

Abstract
The neuropeptide galanin exhibits anticonvulsant effects in experimental epilepsy. Two galanin receptor subtypes, GalR1 and GalR2, are present in the brain. We examined the role of GalR1 in seizures by studying the susceptibility of GalR1 knockout (KO) mice to status epilepticus (SE) and accompanying neuronal injury.
SE was induced in GalR1 KO and wild type (WT) mice by Li-Pilocarpine, 60 min electrical perforant path stimulation (PPS), or systemic kainic acid (KA). Seizures were analyzed using Harmonie software. Cell injury was examined by FluoroJade B- and terminal deoxynucleotidyl transferase-mediated UTP nick end labeling; neurogenesis was studied using bromodeoxyuridine labeling.
Compared to WT littermates, GalR1 KO showed more severe seizures, more profound injury to the CA1 pyramidal cell layer, as well as injury to hilar interneurons and dentate granule cells upon Li-pilocarpine administration. PPS led to more severe seizures in KO, as compared to WT mice. No difference in the extent of neuronal degeneration was observed between the mice of two genotypes in CA1 pyramidal cell layer; however in contrast to WT, GalR1 KO developed mild injury to hilar interneurons on the side of PPS. KA-induced seizures did not differ between GalR1 KO and WT animals, and led to no injury to the hippocampus in either of experimental group.
No differences were found between KO and WT mice in both basal and seizure-induced neuronal progenitor proliferation in all seizure types. Li-Pilocarpine led to more extensive glia proliferation in GalR1 KO than in WT, and in both mouse types in two other SE models.
In conclusion, GalR1 mediate galanin protection from seizures and seizure-induced hippocampal injury in Li-Pilocarpine and PPS models of limbic SE, but not under conditions of KA-induced seizures. The results justify the development and use of GalR1 agonists in the treatment of certain forms of epilepsy.
Keywords: Hippocampus, cell death, pilocarpine, kainic acid, perforant path simulation
Galanin is a neuropeptide with potent anticonvulsant and, as was suggested recently, neuroprotective effects. Both natural and synthetic galanin receptor (GalR) agonists attenuated seizures in several epilepsy models (Bartfai et al., 2004; Mazarati et al., 1992; 1998; Saar et al., 2002)). Mice with functional disruption of galanin gene had increased susceptibility to seizures in experimental models of epilepsy, while mice overexpressing galanin exhibited increased seizure resistance (Kokaia et al., 2001; Mazarati et al., 2000). Transfection of neurons with an adeno-associated virus vector cloned with galanin-encoding sequences mitigated chronic epilepsy which resulted from an episode of status epilepticus (SE) (Haberman et al., 2003; Lin et al., 2003). Both anticonvulsant and putative neuroprotective properties of galanin likely involve inhibition of the release of excitatory neurotransmitter glutamate from neuronal presynaptic terminals (Mazarati et al., 2000; Zini et al., 1993a;b), and have been best described in models of temporal lobe epilepsy, in which epileptic focus is located in the hippocampus.
Three GalR subtypes, belonging to a superfamily of G-protein coupled receptors, have been cloned (Branchek et al., 2000), and two of them GalR1 and GalR2 have been identified in the brain (Branchek et al., 2000; Depczynski et al., 1998; Gustafson et al., 1996; O’ Donnel et al., 1999). Poor pharmacology of GalR complicates studies of their distinctive role in seizures. Only two preferential GalR2 agonists are currently available (Branchek et al., 2000; Hua et al., 2004; Jureus et al., 1997; Kerekes et al., 2003; Liu et al., 2001; Mazarati et al., 2004), while neither selective GalR1 agonists, nor GalR1 and GalR2 antagonists exist. However, genetic and knockdown approaches implicated both GalR subtypes in mediating anticonvulsant effects of galanin. Thus, in vivo administration of peptide nucleic acid antisense targeted at GalR2 into the hippocampus, increased the severity of SE induced by perforant path stimulation in rats (Mazarati et al., 2004). On the other hand, mice of C56bl/6j:129/sv mixed background lacking GalR1 displayed spontaneous seizures with 25% penetrance (Jacoby et al., 2002a;b), as well as changes in the expression of a number of neuropeptides, which has been implicated in seizure regulation (Fetissov et al., 2003). The latter observations, however did not offer definitive conclusions on the contribution of GalR1 in seizure control. The fact that the majority of GalR1 knockout (KO) mice (75%) did not exhibit spontaneous seizure phenotype questions whether this receptor subtype is critical in mediating anticonvulsant properties of galanin. On the other hand, the absence of spontaneous seizures does not mean, that GalR1 do not contribute to seizure control. For instance, galanin KO mice did not exhibit spontaneous seizures, however they had increased susceptibility to seizures induced by pentylenetetrazole, kainic acid (KA), and perforant path stimulation (PPS) (6). Similarly, neither Neuropeptide Y, nor Y5 KO mice exhibited spontaneous seizures, but showed decreased seizure threshold in KA-induced seizure test (Baraban et al., 1997; Marsh et al., 1999).
