

Abstract
Heat shock proteins such as gp96 have the ability to chaperone peptides and activate antigen presenting cells. In this study we tested whether adenovirus (Ad)-mediated overexpression of secreted or membrane-associated forms of gp96 in tumor cells would stimulate an anti-tumor immune response. Studies were carried out in C57Bl/6 mice bearing aggressively growing subcutaneous tumors derived from syngeneic TC-1 cells, a cell line that expresses HPV16 E6 and E7 proteins. We found that secreted gp96 can induce protective and therapeutic anti-tumor immune responses. Our data also indicate that the anti tumor effect sgp96 expression appears to be limited by induction of suppressive regulatory T cells (Tregs). TC-1 tumor transplantation increased the number of splenic and tumor infiltrating Tregs. Importantly, treatment of mice with low-dose cyclophosphamide decreased the number Tregs and enhanced the immunostimulatory effect of sgp96 expression. We also tested whether an oncolytic vector (Ad.IR-E1A/TRAIL), that is able to induce tumor cell apoptosis and, potentially, release cryptic tumor epitopes in immunogenic form, can stimulate anti-tumor immune responses. While tumor cells infected ex vivo with Ad.IR-E1A/TRAIL had no anti-tumor effect when used as a vaccine alone, the additional treatment with low-dose cyclophosphamide resulted in elimination of pre-established tumors. This study gives a rationale for testing approaches that suppress Tregs in combination with oncolytic or immunostimulatory vectors.
Keywords: adenovirus, oncolytic, heat shock protein, gp96, regulatory T-cells, cyclophosphamide
Introduction
Heat shock proteins (HSP) have a unique ability to activate both innate and adaptive immunity attributed to their ability to interact with a number of receptors on dendritic cells (DC) and macrophages, that serve as “professional” antigen-presenting cells (APC) (1). Proteins or peptides bound to HSPs are re-displayed in complexes with MHC class I molecules upon internalization by APCs, triggering the production of proinflammatory cytokines and subsequent activation of T-cell mediated immune responses. These properties make HSPs attractive vehicles for delivery of antigens into antigen-presenting pathways. HSPs capable of providing these functions include hsp70, hsp90, gp96, and calreticulin [for a review see (2)]. There are numerous approaches by which HSPs can enhance specific immunity against infectious agents or cancers. Originally, HSPs directly isolated from tumor tissue were used to elicit anti-tumor immunity. As a variation to this scheme, it has been shown that peptides or proteins complexed or fused with syngeneic or allogeneic (mycobacterial) HSPs can elicit potent antibody and cytotoxic responses. The HSP gp96 genetically modified to be displayed on the surface of or secreted from tumor cells activated potent anti-tumor responses and tumor regression (3, 4).
Since 1997, genetically engineered Ads that selectively replicate in and kill tumor cells have been used for the treatment of cancer [for a review see (5)]. Our lab has previously developed oncolytic vectors based on Ad genomes deleted for E1A and E1B genes. HPV E6 or E7 expressing tumor cells support DNA replication of E1-deleted Ad vectors (6, 7). This tumor-specific replication of viral genomes can be converted into tumor-specific transgene expression utilizing homologous recombination in Ad genomes (Ad.IR system) (8). We have used the Ad.IR system to express Ad E1A (to allow for production of progeny virus) and TRAIL (to induce apoptosis and efficient release of progeny virus and viral spread) (9). The resulting oncolytic Ad vectors (Ad.IR-E1A/TRAIL) was able to eliminate pre-established metastases in xenograft models after a single systemic application (9) and did not cause unspecific toxicity in mice or baboons (10).
Adenovirus vectors and particularly oncolytic adenoviruses can potentially increase the potency of HSP-based immunotherapy strategies. Interaction of Ad capsids with cellular receptors induces expression of pro-inflammatory cytokines/chemokines, such as TNFα, IFNγ, IL-1, IL-6, IL-12, MCP-1 and 2, which results in recruitment of effector cells of the innate and adaptive immune system to the site of infection (11). These cytokines also activate functions of APCs. Furthermore, presentation of Ad proteins of the incoming Ad particle and/or de novo expression of Ad proteins in tumor cells could provide an adjuvant effect on the activation of tumor-specific T-cells. With regard to oncolytic adenoviruses, it is thought that tumor cell lysis has the potential to release tumor-antigens as apoptotic bodies or in complex with tumor-derived or expressed HSPs functioning as chaperons for antigen presentation to dendritic cells and (in the context of Ad infection) to subsequent activation of anti-tumor T-cell responses. This hypothesis is supported by several studies. An oncolytic Ad5-based vector showed a strong anti-tumor efficacy towards rectal carcinomas in immunocompetent mice, which was accompanied by an acute inflammatory reaction (e.g. CD8+T-cell infiltration, increased TNFα and IFNγ levels), while the anti-tumor efficacy of this vector against the same cancer cell line was significantly less in athymic mice (12, 13). We have recently shown that transplantation of mouse breast cancer cells (C3L5), that underwent viral oncolysis upon infection with Ad.IR-E1A/TRAIL, into C3H mice induced a systemic anti-tumor immune response that resulted in tumor rejection. This response was significantly greater than with mock-infected or first-generation control vector infected cell vaccines (14). A recent study demonstrated that HPV E6/E7 expressing TC-1 mouse tumor cells that underwent apoptosis after Herpes Simplex Virus infection increased the efficacy of dendritic cell vaccines more than TC-1 cells that died upon ultraviolet B radiation (15). The latter study underscores the adjuvant effect of viral infection. Conversely, a series of studies argue that tumor cell death via apoptosis and uptake of apoptotic bodies by APCs can cause immunological ignorance to tumor antigens (16). It is thought that phagocytic uptake of apoptotic cells by macrophages/APCs and subsequent signaling results in a decreased ability to efficiently stimulate T effector cell responses (17), increased anti-inflammatory cytokine production (18, 19), decreased pro-inflammatory cytokine production (20), and/or possibly aid the generation of regulatory T-cells (21, 22). Notably, the studies supporting an immunosuppressive role of tumor cell apoptosis were not done in the context of Ad infection.
Tumors employ several mechanisms to evade an immune response, including the downregulation of tumor-selective antigens, MHC, and co-stimulatory molecules. Among these mechanisms, the escape of tumors from immunological control via T regulatory cells (Tregs) is attracting increased attention. Human and murine Tregs are CD4+CD25+ and express a number of other markers including Forkhead P3 (FoxP3), CTLA4, Glucocorticoid-Induced TNFR-Related Protein (GITR), L-selectin (CD62L), Neuropilin-1, and OX40 antigen (CD134). As early as the late 1970s, studies demonstrated that administration of cyclophosphamide (CY) can improve anti-tumor responses. CY is a chemotherapeutic agent used to treat various types of cancer. The high doses (in humans >120mg/kg, in mice >400mg/kg) of drug required for effective chemotherapy cause immunosuppression. However, at low doses (in mice: <100mg/kg), CY treatment results in enhanced immune responses against a variety of antigens (23-27), a property that was attributed to the ability to selectively kill Tregs (28-31). In mouse experiments it was shown that upon CY injection, the numbers of Tregs decreased by day 4. By day 10 the absolute number of Tregs returned to normal indicating that the effects of CY are transient without prolonged reduction of tolerance in the body (31). Administration of immunostimulating modalities during this period of Treg inhibition would theoretically allow for enhanced anti-tumor immune responses with a decreased likelihood of autoimmunity.
The objectives of this study are to analyze, in a model with HPV E6/E7 expressing TC-1 tumor cells, the effect of i) adenovirus-mediated overexpression of membrane-bound or soluble forms of gp96, ii) Ad.IR-E1A/TRAIL induced tumor cell death and a combination of both on the induction of an anti-tumor immune response, and iii) of low-dose CY treatment on the anti-tumor efficacy of our Ad-based approaches.
Materials and Methods
Cell lines and antibodies. TC-1 cells (syngeneic to C57Bl/6 mice) (ATCC# CRL-2785) were used in these studies. Cells were maintained in RPMI 1640 + 10% FBS and supplemented with L-glutamine, penicillin and streptomycin. Antibodies used were a rabbit polyclonal anti-gp96 (Stressgen, Victoria, BC Canada), a rat monoclonal anti-gp96 antibody (Stressgen, Victoria, BC Canada), a mouse anti-rat FITC-conjugated antibody and donkey anti-rat HRP-conjugated antibody (Jackson Labs., Bar Harbor, MN).
