

Abstract

An operationally simple oxidative dearomatization of resorcinol derivatives is reported that employs an inexpensive chiral directing group. The method provides access to a variety of p-quinol derivatives in good yield and diastereoselectivity. A short reductive process affords 4-hydroxy-4-alkylcyclohexenone derivatives in excellent yields and enantiomeric excesses.
Four types of chiral cyclohexadienones have proven extremely useful in natural product synthesis (see Figure 1, A–D).1 Despite their usefulness, there are relatively few ways to prepare or use A–D in an enantioselective format. Fujioka and co-workers prepared and examined the reactivity of a chiral ketal derived from the masked o-benzoquinone A and observed it to undergo diastereoselective reductions.2 o-Quinol building blocks of the general structure B have been examined by a number of groups. Methods for their preparation include the oxidatively triggered ipso cyclization of an oxime reported by Hoshino3 and the bacterial oxidations of phenols described separately by Myers4 and Breitmaier.5 With regard to masked p-benzoquinones C, Corey,6 Wipf,7 and Porco8 have independently investigated epoxidations of chiral ketal derivatives. Taylor, on the other hand, has championed the asymmetric Julia–Colonna epoxidation of these systems,9 while Feringa has studied asymmetric 1,4-additions of diethylzinc.10 Other asymmetric strategies developed by de March,11 Winterfeldt,12 Carreno,13 Wipf,14 and Corey15 utilize enantioselective desymmetrization of various prochiral MPB ketals with an asymmetric Diels–Alder cycloaddition followed by subsequent diastereoselective reactions and a retro-cycloaddition. With regard to p-quinol systems such as D, a similar masking strategy has been employed. Winterfeldt demonstrated that these systems engage in diastereoselective [4 + 2] cycloadditions with an unusual chiral diene,16 while Feringa has reported that symmetric derivatives of D undergo asymmetric 1,4-additions with diethylzinc in the presence of a chiral ligand.17 Despite these many successes, a careful examination of the literature suggests that improvements could be made particularly with regard to the range of derivatives that are prepared by current enantioselective technologies.
Figure 1.
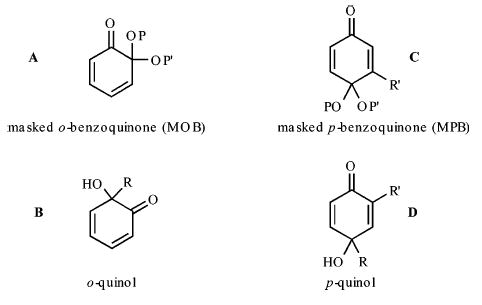
Some useful chiral cyclohexadienones.
Recently, we described a novel oxidative dearomatization for the conversion of 4-alkylated resorcinol derivatives into a lactone intertwined with a p-quinol. We also showed that this p-quinol scaffold is capable of undergoing a variety of diastereoselective reactions, which include 1,2-additions, 1,4-conjugate additions, and [4 + 2] cycloadditions (Scheme 1).18 Furthermore, we demonstrated the usefulness of this intermediate in efficient albeit racemic syntheses of epoxysorbicillinol and bisorbicillinol.18b
Scheme 1.
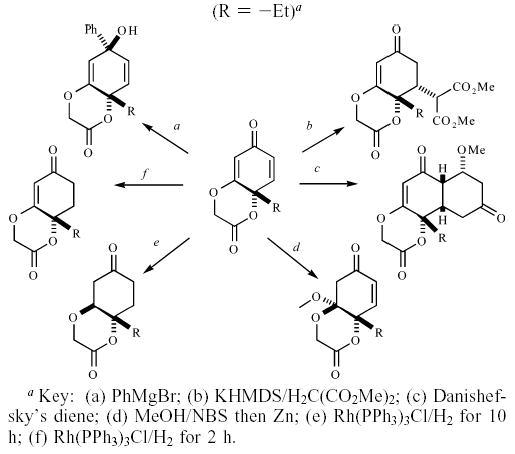
Diastereoselective Reactions of p-Quinols (R = −Et)a
Following a methodical investigation, we are now able to report that the cyclohexadienone scaffolds of type 14–18 can be obtained with excellent enantioselectivity and in respectable yield (Table 1). To the best of our knowledge, this diastereoselective dearomatization of resorcinol derivatives is unique. However, it requires access to 4-alkyl monoprotected resorcinols, which we prepare using o-quinone methide chemistry developed in our group.19
Table 1.