It has not been studied, whether GalR1 KO animals are more prone to epileptic insult, than their wild type (WT) littermates. Therefore, we examined patterns of seizures and seizure-induced neuronal injury to the hippocampus in GalR1 KO mice which had not shown spontaneous seizures, in experimental models of limbic SE. We compared GalR1 KO with their WT littermates in three commonly used models of SE, in which seizures were induced by LiCl and pilocarpine (Li-pilocarpine) (Shilbey and Smith., 2002), PPS with electrical current (PPS) (Mazarati et al., 2000), and systemic KA (Mazarati et al., 2000; McKhan et al., 2003).
In addition, considering recently suggested regulatory role of galanin and GalR2 in seizure-induced neurogenesis in the hippocampus (Mazarati et al., 2004), we studied both basal and postSE neurogenesis (Gould and Tanapat, 1997; Parent et al., 1997) in the dentate gyrus in GalR1 KO and WT mice.
Experimental procedures.
Animals. GalR1 KO mice were originally generated at the Gavan Institute, Sydney, Australia as previously described (Jacoby et al., 2002a,b), where they were backcrossed into the C57BL/6J line for five generations. The line was then re-derived at the Jackson Laboratory (Bar Harbor, ME) and backcrossed into C57BL/6J for two additional generations. We used male homozygous GalR1 KO mice and their WT littermates derived from the crossing of heterozygous breeding pairs. The animals were housed (four per cage) at a 12 hr light/dark cycle with ad libitumaccess to water and food. All experiments were approved by West Los Angeles Institutional Animal Care and Use Committee, and were carried out in accordance with the NIH Guide for the Care and Use of Laboratory animals.
Surgery. Under Isoflurane anesthesia animals were implanted with the recording electrode (Plastics One, Roanoke, VA, USA) into dorsal hippocampus (1.8 mm posterior, and 1.5 mm left from Bregma, 2 mm deep from brain surface). The animals to be subjected to PPS were also implanted with a stimulating electrode (Plastics One) into the left perforant path (0.5 mm anterior and 2.5 mm left from Lambda, 2 mm deep from brain surface, (Mazarati et al., 2000). Electrodes were fixed to the bone with a Michel wound clip and dental cement.
Induction and analysis if status epilepticus. SE was induced by Li-Pilocarpine in 4 WT and 5 GalR1 KO mice (Shilbey and Smith, 2002), PPS in 4 WT and 5 GalR1 KO mice (6), or systemic KA in 4 WT and 4 GalR1 KO mice (Mazarati et al., 2000; McKhann et al., 2003). Since some of GalR1 KO animals were reported to have spontaneous seizures (Jacoby et al., 2002a;b; Fetissov et al., 2003), prior to SE induction all animals had been subjected to electrographic recording for 1 week in order to identify spontaneous seizures. For Li-Pilocarpine SE, animals were injected intraperitoneally. with 3 mEq/kg LiCl, followed 16-24 hrs later with subcutaneous pilocarpine HCl, 150 mg/kg (both from Sigma, St. Louis, MO, USA). Self-sustaining SE was induced by 60 min PPS delivered to free-running mice using Grass S8800 stimulator. Parameters of the stimulation were the following: 5 sec trains (0.1 msec, 10 V, 33 Hz square wave monophasic stimuli) delivered every minute, together with continuous 3 Hz stimulation using the same parameters, for 60 minute (Mazarati et al., 2000). Finally, SE was induced by subcutaneous injection of KA (Sigma), 20 mg/kg. During SE electrographic activity was continuously recorded for 24 hrs using Harmonie Software (Stellate Systems, Montreal, QU). Seizures were analyzed offline by reviewing electrographic recording. The following parameters were used to quantify SE: duration of SE, i.e. time between the occurrence of the first and the last seizure, and cumulative seizure time (total time spent in seizures), by adding the duration of each seizure episode (Mazarati et al., 2000). Seizures were defined as clusters of spikes with the frequency 3Hz or more, amplitude 1 mV or higher, and duration 5 s or longer (examples on Fig. 1A). Assessment of neuronal injury.Four days after SE induction animals were deeply anesthetized with Pentobarbital Sodium, and perfused through ascending aorta with 0.9% NaCl followed by 4% paraformaldehyde. Brains were removed, postfixed in 4% paraformaldehyde, embedded in Paraffin and cut in coronal plane at 10 micron. In addition, two naïve WT and two naïve GalR1 KO mice where used (also used in neurogenesis studies, see below). Sections of the dorsal hippocampus approximately 2 mm posterior from Bregma, as identified by comparison of the section with the images from Mouse Brain atlas (Paxinos and Franklin, 2001), were stained with 0.0004%. Fluoro-Jade B (FJB, Histochem, Jefferson, AR, USA) (Schmued and Hopkins, 2000) and counterstained with 4’-diamidino-2-phenylindole (DAPI, Molecular Probes, Eugene, OR, USA). Sections were examined under the microscope under green (FJB) and blue (DAPI) fluorescence.