Generation of recombinant Ad and Ad.IR vectors expressing a soluble or membrane-anchored form of gp96. The human gp96 cDNA cloned in the plasmid pGEMEX.gp96 (kindly provided by Dr. P. K. Srivastava) was amplified by PCR whereby the KDEL endoplasmatic reticulum (ER) retention signal was eliminated. This modified cDNA was cloned into pAd.RSV (32) to generate pAd.sgp96. To generate pAd.IR.E1A/gp96, the lacZ gene in pAd.IR.E1A/bGal (32) was replaced with the sgp96 gene. To generate the membrane gp96 expressing vectors, the PCR amplified, KDEL-deleted cDNA was cloned into the pDisplay plasmid (Invitrogen, Carlsbad, CA) in frame with the membrane anchor. The resulting mgp96 fragment was released and cloned into pAd.RSV to generate pAd.mgp96, or in replacement of the lacZ cassette, into pAd.IR.E1A/bGal, in order to generate pAd.IR-E1a/mgp96. Ad vectors were produced in 293 cells by recombination with pBHG10 and titrated by OD260 spectometry as well as by quantitative Southern blotting for viral genomes using an 8kb probe corresponding to a region in the backbone of all adenoviral vectors as described elsewhere (33). The infectious titers were determined by plaque tittering on 293 cells. All vectors had a genome to pfu ratio of ∼20:1.
Western blotting. For the detection of mgp96, proteins were extracted from transduced cells, and for detection of the soluble form of gp96 a rabbit polyclonal anti-gp96 antibody (Stressgen, Victoria, BC Canada) and protein G-sepharose beads (Sigma) were used to pull down the complexes. After polyacrylamide electropheresis, proteins were blotted onto nitrocellulose membranes and probed with a rat monoclonal anti-gp96 antibody followed by anti-rat Ig-HPR conjugated antibody.
Immunofluorescence analysis of gp96. Cells were plated in 8-well chamber slides, transduced with Ad vectors expressing the soluble or membrane forms of gp96, or control vectors, and fixed after 48-72 hours. Cells were first incubated with a rat monoclonal anti-gp96 antibody (Stressgen), followed by a mouse anti-rat FITC-conjugated antibody (Jackson). Cells were visualized under UV-light and photographed.
Crystal violet assay. Cells were plated in 24-well plates and transduced at different MOIs with adenoviral vectors in triplicate. Viable cells in each well were stained at the indicated time points with crystal violet as described elsewhere (8). Results were expressed as the percentage of viable cells compared to mock-treated controls.
Animal experiments. C57Bl/6 mice (Jackson Labs, Bar Harbor, ME) were used. Animal experiments were performed in a number of different settings to evaluate the anti-tumor efficacy of gp96 expression as a soluble or membrane form from TC-1 cells. “Vaccination” setting I: Cells were transduced with Ad vectors at an MOI of 100pfu/cell. Sixteen hours later cells were collected, washed, and irradiated with 6000 rads. Mice were injected with 1×106-irradiated cells at days 0 and 14, and challenged with 2×105 untreated tumor cells on day 24. Tumor growth was monitored after that. Vaccination setting II: Mice were injected subcutaneously into the right inguinal flank with 5×104 untransduced TC-1 cells. When tumors reached a diameter of 2 mm, one group of mice was intraperitoneally injected with 500μl of 4mg/ml of CY in PBS; the other group received 500μl of PBS. Four days later, mice were subcutaneously injected (into the left inguinal flank) with 1×106 TC-1 cells transduced ex vivo with the indicated Ad vectors. Following vaccination the volume of the primary tumor was measured twice a week. The tumor volume was calculated using the formula (largest diameter × (smallest diameter)2. Mice were sacrificed when the tumor volume reached a volume of 1000 mm3. “Therapeutic” setting: Two strategies were used. In the first setting, cells were transduced with 100 pfu/cell of the indicated Ad vectors. Sixteen hours later (for Ad.sgp96 and Ad.mgp96 vectors) or 72 hrs later (for Ad.IR vectors), mock- or Ad-transduced cells were collected, washed, and mice were subcutaneously injected into the right inguinal region with 1×105 cells. In another setting, transduced cells were mixed with 20% of non-transduced cells.
Analysis of Tregs. Splenocytes were analyzed by flow cytometry using the following antibodies: rat Mab anti-FoxP3-PE (clone FHK16s, eBioscience), rat Mab anti-CD4-PE, and rat Mab anti-CD25-FITC (all from eBioscience). All samples were treated with Fc-block (CD16/CD32). Corresponding isotope controls yielded no significant staining. Cryosections of TC-1 tumors and spleens were analyzed by immunofluorescence using rabbit anti-FoxP3 antibodies [provided by Dr. Kouji Matsushima (University of Tokyo) (34)] and rat Mab antiCD25 antibodies (clone 16.15, eBioscience). Binding of primary antibodies was visualized with goat anti-rabbit-Alexa fluo 568 (red) and goat anti-rat-Alexa fluo 488 (green) antibodies (Molecular Probes, Eugene, OR). Nuclei are stained with DAPI (Sigma).
ELIspot: Splenocytes of naïve C57Bl/6 mice were pulsed with 10μg of the HPV16 E749-57 carrying the H-2Db restricted peptide (RAHYNIVTF) (35) or an unrelated control peptide. (C57Bl/6 mice are H-2Db and the E749-57 peptide contains a CTL epitope). On day 14 after vaccination with mock infected TC-1 cells or cells transduced with Ad.Co, Ad.sgp96, or AdIR.E1A/TRAIL (with and without CY), vaccinated animals were sacrificed, splenocytes were collected and 1×106 cells were mixed with 1×106 ex vivo pulsed splenocytes for in vitro sensitization. After 24 hours of incubation in 96 well plates, cells were plated in anti-IFNγ-coated wells of ELIspot plates (Millipore, Bedford, Mass). Twenty-four hours later, plates were washed and the spots of IFNγ producing T-cells were counted.
Results
Vector construction. gp96 is an endoplasmic reticulum resident protein that is released upon necrotic cell death or virus infection and subsequently confers antigen-transport to and activation of DCs. To increase the immunostimulatory potency of gp96, we constructed a secreted form of gp96 (sgp96) by deleting the C-terminal KDEL endoplasmic reticulum-retention signal. Notably, gp96 has an N-terminal leader peptide (L) allowing for secretion. For comparison, we also generated a membrane-localized form of gp96 (mgp96) that was recently shown to induce potent anti-tumor responses (4). To create mgp96, a C-terminal PDGFR transmembrane domain was linked to the gp96 ORF deleted for the KDEL signal (Fig.1A). We constructed two sets of vectors for constitutive and replication-activated (tumor-specific) gp96 expression. We hypothesized that restricting gp96 expression to tumor cells would increase the safety profile of our vectors, considering that unrestricted gp96 expression (in non-tumor cells) without cell death can cause autoimmune reactions (36). For constitutive gene expression, E1/E3-deleted Ad5-based vectors expressing mgp96 and sgp96 under the control of the ubiquitously active RSV promoter were generated (Ad.mgp96 and Ad.sgp96). For tumor-specific gp96 expression, we constructed Ad.IR vectors (Ad.IR-E1A/mgp96 and Ad.IR-E1A/sgp96). In addition to gp96, these vectors express E1A in a tumor-specific manner, which will confer high-level transgene expression through replication of vector genomes only in tumor cells. E1A also has the potential for upregulating heat-shock-proteins in tumor cells, which, in turn, would improve antigen presentation to dendritic cells (37). Other control Ad vectors (Ad.Co) included Ad.GFP (14) and Ad.bGal (8). Control Ad IR-vectors included Ad.IR-GFP (14) and Ad.IR-E1A/AP (32).
Figure 1.