Diastereoselective Dearomatization of Resorcinol Derivatives
| entry | starting material | product | Yield | ratio |
|---|---|---|---|---|
| 1 |

|
|
75% | >20:1 |
| 2 |
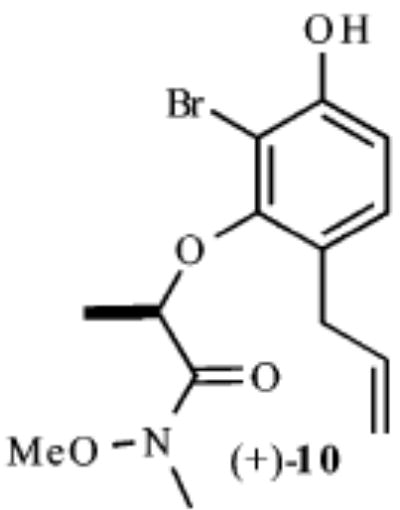
|
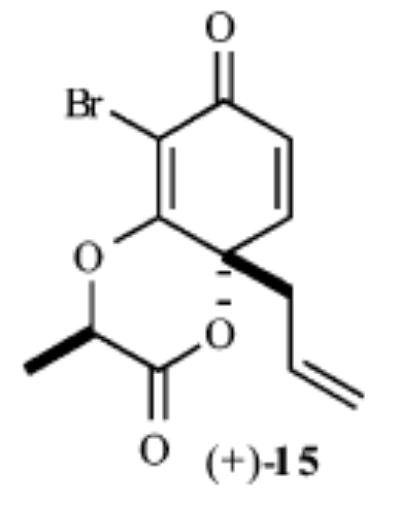
|
61% | >20:1 |
| 3 |
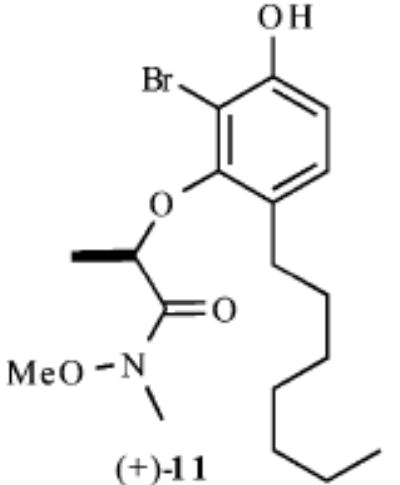
|
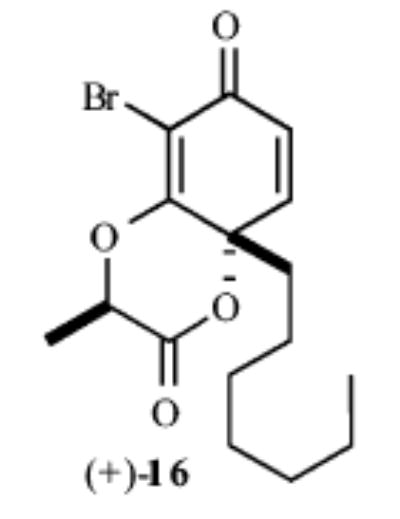
|
65% | >20:1 |
| 4 |

|
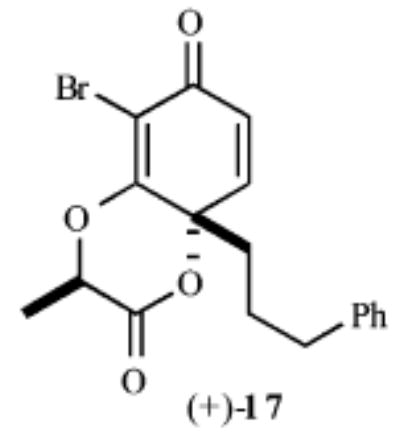
|
72% | >20:1 |
| 5 |
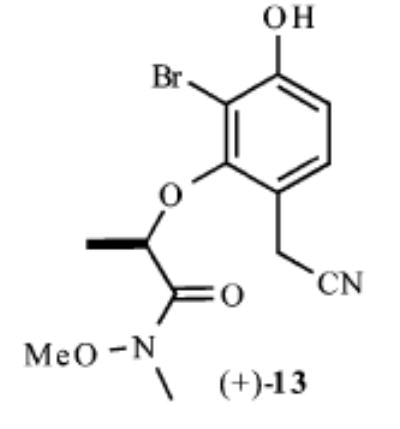
|
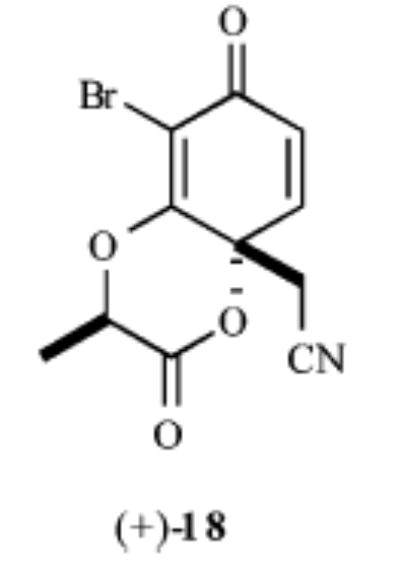
|
63% | >20:1 |
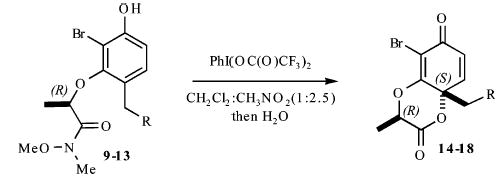
Our synthesis of these substrates begins with bromination20 of the aldehyde 1 and subsequent bis-BOC protection, which affords the bromo aldehyde 2 in greater than 70% yield (Scheme 2). Reduction of the aldehyde 2 with NaBH4 provides the corresponding benzyl alcohol, which upon treatment with an excess of a nucleophile generates an o-quinone methide. A conjugate addition