Fig. 1.

Comparison of three models of status epilepticus in wild type (+/+) and GalR1 knockout (-/-) mice. A. Examples of seizure activity during SE induced by Li-pilocarpine (PILO, top row), perforant path stimulation (PPS) (middle row) and kainic acid (KA, bottom row). Left panels-baseline activity prior to seizure induction; middle panels- seizures, right panels- inter-seizure activity.B. Statistical analysis of SE. Numbers on the X-axis show number of animals in each group. Data are presented as Mean±SEM. *-p<0.05 vs. respective +/+ treatment (Two-Way ANOVA + Bonferroni T-post hoc test). SE induced by both Li-pilocarpine and PPS was more severe in GalR KO, than in wild type mice.
Sections adjacent to those used for FJB labeling, were processed for terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL) (Fujikawa et al., 1999) using FluoresceinFragEL DNA fragmentation detection kit (EMD Biosciences, San Diego, CA, USA), according to the manufacturer’s protocol, and counterstained with DAPI. Sections were examined under the microscope under green (TUNEL) and blue (DAPI) fluorescence.
Neuronal injury was assessed using semi-quantitative method by an unbiased investigator, using Olympus AX70 microscope, in the CA1 pyramidal cell layer, in a pair of adjacent sections from each animal. In Li-pilocarpine and KA-treated animals, left side was used. In PPS animals left and right sides were examined, taking into the account unilateral mode of stimulation. The extent of injury was evaluated in the images acquired by Sony DKC5000 digital camera and Adobe Photoshop 5.01 software under x 40 magnification, by calculating the ratio of FJB- or TUNEL-positive profiles (green fluorescence) to the total number of profiles (DAPI-positive, blue fluorescence) in the area of interest (Mazarati et al., 1004), averaged for the two sections used. The area used is outlined on the inset in Fig. 4. Cell injury was also examined the dentate gyrus and CA3.
Fig. 4.

Analysis of injury resulting from status epilepticus. Quantification of FluroJade B-positive (A, FJB+) and TUNEL-positive (B, TUNEL+) profiles in CA1 pyramidal cell layer in WT (+/+) and GalR1 knockout (-/-) mice after SE induced by LiCl-pilocarpine (Pilo), perforant path stimulation (PPS), or kainic acid (KA). The extent of injury was expressed as percent of FJB-positive or TUNEL-positive profiles from the total number of profiles, identified by DAPI counterstaining. Profile counts were made in the area outlined by the box in the inset. In GalR1 KO mice Li-pilocarpine-induced SE led to significantly more severe CA1 injury, than in WT. No differences between WT and GalR1 KO animals were observed after PPS; KA induced CA1 injury in neither wild type, nor control animals. Inset: from (Paxinos and Franklin, 1991).C. Comparison of the increase of the number of FJB+ and TUNEL+ profiles in Li-pilocarpine (left) and PPS (right)-induced SE, depending on the genetic background. Note, that in Li-pilocarpine-treated GalR1 KO the increase of the number of FJB+ cells was disproportionately larger than the increase in TUNEL, while in PPS animals the two types of labeling parallel. *-p<0.05 vs. WT (Two-Way ANOVA + Bonferroni t-test).