Characterization of gp96 expressing Ad vectors.A) Schematic representation of Ad vectors. The C-terminal KDEL signals was deleted from the gp96 cDNA to produce the secreted form of gp96 (sgp96) or replaced with the PDGF transmembrane domain (TM) to produce mgp96. Gp96 contains an N-terminal leader (L) peptide. Ad.sgp96 and Admgp96 express the soluble form of gp96 and the membrane-anchored form of gp96, respectively, under the control of the RSV promoter. Ad.IR.IR-E1A/sgp96, Ad.IR.IR-E1A/mgp96, Ad.IR.IR-E1A/TRAIL, and Ad.IR-E1A/AP contain the Ad5 E1A gene linked via an IRES to sgp96, mgp96, TRAIL, and alkaline phosphatase (AP) genes, respectively. The bicistonic transgene cassettes are flanked by inverted homology regions (IR). The orientation of the bicistronic transgene cassettes towards the RSV promoters is in 3′→5′ direction. If Ad.IR vectors infect cells that support Ad DNA replication (e.g. TC-1 cells), (replication-dependent) homologous recombination between the IRs links the RSV promoter to the 5′ end of the transgene cassette, which results in gene expression. [For a more detailed explanation of Ad.IR vectors, see ref: (47).] PA: SV40 polyadenylation signal, ITR: inverted terminal repeats.B) Transduction of TC-1 cells by Ad vectors. TC-1 cells were infected with Ad.GFP and the replication-activated Ad.IR-GFP vector (14) at the indicated MOIs. The percentage of GFP expressing cells was analyzed 24 hours after infection by flow cytometry. Shown are the mean values from three experiments. The standard deviation was less then 10% for all groups.C) Analysis of gp96 expression by Western-blotting with gp96 specific antibodies. Cells were infected with the indicated Ad vectors at an MOI of 100pfu/cell and gp96 expression was analyzed 48 hours later in cell lysates and supernatants. For analysis of cell lysates, a total of 10μg of protein was loaded on 10% polyacrylamide/SDS gels and separated (left panel). For analysis of secreted sgp96, 500μl of culture supernatant was immuno-precipitated using anti-gp96 antibodies and protein A sepharose. The precipitated proteins were separated by polyacrylamide gel electropheresis (right panel). The arrow on the left site marks endogenous (ER-localized) gp96. The stars indicate recombinant sgp96 and mgp6.D) Analysis of mgp96 expression by immunofluorescence. Cells were infected with AdbGal (a control vector), Ad.mgp96, and Ad.IR-E1A/mgp96 as described in B) and analyzed by staining with a rat anti-gp96 antibody and an anti-rat-FITC conjugated antibody. Note the membrane localization of gp96.E) In vitro toxicity of gp96 vectors (crystal violet). TC-1 cells were infected with Ad vectors at the indicated MOIs. Cell viability was assessed by crystal violet staining 9 days after infection.
Transgene expression. As a model for our studies we selected TC-1 cells. TC-1 cells are immortalized murine epithelial cells that stably express HPV-16 E6 and E7 proteins, which can support DNA replication of E1-deleted Ad vectors and serve as easily detectable tumor antigens. Upon subcutaneous transplantation into syngeneic C57Bl/6 mice, TC-1 cells form aggressively growing, vascularized tumors. Infection of TC-1 cells with Ad.GFP at an MOI of 30 pfu per cell conferred transgene expression in nearly 100% of cells (Fig. 1A). The level of viral DNA replication of Ad.IR-GFP in TC-1 cells was ∼10-fold lower than in human tumor cells (data not shown). Consequently transgene (GFP) expression levels in TC-1 cells from replication-activated Ad.IR vectors was significantly lower than from Ad vectors that contained the transgene under the direct control of the RSV promoter (Fig. 1A). This is also reflected in Western blot analyses for gp96 expression in TC-1 cells upon infection with Ad.mgp96, Ad.sgp96, Ad.IR-E1A/mgp96, or Ad.IR.E1A/sgp96 (Fig. 1C). This analysis also confirmed that mgp96 is exclusively found in cell lysates, while the vast majority of sgp96 is secreted and detectable in the supernatant. Furthermore, Figure 1C shows that TC-1 cells express endogenous gp96 at high levels, and that this is not triggered by Ad infection. Membrane-localization of mgp96 expression was demonstrated by immunofluorescence of Ad transduced TC-1 cells using gp96-specific antibodies (Fig. 1D).
Effect of gp96 expression on TC-1 cells in vitro. We first tested in vitro whether expression of gp96 affects TC-1 viability (Fig. 1E). In CPE assays there was no difference in cytotoxicity between control Ad infected and Ad or Ad.IR vectors expressing either gp96. This indicates that gp96 overexpression per se does not exert an anti-tumor effect. Notably, compared to Ad.sgp96 and Ad.mgp96, all vectors containing Ad E1A displayed slightly more cytotoxicity when infected onto TC-1 cells at an MOI of 100pfu/cell.
Gp96-mediated stimulation of anti-tumor immune responses. Next we analyzed the effect of gp96 expression in TC-1 cells on growth of subcutaneous tumors. In an initial study, TC-1 cells were infected ex vivo at an MOI of 100pfu/cell. A total of 2×105 infected cells were then transplanted subcutaneously into C57Bl/6 mice and tumor volume was measured over a period of 25 days (Fig. 2A). While tumor growth of Ad.mgp96 infected TC-1 cells was comparable to mock- and Ad.bGal infected cells, tumors derived from Ad.sgp96 infected cells grew significantly slower (P<0.01). In a first attempt to assess whether gp96 expression can induce an anti-tumor response to TC-1 cells that are not transduced with Ad, we transplanted Ad infected cells together with a mixture (20%) of non-transduced cells (Fig. 2B). The outcome was comparable to that seen with 100% transduced cells. To corroborate these results, we conducted the following vaccination experiment. We injected TC-1 cells transduced ex vivo with Ad vectors, repeated the vaccination two weeks later and challenged the mice with 2×105 untransduced TC-1 cells 14 days after the second vaccination. (In this experiment, vaccination cells were sterilized by γ-irradiation (4000 rad) to avoid outgrowth of tumors while providing gp96 expression). The tumor volume was measured and survival recorded (Fig. 2C). Mice vaccinated with Ad.sgp96 had a slightly better survival but the differences between groups vaccinated with Ad.bGal, Ad.mgp96, and Ad.sgp96 infected cells were not significant (p=0.42 for Ad.bGal vs Ad.sgp96).
Figure 2.
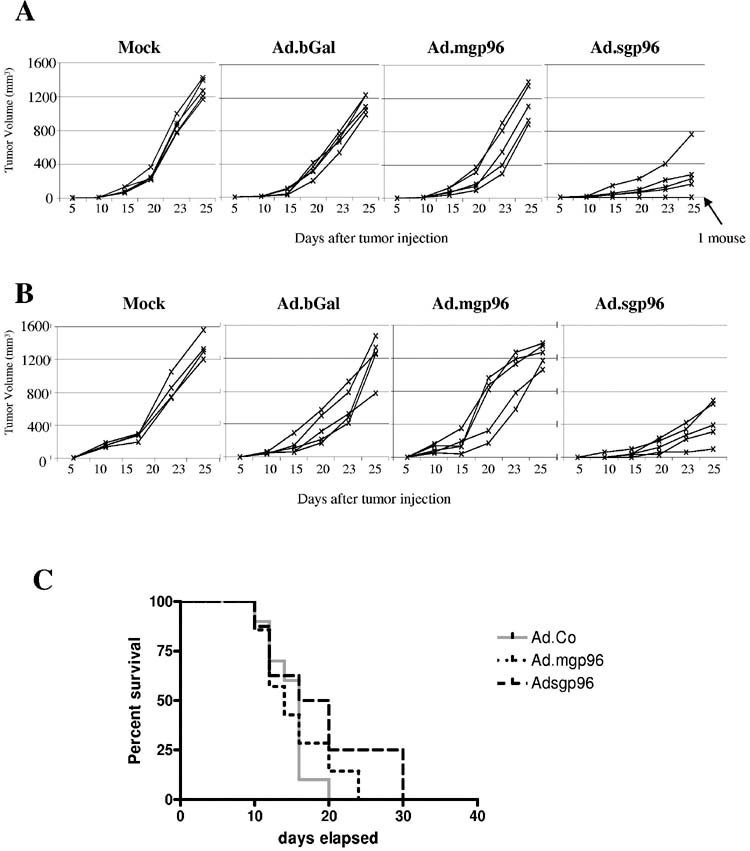
In vivo studies with Ad.mgp96 and Ad.sgp96 to assess anti-tumor immune responses upon gp96 overexpression.A) TC-1 cells were transduced with the indicated Ad vectors at an MOI of 100pfu/cell. Sixteen hours later mock- and Ad-transduced cells were collected, washed and a total of 1×105 cells were subcutaneously injected into C57Bl/6 mice. Tumor volumes were measured at the indicated tumor points. Each line represents one tumor.B) TC-1 cells were transduced as described in A). Transduced cells (8×104) were mixed with 2×104 non-transduced cells and subcutaneously injected into mice.C) TC-1 cells were transduced as in A). Sixteen hours after Ad transduction, cells were collected, washed, and irradiated with 6000 rads. Mice were vaccinated with 1×106-irradiated cells. The vaccination was repeated 14 days later. Ten days after the second vaccination mice were challenged with 2×105 untreated TC-1 cells. Tumor growth was monitored. The time when tumors reached 1000mm3 was recorded and survival was expressed as a Kaplan Meier survival curve. N=5 Each line represents one mouse.