ensues and provides the monoprotected 4-alkylated resorcinol derivatives 3–7 in good yields (59–89%). These phenols undergo Mitsunobu coupling with 2S-hydroxy-N-methoxy-N-methylpropionamide (−)-821 and thereby afford phenols (9–13) upon deprotection with ZnBr2 (4 equiv, 0.1 M in CH3NO2) in >75% yield over the three steps.
Scheme 2.
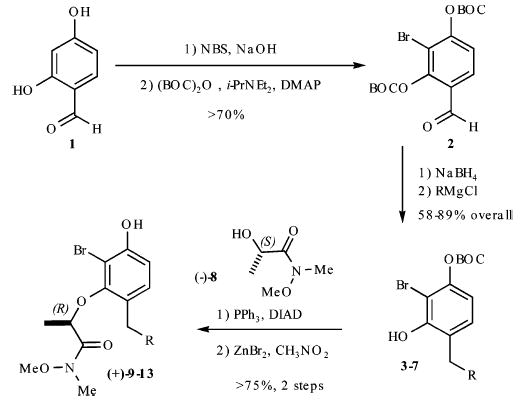
Preparation of Resorcinol Precursors
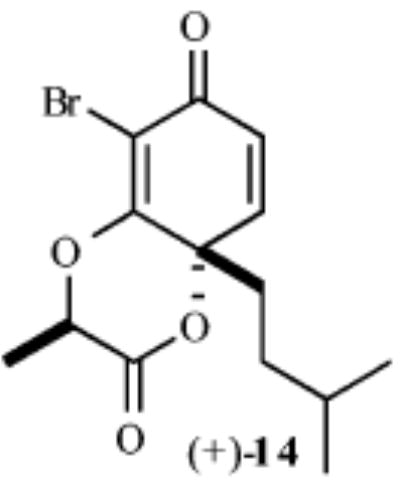
As shown in Table 1, exposure of these readily prepared chiral resorcinols (9–13) to 1.1 equiv of PhI(OCOCF3)2 (0.3 h, 0.1 M CH2Cl2:CH3NO2/1:2.5, 0 °C) followed by an aqueous workup affords a series of chiral lactone scaffolds (14–18) in good yields with excellent diastereoselectivity. Because the protocol proves diastereoselective with a variety of differently sized C4-substituents, the magnitude of asymmetric induction afforded by the chiral amide appears independent of the steric bulk of the C-4 substituent.