Studies of neural progenitor proliferation. All mice used for SE induction, plus two naïve WT and two GalR1 KO mice received 8 i.p. injections of 5-bromodeoxyuridine (BRDU, Sigma, 100 mg/kg, 4 injections/day 2 hrs apart for 2 days). Post-SE mice received BRDU injections on days 2 and 3 after SE (Zhao et al., 2003). All mice were euthanized 24 hrs after the last BRDU injection, and the brains were processed as described in the Assessment of neuronal injuryabove. Sections were subjected to antigen retrieval using CitraPlus antigen retrieval solution (Biogenex, San Ramon, CA, USA) according to the manufacturer’s protocol. Sections were then labeled with anti-BRDU monoclonal FITC-conjugated rat antibodies (1:500, Accurate Chemical Westburry, NY, USA) and, to identify proliferating astrocytes, with anti-Glial Fibrillary Acidic Protein (GFAP) polyclonal rabbit antibodies (1:1000, Chemicon, Temecula, CA, USA), which were revealed with Alexa fluor 594 (red) anti-rabbit secondary antibodies (1:400, Molecular probes). Proliferating neuronal progenitors or postmitotic neurons were identified as BRDU-positive, GFAP-negative profiles (example on Fig. 6E). Proliferating astrocytes were identified as profiles with BRDU-positive nucleus surrounded by GFAP-positive cytoplasm (example on Fig. 6F). For each animal the numbers of both types of profiles were counted in a pair of sections similar to those used for FJB or TUNEL staining, along the dentate granule cell layer, in the hilus, and in the area outside of the dentate gyrus, as outlined by the box on the inset, Fig. 6. The data were averaged between the two sections used.
Fig. 6.
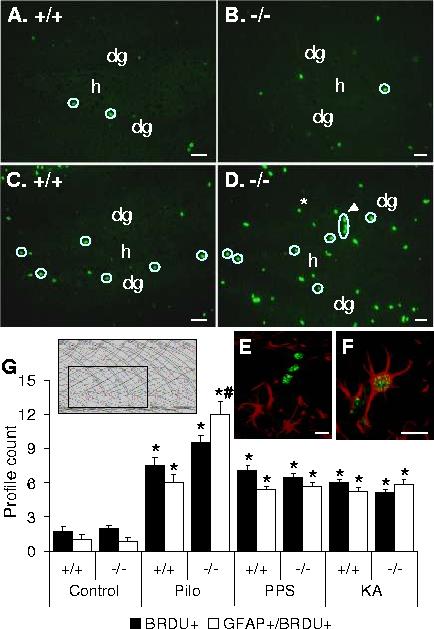
Neurogenesis in the dentate gyrus after status epilepticus. Animals received 8 BRDU injections (100 mg/kg) over 2 days, starting on the next day after LiCl-Pilocarpine SE, and were euthanized 24 hrs after the last BRDU injection. Neurogenesis in the dentate gyrus was assessed by counting BRDU-positive profiles (green) as outlined by the box on the inset, from each animal which underwent SE, and from 2 wild type and two GalR1 KO naïve mice. Proliferating glial cells were identified as profiles with GFAP-positive cytoplasm (red) and BRDU-positive nuclei (green) (examples on E,F). BRDU-positive, GFAP-negative profiles are outlined by circles on AD. A,C: Wild type (+/+); B, D-GalR1 KO (-/-). No differences were observed in basal neurogenesis in naïve animals (A,B,G). E: an example of BRDU-positive (green)/GFAP-negative profile. This cluster of three nuclei extends from subgranular zone into the dentate granule cell layer, and is indicated by an arrowhead on D. F: an example of BRDU-positive/GFAP-positive profile; this cell is located outside dentate gyrus, and indicated by an asterisk on D. Scale bar: AD 50 micron, E,F-20 micron. G. Statistical analysis. Proliferation of both neuronal progenitors and glia was observed in all three models of SE without significant differences among the models and between mice of two genotypes. LiCl-pilocarpine SE led to more pronounced glial proliferation on GalR1 KO mice (GFAP+/BRDU+).dg-dentate granule cell layer; h-hilus. *p<0.05 vs. control (naïve) mice; #- p<0.05 vs. respective wild type (Two-way ANOVA + Bonferoni t-test). Inset-from (Paxinos and Franklin, 1991).
Statistics. All data were analyzed using SigmaStat software (SPSS, Chicago, IL, USA), by means of Two-Way ANOVA withpost hoc Bonferroni t-test (quantification of cumulative seizure time and SE duration; assessment of FJB and TUNEL; counting of BRDU-positive/GFAP-negative and BRDU-positive/GFAP-positive profiles), upon verification of normal distribution; and by Pearson Product Moment Correlation (correlation between BRDU labeling and electrographic seizure parameters). P <0.05 was accepted for statistical significance.
Results
Seizures. None of WT or GalR1KO animals displayed either spontaneous seizures, or spikes during a 1 week observation period. All three SE protocols led to the development of seizures in both WT and GalR1 KO mice. Electrographic seizures (Fig. 1A, left panel) correlated with the body and forelimb clonus (rearing and/or rearing and falling). Between electrographic seizures animals displayed spikes of varying amplitude (>0.5 mV),(Fig. 1A, right panel).which correlated with facial, forelimbs, or body clonus.
Injection of pilocarpine to WT mice led to the development of seizures, which lasted between 4 and 5 hours with cumulative seizure time between 2 and 3 hrs. Pilocarpine-induce SE in GalR1 KO animals lasted for 10-12 hours, with 4-5 hours spent in seizures. Both parameters were significantly different from those in WT (p<0.05 Fig. 1B).