We hypothesized that the failure of vaccination to generate effective protective immune responses could have been due to a suppressive effect of Treg cells that are induced by TC-1 tumors. We therefore studied the presence of Tregs in our model and evaluated whether Tregs can be killed by low-dose CY treatment.
Splenocytes of TC-1 tumor-bearing or naïve mice with and without CY treatment (2mg/mouse) were analyzed by flow cytometry for the presence of CD4+, CD25+, and FoxP3+ cells (Fig. 3). In control mice (without tumors and without CY treatment), the percentage of CD4+, CD4+CD25+, and FoxP3+ cells in the spleen was 18, 2, and 3.2%, respectively. The presence of subcutaneous TC-1 tumors significantly increased the percentage of splenic FoxP3+ and CD25+ cells within the CD4+ cell population in mice that were not treated with CY (p=0.018 for FoxP3+, p=0.05 for CD25+/CD4+ and p=.0.004 for FoxP3+/CD4+) (Fig. 3A, right panel). Four days after CY injection, the percentage of CD4+ in the spleen was higher compared to control mice without CY treatment (Fig. 3A, left panel). However, the amount of Tregs, as the percentage of CD25+ cell within the CD4+ population decreased after CY treatment (Fig. 3A, middle panel) (p=0.006). Furthermore, the percentage of FoxP3+ and FoxP3+/CD4+ cells in the spleen was significantly decreased in CY-treated mice (P<0.000001) (Fig. 3A, right panel). Sections of tumors were analyzed by immunofluorescence for FoxP3 and CD25 (Fig. 3B). These analyses showed markedly less FoxP3+ and CD25+ cells in TC-1 tumors of CY-treated mice. Taken together, our data indicate that TC-1 tumors attract Tregs and increase the number of Tregs in spleen and that low-dose CY treatment specifically kills Tregs.
Figure 3.
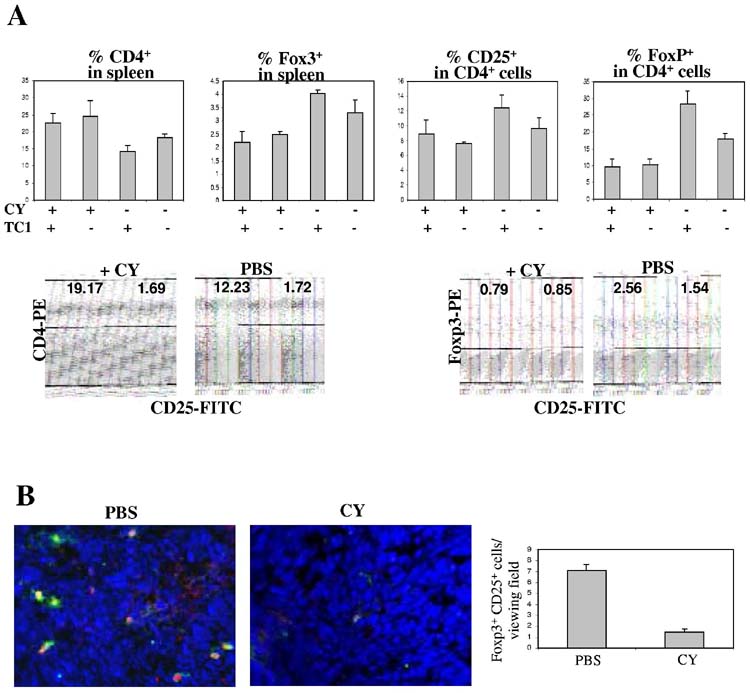
CY treatment reduces the number of Tregs in spleen and tumor.C57Bl/6 mice were injected with TC-1 cells or PBS. Ten days later, half of the mice received an intraperitoneal injection of 2mg cyclophosphamide (CY). At day 4 post CY injection, mice were sacrificed and tumors and spleens were harvested.A) Splenocytes were analyzed by flow cytometry using antiFoxP3–PE, anti-CD4-PE, and anti-CD25-FITC antibodies. Shown is the percentage of CD4+ and FoxP3++ cells as well as the percentage of CD4+ cells that are positive for CD25. Corresponding isotope controls yielded no significant staining.Characteristic analyses of splenocytes from mice with and without CY treatment are shown below the bar graphs.B) Cryosections of TC-1 tumors were analyzed by immunofluorescence. Characteristic sections are shown (magnification: 40x). FoxP3 staining appears red and is mostly localized to the cell center. CD25 staining appears green and mostly localized to the cell periphery. Note that most cells are FoxP3+CD25+. The table on the right shows the average number of positive cells per viewing field (from 10 random fields).
Having established that low-dose CY treatment can eliminate Tregs in spleen and tumors, we performed the following vaccination study: Mice were injected with TC-1 cells. When subcutaneous tumors reached measurable size, half of the mice were injected with low-dose CY or PBS, followed by cell vaccinations 4 days later. Cell based vaccines were TC-1 cells mock-treated, infected ex vivo with a control Ad vector expressing GFP (Ad.Co) and with Ad.mgp96 and Ad.sgp96 at an MOI of 500pfu/cell. No significant difference was seen between the groups of mice that were not treated with low-dose CY (Fig. 4A). CY injection alone slightly accelerated tumor growth (Fig. 4B “Mock” + CY). Importantly, tumor growth was greatly delayed in mice vaccinated with Ad infected TC-1 cells (Fig. 4B), whereby there was a significantly stronger delay in tumor growth (and prolonged survival) in mice vaccinated with sgp96 expressing TC-1 cells (p<0.01 for Ad.Co+CY vs Ad.sgp96+CY) (Figs. 4C and D). A similar outcome was seen in studies performed with Ad.IR vectors expressing sgp96 (data not shown).
Figure 4.