Hydrogenation of p-quinol (+)-14 over rhodium (Rh/Al, 150 psi H2, THF) chemoselectively reduces the enone functionality and affords the vinylogous ester (+)-20 in 70% yield (Scheme 3). Subsequent hydrogenolysis of the bromine atom and hydrogenation of the vinylogous ester in (+)-20 is accomplished with Pd/C (1 atm, 1.0 equiv of Hünig’s base, 0.1 M EtOAc) and affords the lactone (+)-19 in quantitative yield. The relative stereochemistry of the protons (Ha–Hd) in (+)-19 was established by 2D-NOESY experiment. In our experience, a two-step reduction protocol is preferred to a single-pot reduction of (+)-13 with PtO2 (4 h, 1 atm H2) because the yields of the latter procedure prove quite variable (40–80%) and afford significant quantities of rearomatized materials. Addition of solid KOTMS to (+)-19 (1.1 equiv, 0.1 M Et2O, rt) affords the enantiomerically enriched ketone (+)-21 in >95% yield. Comparison with a racemic standard by chiral GC analysis demonstrated that (+)-20 is obtained in >99% ee by this eight-pot process. It should be noted that β-elimination, which is necessary for formation of the enone (+)-21, most likely does not occur until the lactone undergoes ring opening and a subsequent ring-flip.
Scheme 3.
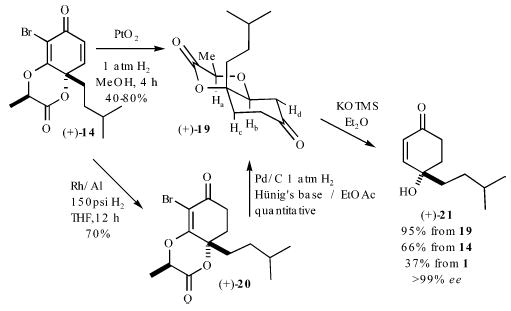
Cleavage of Directing Group
With regard to the diastereoselectivity of this process, it appears that the major isomer arises from the boat transition state B, while the chair transition state A leads to the minor diastereomer (Figure 2). The boat transition state is not objectionable because most sites in the forming and neighboring six-membered ring are sp2 hybridized. The Weinreb amide affords much higher diastereoselectivity than other amides. Presumably, there is less steric interaction between the –OMe and –Br in the Weinreb amide than the corresponding interaction between an alkyl and –Br substituent in other amides in the transition state. The >25% improvement in yields and reproducibility in this dearomatization process as compared with earlier des-bromo des-methyl examples18a can be attributed to a favorable steric interaction that facilitates lactonization, as well as prevention of undesired aryl couplings with the oxidant.18a,22 It appears that a C2 substituent, such as bromine, limits the degrees of freedom and assists in placing the amide carbonyl in closer proximity to the intended site of reaction.
Figure 2.
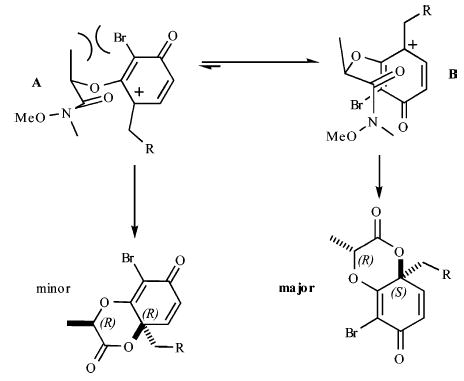
Boatlike transition state is preferred.18b
This method provides a flexible chiral scaffold, which subsequently allows access to a variety of p-quinols and their derivatives in good yield and enantioselectivity. On the basis of our previous observations with related systems,18 easy access to a great many types of chiral ensembles can be imagined. We expect this enantioselective dearomatization process may prove useful in syntheses of nonracemic molecules.
Acknowledgments
Research Grants from the National Science Foundation (Career-0135031) and National Institutes of Health (GM-64831) are greatly appreciated. L.H.M. gratefully acknowledges additional financial support provided by J. H. Tokuyama and a minority supplement (GM-64831) for furthering his research. We also thank the patient editors of the American Chemical Society, who strive to do an outstanding job.