After completion of PPS, WT animals displayed self-sustaining seizures for 3-4 hours, with cumulative seizure time 1.5-2 hours. SE duration in GalR1 KO mice was 5.5-6.5 hours, with 3.5-4 hours spent in seizures (both parameters statistically different from WT, Fig. 1B).
Upon injection of KA seizures started between 15 and 30 min in both WT and GalR1 KO mice. In both types of animals seizures lasted 3.5-4.5 hours, and time spent in seizures was between 2.5 and 3 hours (p>0.05, Fig. 1B).
Neuronal injury. None of naïve GalR1 KO, or WT mice showed neuronal injury (not shown). Both WT and GalR1 KO mice which had undergone Li-pilocarpine-induced SE, developed injury to CA1 pyramidal cell layer of the hippocampus (Fig 2A,B, Fig,3 A,B). In WT animals 42+2% of total counted profiles were FJB-positive (Fig. 4A). GalR1 KO mice showed significantly more severe CA1 injury (p<0.05), with 86+2% injured profiled observed in CA1 (Fig. 2B, 4A).
Fig. 2.

FluoroJade B staining of the hippocampus in wild type and GalR1 KO mice following SE.Animals were euthanized 4 days after SE. Injured FluoroJade B-positive cells appear bright white. A, C, E, G, I- WT (+/+); B, D, F, H, J- GalR1 KO (-/-) mice. LiCl-Pilocarpine SE (A-D) led to moderate injury in the CA1 pyramidal cell layer in WT (A), and significantly more severe injury in GalR1 KO mice (B see also Fig. 4A). No injury to hilus of the dentate gyrus was observed in WT (c), while GalR1 KO animals showed moderate- to severe injury to hilar interneurons (D). Occasional injured dentate granule cells were also seen, as indicated by arrows on D. PPS-induced SE (E-J) led to moderate injury to CA1 in both WT and GalR1 KO mice with no differences between the genotypes (E,F,also see Fig. 4A). No hilar injury was observed on sides contralateral (contra, G) and ipsilateral (ipsi, I) to PPS in WT animals. In GalR1 KO mice mild injury to hilar interneurons was seen on the PPS side (ipsi, J), but not on the contralateral side (contra, H). Scale bar 50 micron.
Fig. 3.

TUNEL staining of the hippocampus in wild type and GalR1 KO mice following SE. Animals were euthanized 4 days after SE. TUNEL-positive cells appear green, DAPI-counterstained nuclei are blue. A, C, E, G, I- Wild type (+/+); B, D, F, H, J- GalR1 KO (-/-) mice. Li-pilocarpine SE (A-D) led to moderate injury in the CA1 pyramidal cell layer in WT animals (A), and significantly more severe injury in GalR1 KO mice (B see also Fig. 4b). No injury to hilus of the dentate gyrus was observed in WT (C), while GalR1 KO animals showed moderate-to severe injury to hilar interneurons; in addition few cells located in dentate granule cell layer (arrows) were TUNEL-positive (d). PPS-induced SE (E-J) led to moderate injury to CA1 in both wild type and GalR1 KO mice D no differences between the groups (E,F, also see Fig. 4B. No hilar injury was observed on sides contralateral (contra, G) and ipsilateral (ipsi, I) to PPS in WT. In GalR1 KO mice mild injury to hilar interneurons was seen on the PPS side (ipsi, J), but not on the contralateral side (contra, H). Scale bar 50 micron.
About one third of CA1 pyramidal cells was TUNEL-positive in WT mice, while half of counted pyramidal CA1 cells was TUNEL-positive in GalR1 KO (p<0.05, Fig. 3A,B, Fig. 4B). The ratio of TUNEL-positive to FJB-positive profiles was 69% in WT and 56% in GalR1 KO mice (p<0.05); thus the increase of the number of FJB-positive cells in GalR1 KO versus WT was steeper, than the increase in the number of TUNEL-positive nuclei (Fig. 4C).
None of WT mice showed injury to hilar interneurons (Fig. 2C, 3C). All GalR1 KO animals had consistent moderate- to severe injury to the hilus (35-50% of total number of DAPI-positive profiles in hilus), as identified by both FluroJade B and TUNEL (Fig. 2D, 3D). In all animals occasional injured dentate granule cells were also found (1-2 cells/section, indicated by arrows on Fig 2D and 3D).