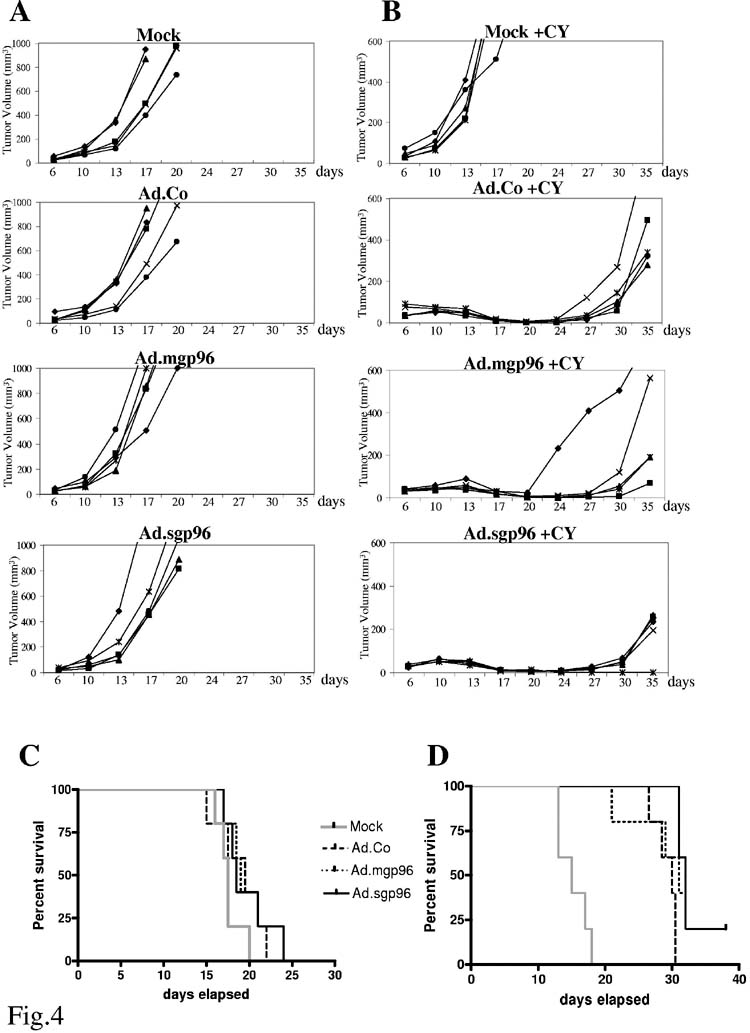
Effect of cyclophosphamide upon vaccination with TC-1 cells that overexpress gp96.C57Bl/6 mice were subcutaneously injected with 2×104 TC-1 cells. When tumors reached a diameter of 2mm, one group of mice (B) received an intraperitoneal injection of 2mg cyclophosphamide (CY). The other group (A) received PBS injections. Four days later, mice were vaccinated with 1×106 TC-cells, mock-infected or infected ex vivo with Ad.Co (a bGal-expressing first-generation vector), Ad.mgp96, or Adsgp96 at an MOI of 500 pfu/cell. Growth of primary tumors was recorded over a period of 35 days. Mice were sacrificed when tumors reached a volume of 1000mm3 (for PBS injected groups) (C) or 200mm3 (for CY treated groups) and survival was recorded (D). The relatively high MOI of 500pfu/cell was chosen for TC-1 cell infection to inhibit outgrowth of vaccination tumors. Notably, Ad.Co infection at this MOI caused cell death within 5 days in cultured TC-1 cells.N=5 (Each line represents one mouse/tumor).
In conclusion, the secreted form of sgp96 is able to induce an anti-tumor immune response that slows outgrow of wild-type subcutaneous tumors. This effect can be greatly enhanced by pre-injection of low-dose CY which has the potential to selectively kill regulatory T-cells.
Induction of an immune response of TC-1 cells infected with Ad.IR-E1A/TRAIL. One of the goals of our study was to test whether Ad.IR-E1A/TRAIL-mediated oncolysis in combination with sgp96 expression triggered synergistic anti-tumor immune responses. Towards this goal we evaluated first whether vaccination with TC-1 cells infected with Ad.IR-E1A/TRAIL can protect against tumor cell challenge. Upon infection of cultured TC-1 cells at an MOI of 100pfu/cell, the oncolytic vector, Ad.IR.E1A/TRAIL, efficiently killed TC-1 cells by apoptosis as verified in capase 3 activation assays (data not shown). Vaccination studies were conducted as described for Figure 4. Cell based vaccines were TC-1 cells infected ex vivo with a control Ad vector or Ad.IR-E1A/TRAIL at an MOI of 500pfu/cell (Fig. 5). (For mock controls, see Figure 4). No significant difference was seen between the two groups of mice that were not treated with CY. Importantly, in 3 out of 5 CY-treated mice vaccinated with Ad.IR-E1A/TRAIL infected TC-1 cells, tumor growth was completely suppressed for 30 days. Suppression of tumor growth upon Ad.IR-E1A/TRAIL infection was significantly greater than upon Ad.Co infection, indicating that TC-1 cells that die by TRAIL mediated apoptosis can act as strong vaccines. Interestingly, TC-1 cells infected with Ad.Co at an MOI of 500 pfu/cell also demonstrated a vaccination effect in CY-treated mice, which could be due to expression of viral proteins and/or virus mediated cell death.
Figure 5.
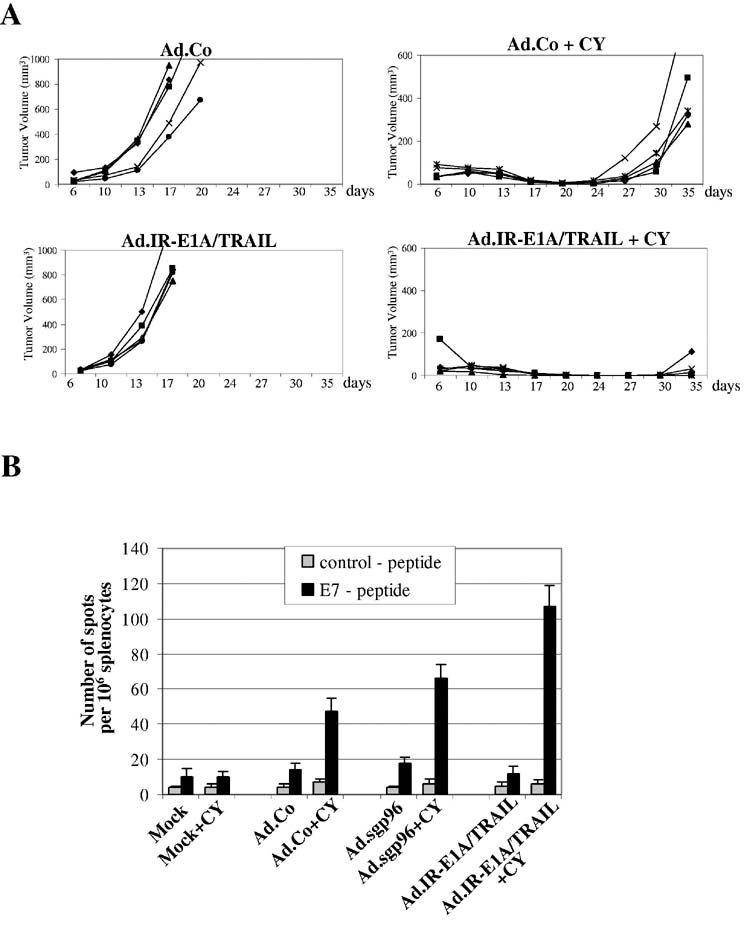
Induction of immune response upon oncolysis with Ad.IR-E1A/TRAIL.A) Mice were subcutaneously injected with 2×104 TC-1 cells. When tumors reached a diameter of 2mm, one group of mice (right panel) received an intraperitoneal injection of 2mg cyclophosphamide (CY). The other group (left panel) received PBS injections. Four days later, mice were vaccinated with 1×106 TC-1 cells, mock-infected or infected ex vivo with Ad.Co or Ad.IR-E1A/TRAIL at an MOI of 500 pfu/cell. Growth of primary tumors was recorded over a period of 35 days. N=5B) Analysis of frequencies of IFNγ-producing E7 specific T-cells. Shown are the frequencies of IFNγ producing T-cells specific to the HPV16 E749-57 carrying the H-2Db restricted peptide (RAHYNIVTF) or an unrelated control peptide. N=3 animals per group.
To further characterize anti-tumor immune responses seen in Figures 4 and 5A, splenocytes of vaccinated mice were analyzed by ELIspot assay for the frequency of IFNγ producing T-cells specific to the HPV16 E749-57 peptide (RAHYNIVTF) carrying an H-2Db restricted epitope (35) (Fig. 5B). Overall, this study indicated that the number of E7 specific T-cells correlated with the tumor-destructive immune responses described above. The fraction of E7-specific IFN-γ producing cells is larger in CY-treated animals vaccinated with Ad.IR-E1A/TRAIL or Ad.sgp96 transduced TC-1 cells than with mock- or Ad.Co-infected TC-1 cells.
In summary, the oncolytic vector, Ad.IR-E1A/TRAIL, that induces TRAIL mediated apoptosis in the context of Ad infection, can trigger therapeutic T-cell responses if mice are pre-treated with low-dose CY.
