Simonsen KB. Chem Eur J. 1999;5:3651–3665. [Google Scholar]; (e) Wood, J. L.; Thompson, B. D.; Yusuff, N.; Pflum, D. A. Abstracts of Papers, 219th National Meeting of the American Chemical Society, San Francisco, CA, March 26–30, 2000; American Chemical Society: Washington, DC, 2000; ORGN 848.
- 4.A [4 + 2] cycloaddition of 4 produces bisorbicillinol 2, while a net [4 + 4] cycloaddition of 4 results in trichodimerol, see refs 3a,b.
- 5.Van De Water RW, Magdziak D, Chau J, Pettus TRR. J Am Chem Soc. 2000;122:6502–6503. [Google Scholar]
- 6.Morrow GW, Schwind B. Synth Commun. 1995;25:269–276. [Google Scholar]
- 7.Schultz AG, Antoulinakis EG. J Org Chem. 1996;61:4555–4559. doi: 10.1021/jo952240g. [DOI] [PubMed] [Google Scholar]
- 8.Robin S, Rousseau G. Tetrahedron. 1998;54:13681–13736. [Google Scholar]
- 9.There is but one example of an oxidative cyclization occurring with an oxygen nucleophile tethered ortho to the site of cyclization, see: Pelter A, Ward RS.Tetrahedron 200157273–282. [Google Scholar]; For an ipso example, see: Wipf P, Kim Y, Fritch PC.J Org Chem 1993587195–7203. [Google Scholar]
- 10.Antonsson, T.; Gustafsson, D.; Hoffmann, K.-J.; Nystrom, J.-E.; Sorensen, H.; Sellen, M. PCT Int. Appl. WO 9723499 A1 19970703, 1997, 94 pp.
- 11.The coefficient of both the C-2 and C-4 sites in the cation are sizable and within striking distance of the tether. However, the coefficient at C-2 is significantly reduced by its proximity to the adjacent carbonyl.
- 12.Moriarity RM, Gupta SC, Hu H, Berenschot DR, White KB. J Am Chem Soc. 1981;103:686–688. [Google Scholar]
- 13.Compound 16 was synthesized in a fashion analogous to that of 13. The X-ray data for 16 has been submitted to the Cambridge Crystallographic database.
- 14.a Obara H, Machida Y, Namazi S, Onodera J. Chem Lett. 1985:1393–1394. [Google Scholar]; b Johns SR, Lamberton JA, Morton TC, Suars H, Wiling R. Aust J Chem. 1987;40:79–96. [Google Scholar]
- 15.Basha A, Lipton M, Weinreb S. Tetrahedron Lett. 1977:4171–4173. [Google Scholar]
- 16.We were unable to purify 15; however, replacing the sorbyl side chain with an acetyl residue furnished a mixed ketal presumed to be an analogue of 15.
- 17.The optical purity was determined by comparison of the [α]D of synthetic and natural 2 at similar concentrations.
- 18.Sevin A. J Org Chem. 1986;51:2671–2675. [Google Scholar]
- 19.Ley SV, Finch H, Thomas A. J Chem Soc, Perkin Trans 1. 1999;10:669–671. [Google Scholar]