PPS resulted in CA1 injury in WT animals which was comparable to that observed in WT mice after pilocarpine treatment (Fig. 2E, 3E, 4A,B). No differences were observed between the left and the right sides (not shown). On the side of stimulation CA1 pyramidal cell layer contained 46+2% of FJB-positive, and 32+2% of TUNEL-positive profiles. In GalR1 KO animals 54+1.5% of counted CA1 profiles were FJB-positive, and 35+2%-TUNEL-positive (p>0.05 vs. WT mice, Fig. 2F, 3F, 4B). The proportion of TUNEL-positive profiles to FJB-positive profiles was 69% in WT and 64% in GalR1 KO mice, that is did not depend on the genotype (p>0.05, Fig. 4Cc). PPS did not result in hilar injury in WT animals (Fig. 2G,I, 3G,I), and in GalR1 KO animals on the side contralateral to PPS (Fig. 2H, 3H). However, all GalR1 KO mice had mild injury (5-12%) to hilus of dentate gyrus on the side of PPS (Fig. 2J, 3J).
No differences between the proportions of TUNEL-positive of FJB-positive profiles were found in two models of SE and between WT and GalR KO mice Even though KA led to the development of seizures, no injury was observed in CA1 and dentate gyrus in both WT and GalR1 KO animals (Fig. 4). The only brain area in which injury was found in mice of both genotypes, were ventro-medial and ventro-lateral thalamic nuclei. The pattern and the severity of the injury to the thalamus did not differ between GalR1 KO and WT animals in all three models of seizures. Fig. 5D shows an example of thalamic injury in a WT mouse after KA-induced convulsions.
Fig. 5.
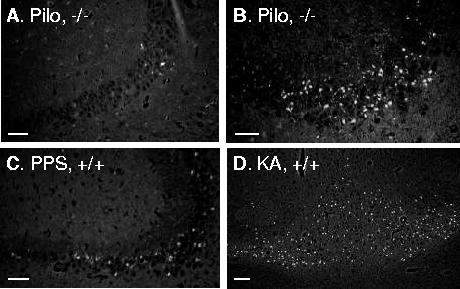
FluoroJade B staining of CA3 and thalamus. Examples of FJB staining of CA3 4 days after SE induced by Li-pilocarpine (Pilo, A, B) and PPS (C) in mice of different genotypes. D. Injury to ventrolateral and ventromedial thalamic nuclei after KA-induced SE, which did not produce hippocampal damage. Scale bar 50 micron.
In addition to CA1 and dentate gyrus, CA3 injury was observed in Li-pilocarpine and PPS models in both GalR1 KO and WT mice. CA3 injury, however, was highly variable and extended from very mild (Fig. 5A) to severe (Fig. 5B), which precluded the possibility of the analysis. The extent of CA3 injury could not be linked to either SE model, of mouse genotype.
In Li-pilocarpine model of SE, which resulted in the most robust seizures and neuronal injury in GalR1 KO, we studied seizure response and a pattern of hippocampal cell damage in GalR1 heterozygotes (n=4). In these animals both seizures, and pattern and the severity of neuronal injury were not different from WT mice (p>0.05 for all indices, not shown).
Neurogenesis in the dentate gyrus.In our previous study (Mazarati et al., 2004) we found that seizure-induced increase in neurogenesis in the dentate gyrus was regulated by GalR2 subtype. Therefore, we studied, whether GalR1 are involved in regulating neurogenesis, by looking into both basal, and seizure-induced neurogenesis in GalR1 KO mice. Basal neurogenesis in the dentate gyrus was not different between WT and GalR2 KO animals, with few BRDU-positive cells, apparent neuronal progenitors (GFAP-negative, example on Fig. 6E) located along the blades of dentate gyrus in subgranular zone (Fig. 6 A,B,G). In addition, scattered BRDU-positive GFAP-positive cells (astrocytes, example on Fig. 6F) were found outside subgranular zone in mice of both genotypes. All types of seizures resulted in the 2.5-3 fold increase of the number of BRDU-positive nuclei in SZG and dentate granule cell layer in both WT and GalR1 KO mice (Fig. 6A,D,G, p<0.05 for each treatment for both Wt and GalR1 KO). No significant differences were found among various treatments and between mice of two genotypes. Along with increased proliferation of apparent neuronal progenitors, SE was accompanied by glial proliferation, evident as an increase of BRDU-positive, GFAP-positive profiles (Fig. 6C,D,G). Glial proliferation was most pronounced after pilocarpine treatment in GalR1 KO mice (p<0.05, Fig. 6G). In animals subjected to PPS, no differences in BRDU labeling were observed between stimulated and contralateral sides (not shown). Across all groups and treatments, the extent of glia proliferation (number of BRDU/GFAP-positive cells) positively correlated with each of the two parameters of seizures, cumulative seizure time and SE duration, (r>+0.8, p<0.05), while the number of BRDU-positive, GFAP-negative profiles did not correlate with seizures indices during SE (r<+0.8, p>0.05).