Analysis of in vitro synergy of gp96 expression and viral oncolysis. A recent report suggested that overexpression of HSP70 supports the oncolytic effect of a replication-competent human Ad vector in vitro (38). To evaluate whether we would see a similar effect in our system, we infected TC-1 cells with Ad.IR vectors expressing E1A + gp96, E1A + TRAIL, and E1A + alkaline phosphatase (AP) (as a control). The oncolytic vector Ad.IR.E1A/TRAIL efficiently killed TC-1 cells (Fig. 6A). When combined with Ad.IR-E1A/sgp96, however, we could not detect a stronger cytolytic effect of Ad.IR-E1A/TRAIL or Ad.IR-E1A/AP in this cell line (Fig. 6A) as well as in several other cell lines tested (data not shown). The discrepancy with the study by Haviv et al. might be due to the use of different HSP's (hsp70 vs gp96) or due to different HSP expression levels.
Figure 6.
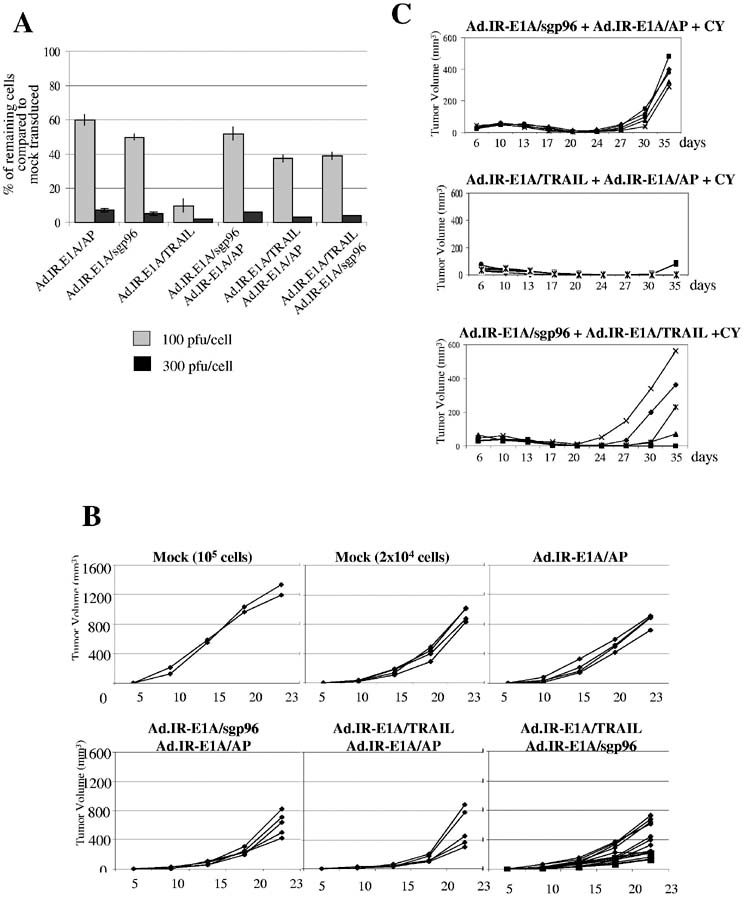
In vivo analysis of a potential synergy between Ad.IR-E1A/sgp96 and Ad.IR-E1A/TRAIL.A) Cytolytic effect of the oncolytic Ad vector Ad.IR-E1A/TRAIL upon infection of TC-1 cells alone or in combination with gp96 expressing Ad vectors. Confluent TC-1 cells (in 24 well plates) were infected at the indicated MOIs and cytotoxicity was assessed by crystal violet staining three days after infection. The remaining (stained) cells were destained with 20% acetic acid, lysed and the absorbance of the lysate was measured at OD570. Results were expressed as % of cells remaining compared to untreated controls. (If two vectors were used for infection, the MOI of each vector was 50 or 150pfu per cell, respectively.) N=3B) TC-1 cells were transduced at a total MOI of 500 pfu per cell. Sixteen hours later Adtransduced cells were harvested, washed and 8×104 transduced cells mixed with 2×104 nontransduced cells were subcutaneously injected into mice. As controls, two groups of mice received 8×104 or 2×104 non-transduced cells. Tumor growth was monitored at the indicated time points. Each line represents one tumor. If two vectors were used, the MOI of each vector was 150pfu/cell.C) Mice were subcutaneously injected with 2×104 TC-1 cells. When tumors reached a diameter of 2mm, all mice received an intraperitoneal injection of 2mg cyclophosphamide (CY). Four days later, mice were vaccinated with 1×106 TC-cells, mock-infected or infected ex vivo with the indicated Ad vectors at a total MOI of 500 pfu/cell. Growth of primary tumors was recorded over a period of 35 days. N=5, except for the Ad.IR-E1A/TRAIL+Ad/IR-E1A/sgp96 group, where 10 mice were analyzed. Each line represents one tumor.
Analysis of anti-tumor synergy between Ad.IR-E1A/sgp96 and Ad.IR-E1A/TRAIL. To study a potential synergy between sgp96 expression and viral oncolysis in inducing anti-tumor immune responses, we performed two studies. First, we injected mice with a mixture of TC-1 cells infected ex-vivo with Ad vectors and non-infected cells (80:20) and measured tumor outgrowth (Fig. 6B). Cells were infected at a total MOI of 500pfu/cell with Ad.IR-E1A/AP, Ad.IR-E1A/sgp96+Ad.IR-E1A/AP, Ad.IR-E1A/TRAIL+Ad.IR-E1A/AP, and Ad.IR-E1A/sgp96+Ad.IR-E1A/TRAIL (Fig. 6B). While the combination of the oncolytic vector and the sgp96 expressing vector slightly slowed tumor growth, this effect was not significant compared to the other groups (Ad.IR-E1A/sgp96+Ad.IR-E1A/TRAIL vs Ad.IR-E1A/sgp96+Ad.IR-E1A/AP: p=0.021; Ad.IR-E1A/sgp96+Ad.IR-E1A/TRAIL vs Ad.IR-E1A/TRAIL+Ad.IR-E1A/AP: p=0.052). Secondly, in a vaccination scheme, TC-1 cells were infected ex vivo at an MOI of 500 pfu/cell with Ad.IR-E1A/sgp96+Ad.IR-E1A/AP, Ad.IR-E1A/TRAIL+Ad.IR-E1A/AP, and Ad.IR-E1A/sgp96+Ad.IR-E1A/TRAIL and injected into mice with pre-established TC-1 tumors 4 days after treatment with low-dose CY (Fig. 6C). As in Figure 5, Ad.IR-E1A/TRAIL mediated oncolysis had a strong vaccination effect. Surprisingly, sgp96 expression reduced the vaccination effect of Ad.IR-E1A/TRAIL infected TC-1 cells.
In conclusion, TC-1 cells infected with Ad.IR-E1A/sgp96induce a tumor-destructive immune response in CY-treated mice. This immune response was not increased by additional infection with Ad.IR-E1A/TRAIL.
Discussion
The goal of this study was to overexpress an HSP from Ad vectors in tumor cells and test whether this alone or in combination with TRAIL-mediated tumor cell apoptosis (from Ad.IR-E1A/TRAIL) would stimulate an anti-tumor immune response. As outlined earlier, we hypothesized that HSP would efficiently chaperone cryptic tumor cells antigens (released upon oncolysis) to APCs and that this in the context of Ad-induced release of proinflammatory cytokines would induce a strong T-cell response.
Based on two earlier reports demonstrating the successful application of a gp96 genetically modified to be displayed on the surface of expressing cells for cancer immunotherapy (4), we focused on gp96 as a test HSP. In addition, to a membrane-anchored gp96, we also expressed a secreted form of gp96 to enable presentation of tumor antigens to APCs that are not directly in contact with infected tumor cells. It is known that endogenous gp96 is abundant in tumor cells and our data shown in Figure 1C confirm this for TC-1 cells. Furthermore, endogenous gp96 expression could theoretically be further upregulated by Ad infection and/or E1A expression from Ad.IR-E1A/TRAIL (37). In spite of this, we found that TC-1 cells mock-infected or infected with control vectors (Ad.bGal or Ad.IR-E1A/AP) could not induce anti-tumor immune responses in therapy or vaccination schemes (Figs. 4 and 6B). Furthermore, we were unable to detect a significant anti-tumor effect if a membrane-anchored form of gp96 was expressed. However, the secreted gp96 form when overexpressed from an Ad vector could induce an in vivo response capable of significantly delaying tumor growth. Several reasons could account for the lack of a response upon Ad.mgp96 treatment. The secreted gp96 form might facilitate the transport of tumor antigens to regional lymph nodes and its uptake by APCs, while membrane gp96 can only interact with APCs residing in the tumor. Furthermore, as outlined in more detail below, TC-1 cells attract Tregs [through expression of specific cytokines as suggested for human tumors (39)] and these tumor associated Tregs could interfere with T-cell activation at the tumor site.