Footnotes
Supporting Information Available: Experimental procedures and full spectral data for compounds 2–21. This material is provided free of charge via the Internet at http://pubs.acs.org
References
- 1.Magdziak D, Meek S, Pettus TRR. Chem Rev. 2004;104:1383–1429. doi: 10.1021/cr0306900. [DOI] [PubMed] [Google Scholar]
- 2.Fujioka H, Annoura H, Murano K, Kita Y, Tamura Y. Chem Pharm Bull. 1989;37:2047–2052. [Google Scholar]
- 3.Murakata M, Yamada K, Hoshino O. J Chem Soc, Chem Commun. 1994:443–444. [Google Scholar]
- 4.Myers AG, Siegel DR, Buzzard DJ, Charest MG. Org Lett. 2001;3:2923–2926. doi: 10.1021/ol010151m. [DOI] [PubMed] [Google Scholar]
- 5.Kneifel H, Poszich-Buscher C, Rittich S, Breitmaier E. Angew Chem, Int Ed Engl. 1991;30:202–203. [Google Scholar]
- 6.Corey EJ, Wu LI. J Am Chem Soc. 1993;115:9327–9328. [Google Scholar]
- 7.Wipf P, Kim Y, Jahn H. Synthesis. 1995:1549–1561. [Google Scholar]
- 8.Hu Y, Li C, Kulkarni BA, Strobel G, Lobkovsky E, Torczynski RM, Porco JA., Jr Org Lett. 2001;3:1649–1652. doi: 10.1021/ol0159367. [DOI] [PubMed] [Google Scholar]
- 9.Genski T, Macdonald G, Wei X, Lewis N, Taylor RJK. Synlett. 1999:795–797. [Google Scholar]
- 10.Imbos R, Minnaard AJ, Feringa BL. Tetrahedron. 2001;57:2485–2489. [Google Scholar]
- 11.de March P, Escoda M, Figueredo M, Font J, Garcia-Garcia E, Rodriguez S. Tetrahedron: Asymmetry. 2000;11:4473–4483. [Google Scholar]
- 12.Wolter M, Borm C, Merten E, Wartchow R, Winterfeldt E. Eur J Org Chem. 2001:4051–4060. [Google Scholar]
- 13.Carreno MC, Gonzalez MP, Fisher J. Tetrahedron Lett. 1995;36:4893–4896. [Google Scholar]
- 14.Wipf P, Jung JK. J Org Chem. 2000;65:6319–6337. doi: 10.1021/jo000684t. [DOI] [PubMed] [Google Scholar]
- 15.Breuning M, Corey EJ. Org Lett. 2001;3:1559–1562. [Google Scholar]
- 16.Gerstenberger I, Hansen M, Mauvais A, Wartchow R, Winterfeldt E. Eur J Org Chem. 1998;643:3–650. [Google Scholar]
- 17.Imbos R, Brilman MHG, Pineschi M, Feringa BL. Org Lett. 1999;1:623–625. [Google Scholar]
- 18.a Van De Water RW, Hoarau C, Pettus TRR.Tetrahedron Lett 2003445109–5113.16957784 [Google Scholar]; b Pettus LH, Van De Water RW, Pettus TRR. Org Lett. 2001;3:905–908. doi: 10.1021/ol0155438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a Van De Water RW, Magdziak DJ, Chau JN, Pettus TRR. J Am Chem Soc. 2000;122:6502–6503. [Google Scholar]; b Jones R, Van De Water RW, Lindsey C, Hoarau C, Ung T, Pettus TRR. J Org Chem. 2001;66:3435–3441. doi: 10.1021/jo001752e. [DOI] [PubMed] [Google Scholar]; c Hoarau C, Pettus TRR. Synlett. 2003:127. doi: 10.1055/s-2003-36234. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tuttle K, Pettus TRR. Synlett. 2003:2234–2236. [Google Scholar]; e Lindsey C, Gomez-Diaz C, Villalba JM, Pettus TRR. Tetrahedron. 2002;58:4559–4565. [Google Scholar]
- 20.Bui E, Bayle JP, Perez F, Liebert L, Courtieu J. Liquid Crystals. 1990;8:513–526. [Google Scholar]
- 21.a Paterson I, Wallace DJ, Cowden CJ. Synthesis. 1998:639–652. [Google Scholar]; b Less SL, Leadlay PF, Dutton CJ, Staunton J. Tetrahedron Lett. 1996;37:3519–3520. [Google Scholar]
- 22.Murakata M, Yamada K, Hoshino O. J Chem Soc, Chem Commun. 1994:443–445. [Google Scholar]