Discussion
Our studies found enhanced susceptibility to seizures and seizure-induced neuronal injury of GalR1 KO mice in limbic SE. The extent of the differences between KO and WT animals depended on the model of SE. Li-Pilocarpine SE was significantly more severe, and was accompanied by more severe and extensive neuronal injury in mutants. GalR1 KO mice developed more severe seizures following PPS, than their WT littermates, but no difference in neuronal injury between the two genotypes was found. Finally, no differences in both seizure and neuronal injury patterns were observed following KA administration.
In contrast to previous reports (Jacoby et al., 2002a;b; Fetissov et al., 2003), we did not observe spontaneous seizures in GalR1 KO, which may be attributed to the relatively low number of animals used (n=16), and reported relatively low penetrance (25%) (Jacoby et al., 2002a). Nevertheless, our data suggest that the inactivation of GalR1 gene has profound effects on seizure susceptibility and neuronal vulnerability under conditions of precipitating insult even in spontaneous seizure-free mutants.
As mentioned above, the most dramatic differences in seizures and neuronal injury were observed in Li-pilocarpine model of SE. Initiation phase of Li-pilocarpine SE depends on the activation Mcholinoreceptors (Morrisett et al., 1987). Pilocarpine, a M-cholinomimetic alkaloid, directly activates M-cholinoreceptors in the hippocampus. Furthermore, the onset of seizures during LiPilocarpine SE coincides with the dramatic increase of acetylcholine content in the hippocampus and cortex (Jope et al., 1987). Galanin coexists with acetylcholine in septum/diagonal band complex, an area which sends cholinergic/galaninergic projections to the hippocampus (Melander et al., 1986; Consolo et al., 1994). In the hippocampus galanin inhibits both acetylcholine release (Dutar et al., 1989; Fisone et al., 1987), and postsynaptic cholinergic functions (Consolo et al., 1991). Inactivation of GalR1 in KO mice may facilitate both acetylcholine release and postsynaptic effects of pilocarpine and acetylcholine, thus promoting both components which contribute to the development of Li-Pilocarpine SE.
In KA-induced SE seizures were milder in GalR1 KO, than in two other models. C57Bl mice are known to have increased resistance to KA-induced seizures and neuronal injury (McKhan et al., 2003). Our results emphasize that strain-related seizure-resistance (as well as neuronal vulnerability to excitotoxic injury, see below) is not absolute, since both Li-pilocarpine and PPS induced substantial seizures even in WT mice. More important, however, is that in contrast to Lipilocarpine and PPS, we found no differences between WT and GalR1 KO mice after KA administration. This suggests that GalR1 does not interfere with the seizure-inducing mechanisms of KA. Postsynaptic kainate receptors are excitatory ionotropic glutamate receptors and their activation leads to seizures and neurotoxicity (Hampson and Manalo, 1998; Lerma et al., 2001). Presynaptic kainate receptors are metabotropic receptors, and their activation inhibits glutamatergic transmission (Lerma et al., 2001; Chittajallu et al., 1996; Kamiya and Ozawa, 1998). Taken into the account the hypothesis, that galanin inhibits seizures through presynaptic inhibition of glutamate release (Mazarati et al., 2000; Zini et al., 1993a;b), rather than through postsynaptic mechanisms, the absence of differences between WT and GalR1 KO mice in KA seizures are not surprising.
Self-sustaining SE induced by PPS strongly depends on the activation of N-methyl-D-aspartate (NMDA) receptors during its maintenance phase (Mazarati and Wasterlain, 1999). Under these conditions, attenuation of galanin-mediated block of glutamate release from perforant path and subsequent activation of NMDA receptors of dentate granule cells would promote maintenance of SE, as it was observed in GalR1 KO animals.
In parallel with the pattern of seizures, neuronal injury in the CA1area of GalR1 KO after Lipilocarpine SE was twice as severe, than in WT. In addition, while WT mice showed absolute resistance of hilar and dentate granule cells, GalR1 KO had moderate/severe and mild injury in these cell populations respectively. The observed differences in post-SE neuronal injury may be attributed to the longer-lasting and more severe seizures in GalR1 KO mice. On the other hand galanin has been suggested to possess neuroprotective properties aside from its anticonvulsant effects (Elliott-Hunt et al., 2004; Haberman et al., 2003), which may be a factor contributing to the enhanced neuronal vulnerability in GalR1 KO animals.