We also reasoned that it would be beneficial if a secreted from of gp96 is exclusively expressed by tumor cells that undergo apoptosis and release antigen, which, with gp96 acting as a chaperone, is taken up by APCs. We therefore employed the replication activated Ad.IR system that selectively activates transgene expression in tumor cells. Although TC-1 cells support replication of E1-deleted Ad vectors and allow for transgene expression from Ad.IR vectors, the levels of both replication and transgene expression are lower than in human cells. This is not surprising as mouse cells are less permissive for Ad replication than human cells. [While 7 days after infection with first generation adenovirus at an MOI of 500pfu/cell, about 5,000 pfu/cell of de novo produced Ad particles were found in HeLa cell lysates, less than 5 pfu were found in TC-1 cells (data not shown)]. Therefore, experiments performed with human replicating Ad vectors, including Ad.IR vectors, in mouse tumor cells underestimate the potential that the same vectors might have in human tumor cells. Overall, however, the ability of sgp96 expressed from Ad.gp96 or Ad.IR-E1A/gp96 to stimulate an effective prophylactic vaccination in the TC-1 cell model was disappointing compared to studies with HSP's in other tumor models (4). One explanation for this outcome is that a T-cell response against TC-1 cells is suppressed in vivo. Several mechanisms could account for this, including the activation of Tregs (21, 22). A role of Tregs is indicated by our studies showing that i) TC-1 tumor transplantation increases the number of splenic Tregs, ii) infiltrating Tregs are present in TC-1 tumors, iii) low-dose CY treatment decreases the number of tumor infiltrating and splenic Tregs, and iv) CY treatment greatly enhances the effect of sgp96 to trigger a protective immune response. Although it is thought that low-dose CY acts through a mechanism of selective toxicity against Tregs (40), other effects of low-dose CY on T cell homeostasis are likely. Notably, in the period of observation (5 weeks) we did not observe signs of autoimmunity such as hypopigmentation of the coat.
Our hypothesis, that immunogenicity of tumor cells can be increased by viral oncolysis goes back to studies by Lindenmann and Klein in 1967 (41). The underlying mechanisms, include the release of pro-inflammatory cytokines upon Ad uptake into tumor cells or cells of the immune system (including APCs and macrophages), the ability to release cryptic antigens as apoptotic bodies, and the co-expression of viral proteins in tumor cells which are potent immunogens. (CTL specific to Ad proteins might help liberate tumor antigens for cross-priming of APCs.) A number of studies have supported this hypothesis (12-14). We demonstrated here that a vaccination effect of Ad.IR-E1A/TRAIL can be achieved by treatment of mice with low-dose CY and that cells that die from Ad.IR-E1A/TRAIL infection stimulate a stronger immune response than cells that die from Ad-mediated toxicity (after Ad.Co infection). The latter indicates that TRAIL-mediated apoptosis (in the context of Ad infection) is able to release antigens in a more immunogenic form.
Earlier studies reported a synergistic effect of hsp70 and viral oncolysis (42, 43). In our experiment we did not see an additive effect of sgp96 expression and Ad.IR-E1A/TRAIL mediated oncolysis. The reason for the finding that tumor cells, which overexpress sgp96 and die by apoptosis are less immunogenic is unclear. We speculate that the combination of antigen released by apoptosis and antigen chaperoned to APC by sgp96 may overload the ability of the APC in regional lymph nodes to process and present antigen. There may be a relative lack of maturation signals when more antigen is present in immature APCs and this might lead to tolerance/anergy [for a review see (44)]. The “overload” hypothesis is further supported by studies with recombinant gp96 and tumor derived peptides showing that that the immunostimulatory effect of gp96 is dose dependent; low doses of gp96 generate immunity, while doses 10 times the immunizing dose do not (45). Clearly, more experiments with vectors that express different sgp96 levels and studies in other tumor models are required to support our preliminary observation of a lack of synergy between Ad.IR-E1A/sgp96 and Ad.IR.E1A/TRAIL. The conflict between our data and data by Chen's group (42, 43) might also be related to the use of different heat-shock proteins (hsp70 vs gp96). These HSP's can use different receptors to mediate their effects. Hsp70 binds to CD14 or Toll-like receptors which may function to enhance innate immunity via APC activation, while gp96 binds to CD91 which may interfere with phagocytosis of apoptotic cells (46).
In conclusion, this study shows that a secreted form of gp96 has immunostimulatory activity. Our data indicate that the effect of viral oncolysis and Ad-mediated gp96 expression depends on the tumor cell type and the mechanism by which tumor cells die. The finding that an oncolytic or immunostimulatory vector in combination with cyclophospamide treatment induces an immune response that is able to eliminate pre-established tumors has important implications for tumor gene therapy trials.
References
- 1.Asea A, Kraeft SK, Kurt-Jones EA, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6(4):435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 2.Srivastava PK, Amato RJ. Heat shock proteins: the ‘Swiss Army Knife’ vaccines against cancers and infectious agents. Vaccine. 2001;19(1719):2590–2597. doi: 10.1016/s0264-410x(00)00492-8. [DOI] [PubMed] [Google Scholar]
- 3.Yamazaki K, Nguyen T, Podack ER. Cutting edge: tumor secreted heat shock-fusion protein elicits CD8 cells for rejection. J Immunol. 1999;163(10):5178–5182. [PubMed] [Google Scholar]
- 4.Zheng H, Dai J, Stoilova D, Li Z. Cell surface targeting of heat shock protein gp96 induces dendritic cell maturation and antitumor immunity. J Immunol. 2001;167(12):6731–6735. doi: 10.4049/jimmunol.167.12.6731. [DOI] [PubMed] [Google Scholar]
- 5.Kruyt FA, Curiel DT. Toward a new generation of conditionally replicating adenoviruses: pairing tumor selectivity with maximal oncolysis. Hum Gene Ther. 2002;13(4):485–495. doi: 10.1089/10430340252809784. [DOI] [PubMed] [Google Scholar]
- 6.Steinwaerder DK, Carlson CA, Lieber A. DNA replication of first-generation adenovirus vectors in tumor cells. Human Gene Therapy. 2000;11:1933–1948. doi: 10.1089/10430340050129549. [DOI] [PubMed] [Google Scholar]
- 7.Steinwaerder DS, Carlson CA, Lieber A. Human papilloma virus E6 and E7 proteins support DNA replication of adenoviruses deleted for the E1A and E1B genes. Mol Ther. 2001;4(3):211–216. doi: 10.1006/mthe.2001.0447. [DOI] [PubMed] [Google Scholar]
- 8.Steinwaerder DS, Carlson CA, Otto DL, Li ZY, Ni S, Lieber A. Tumor-specific gene expression in hepatic metastases by a replication- activated adenovirus vector. Nat Med. 2001;7(2):240–243. doi: 10.1038/84696. [DOI] [PubMed] [Google Scholar]
- 9.Sova P, Ni S, Ren X-W, Bernt K, Lieber A. Efficacy of tumor-targeted and conditionally replicating oncolytic adenovirus expressing Apo2/TRAIL in treatment of liver metastases in a mouse xenograft model. Molecular Therapy. 2004;9:496–509. doi: 10.1016/j.ymthe.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Ni S, Bernt K, Gaggar A, Li ZY, Kiem HP, Lieber A. Evaluation of biodistribution and safety of adenovirus vectors containing group B fibers after intravenous injection into baboons. Hum Gene Ther. 2005;16(6):664–677. doi: 10.1089/hum.2005.16.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muruve DA, Barnes MJ, Stillman IE, Libermann TA. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum Gene Ther. 1999;10(6):965–976. doi: 10.1089/10430349950018364. [DOI] [PubMed] [Google Scholar]
- 12.Hallden G, Hill R, Wang Y, et al. Novel immunocompetent murine tumor models for the assessment of replication-competent oncolytic adenovirus efficacy. Mol Ther. 2003;8(3):412–424. doi: 10.1016/s1525-0016(03)00199-0. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Hallden G, Hill R, et al. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat Biotechnol. 2003;21(11):1328–1335. doi: 10.1038/nbt887. [DOI] [PubMed] [Google Scholar]
- 14.Bernt K, Ni S, Tieu A, Lieber A. Assessment of a combined, adenovirus-mediated and immo-stimulatory tumor therapy. Cancer Research. 2005:4343–4352. doi: 10.1158/0008-5472.CAN-04-3527. [DOI] [PubMed] [Google Scholar]
- 15.Courreges MC, Benencia F, Conejo-Garcia JR, Zhang L, Coukos G. Preparation of apoptotic tumor cells with replication-incompetent HSV augments the efficacy of dendritic cell vaccines. Cancer Gene Ther. 2005 doi: 10.1038/sj.cgt.7700888. [DOI] [PubMed] [Google Scholar]
- 16.Gough MJ, Melcher AA, Ahmed A, et al. Macrophages orchestrate the immune response to tumor cell death. Cancer Res. 2001;61(19):7240–7247. [PubMed] [Google Scholar]
- 17.Barker RN, Erwig LP, Hill KS, Devine A, Pearce WP, Rees AJ. Antigen presentation by macrophages is enhanced by the uptake of necrotic, but not apoptotic, cells. Clin Exp Immunol. 2002;127(2):220–225. doi: 10.1046/j.1365-2249.2002.01774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fadok VA, Bratton DL, Guthrie L, Henson PM. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J Immunol. 2001;166(11):6847–6854. doi: 10.4049/jimmunol.166.11.6847. [DOI] [PubMed] [Google Scholar]
- 19.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390(6658):350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 20.Morelli AE, Larregina AT, Shufesky WJ, et al. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood. 2003;101(2):611–620. doi: 10.1182/blood-2002-06-1769. [DOI] [PubMed] [Google Scholar]
- 21.Maeda A, Schwarz A, Kernebeck K, et al. Intravenous infusion of syngeneic apoptotic cells by photopheresis induces antigen-specific regulatory T cells. J Immunol. 2005;174(10):5968–5976. doi: 10.4049/jimmunol.174.10.5968. [DOI] [PubMed] [Google Scholar]
- 22.Mahnke K, Knop J, Enk AH. Induction of tolerogenic DCs: ‘you are what you eat’. Trends Immunol. 2003;24(12):646–651. doi: 10.1016/j.it.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Glaser M. Augmentation of specific immune response against a syngeneic SV40-induced sarcoma in mice by depletion of suppressor T cells with cyclophosphamide. Cell Immunol. 1979;48(2):339–345. doi: 10.1016/0008-8749(79)90128-x. [DOI] [PubMed] [Google Scholar]
- 24.Polak L, Geleick H, Turk JL. Reversal by cyclophosphamide of tolerance in contact sensitization. Tolerance induced by prior feeding with DNCB. Immunology. 1975;28(5):939–942. [PMC free article] [PubMed] [Google Scholar]
- 25.Rollinghoff M, Starzinski-Powitz A, Pfizenmaier K, Wagner H. Cyclophosphamide-sensitive T lymphocytes suppress the in vivo generation of antigen-specific cytotoxic T lymphocytes. J Exp Med. 1977;145(2):455–459. doi: 10.1084/jem.145.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida S, Nomoto K, Himeno K, Takeya K. Immune response to syngeneic or autologous testicular cells in mice. I. Augmented delayed footpad reaction in cyclophosphamide-treated mice. Clin Exp Immunol. 1979;38(2):211–217. [PMC free article] [PubMed] [Google Scholar]
- 27.North RJ. Down-regulation of the antitumor immune response. Adv Cancer Res. 1985;45:1–43. doi: 10.1016/s0065-230x(08)60265-1. [DOI] [PubMed] [Google Scholar]
- 28.Beyer M, Kochanek M, Darabi K, et al. Reduced frequencies and suppressive function of CD4+ CD25high regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005 doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- 29.Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34(2):336–344. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- 30.Ikezawa Y, Nakazawa M, Tamura C, Takahashi K, Minami M, Ikezawa Z. Cyclophosphamide decreases the number, percentage and the function of CD25(+)CD4(+) regulatory T cells, which suppress induction of contact hypersensitivity. J Dermatol Sci. 2005;39(2):105–112. doi: 10.1016/j.jdermsci.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105(7):2862–2868. doi: 10.1182/blood-2004-06-2410. [DOI] [PubMed] [Google Scholar]
- 32.Bernt KM, Liang M, Ye X, et al. A new type of adenovirus vector that utilizes homologous recombination to achieve tumor-specific replication. J. Virol. 2002;76:10994–11002. doi: 10.1128/JVI.76.21.10994-11002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shayakhmetov DM, Li ZY, Gaggar A, et al. Genome size and structure determine efficiency of postinternalization steps and gene transfer of capsid-modified adenovirus vectors in a cell-type-specific manner. J Virol. 2004;78(18):10009–10022. doi: 10.1128/JVI.78.18.10009-10022.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hontsu S, Yoneyama H, Ueha S, et al. Visualization of naturally occurring Foxp3+ regulatory T cells in normal and tumor-bearing mice. Int Immunopharmacol. 2004;4(14):1785–1793. doi: 10.1016/j.intimp.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 35.Feltkamp MC, Smits HL, Vierboom MP, et al. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur J Immunol. 1993;23(9):2242–2249. doi: 10.1002/eji.1830230929. [DOI] [PubMed] [Google Scholar]
- 36.Liu B, Dai J, Zheng H, Stoilova D, Sun S, Li Z. Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc Natl Acad Sci U S A. 2003;100(26):15824–15829. doi: 10.1073/pnas.2635458100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melcher A, Murphy S, Vile R. Heat shock protein expression in target cells infected with low levels of replication-competent virus contributes to the immunogenicity of adenoviral vectors. Hum Gene Ther. 1999;10(9):1431–1442. doi: 10.1089/10430349950017770. [DOI] [PubMed] [Google Scholar]
- 38.Haviv YS, Blackwell JL, Li H, Wang M, Lei X, Curiel DT. Heat shock and heat shock protein 70i enhance the oncolytic effect of replicative adenovirus. Cancer Res. 2001;61(23):8361–8365. [PubMed] [Google Scholar]
- 39.Iellem A, Mariani M, Lang R, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2001;194(6):847–853. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J.Exp. Med. 2004;200:771–782. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindenmann J, Klein PA. Viral oncolysis: increased immunogenicity of host cell antigen associated with influenza virus. J Exp Med. 1967;126(1):93–108. doi: 10.1084/jem.126.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang XF, Ren W, Rollins L, et al. A broadly applicable, personalized heat shock protein-mediated oncolytic tumor vaccine. Cancer Res. 2003;63(21):7321–7329. [PubMed] [Google Scholar]
- 43.Ren W, Strube R, Zhang X, Chen SY, Huang XF. Potent tumor-specific immunity induced by an in vivo heat shock protein-suicide gene-based tumor vaccine. Cancer Res. 2004;64(18):6645–6651. doi: 10.1158/0008-5472.CAN-04-1084. [DOI] [PubMed] [Google Scholar]
- 44.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 45.Chandawarkar RY, Wagh MS, Srivastava PK. The dual nature of specific immunological activity of tumor-derived gp96 preparations. J Exp Med. 1999;189(9):1437–1442. doi: 10.1084/jem.189.9.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng H, Zeng Y, Graner MW, Likhacheva A, Katsanis E. Exogenous stress proteins enhance the immunogenicity of apoptotic tumor cells and stimulate antitumor immunity. Blood. 2003;101(1):245–252. doi: 10.1182/blood-2002-05-1580. [DOI] [PubMed] [Google Scholar]
- 47.Carlson CA, Steinwaerder DS, Stecher H, Shayakhmetov DM, Lieber A. Rearrangements in adenoviral genomes mediated by inverted repeats. Methods Enzymol. 2002;346:277–292. doi: 10.1016/s0076-6879(02)46061-2. [DOI] [PubMed] [Google Scholar]