This vulnerability, however, was model-, and area-specific. There was no difference to CA1 damage between GalR1 KO and WT mice after PPS-induced SE, despite the fact that SE was more severe in GalR1 KO. On the other hand, hilus of dentate gyrus in GalR1 KO animals had few injured cells only on the side of the stimulation. The reason for such differences between the two models requires further studies. Even though PPS-induced SE was more severe in KO than in WT, in the animals of both genotypes it was significantly milder, than Li-pilocarpine SE in KO animals. Therefore, in these two models neuronal injury might reflect the severity of the insult, rather than the mode of stimulation, and certain severity of seizures is required in order for them to translate into more pronounced neuronal damage. Alternatively (or in addition), differences in SE circuitries might contribute to the differences in pattern and the degree if neuronal injury.
We found that the percentage of TUNEL positive nuclei was lower than the percentage of FJB-positive cells in the same area of the hippocampus: TUNEL-positive profiles constituted 56-69% of FJB-positive cells in different groups. Similar observation applied to Li-pilocarpine model of SE in rats, in which hematoxylin and eosin staining was used instead of FJB (Fujikawa et al., 1999). TUNEL labels nuclei with DNA fragmentation, a process, which is indicative of programmed cell death (Fujikawa et al., 1999), while FJB (similar to hematoxylin and eosin, Schmued et al., 1997) labels degenerating neurons despite the mode of death (both programmed and non-programmed, although in both cases damaged neurons have necrotic morphology, Fujikawa et al., 1999). In Li-pilocarpine SE the increase of FJB-positive neurons in GalR1 KO versus WT animals was disproportionately larger, than the increase of TUNEL-positive nuclei, while in PPS-induced SE the ratio of TUNEL to FJB-positive profiles did not depend on the animal genotype. This implies that while in Li-pilocarpine SE the augmentation of cell death in GalR1 KO mice happens at the expense of both programmed and non-programmed mechanisms, the contribution of the latter is more significant, than in WT animals.
Absence of neuronal damage to the hippocampus in both KO and WT mice after KA is not surprising. C57bl mice are known to be highly resistant to KA-induced neuronal injury (Schauwecker and Steward, 1997). It should be emphasized, however, that neuronal resistance to KA was not absolute, since we observed damage to thalamic neuronal populations in the animals of both genotypes. Here, however, we did not find any differences between the degrees of the injury.
Our previous studies implicated GalR2 in regulating seizure-induced neurogenesis in the dentate gyrus (Mazarati et al., 2004). Present report failed to reveal any differences in both basal and seizure-induced neuronal progenitor proliferation between GalR1 and WT mice, suggesting this GalR1, in contrast to GalR2, is not involved in regulating this form of neuronal plasticity. Such conclusion, however, cannot be deemed absolute, considering differences in methodologies between the present and previous study. Semi-acute mode of knockdown of GalR subtype (Mazarati et al., 2004) may preclude the triggering of compensatory response, while in GalR KO such compensation may occur, and thus, may regulate neurogenesis by employing other mechanisms.
Interestingly, we did not find any correlation between the degree of neuronal injury in the hippocampus, and the increase of neuronal progenitor proliferation; even mice subjected to KASE showed BRDU labeling comparable to that in two other SE models. This means, that neurogenesis is not a compensatory response to neuronal injury, but is rather a reaction to continuous hippocampal activation during SE. In fact, this suggestion is in line with our previous observation, when we found that neuronal damage can be dissociated from neurogenesis (Mazarati et al., 2004).
Neuronal proliferation, however, does not reflect the severity of seizures, since it was comparable across all SE models and animal genotypes. On the other hand, we found, that glial proliferation, which commonly accompanies SE (Mazarati et al., 2004; Parent et al., 1997; Niquet et al., 1994), was most pronounced in Li-pilocarpine-treated GalR1 KO. Overall, across all groups, the extent of glial proliferation, evident as the increase of the number of BRDU/GFAP-positive profiles, positively correlated with the severity of seizures.
In conclusion, the present reports revealed the important role of GalR1 in the hippocampus in mediating anticonvulsant and neuroprotective effects of galanin in limbic SE. During the past several years, GalR pharmacology made significant progress: the first non-peptide low molecular weight GalR agonist galnon was shown to be a potent anticonvulsant (Saar et al., 2002).
Recently, another non-peptide low molecular weight preferential GalR1 agonist was synthesized; this compound also exhibited strong anticonvulsant effects in a model of SE (Bartfai et al., 2004). Increased seizure susceptibility of GalR1 KO animals justifies future use of GalR1 agonists as antiepileptic drugs in certain forms of epilepsy.
Acknowledgements.
Supported by NIH grant NS 43409
Abbreviations
- BRDU:
5-bromodeoxyuridine
- DAPI:
4’-diamidino-2-phenylindole
- FJB:
FluoroJade B
- GalR:
galanin receptors
- GFAP:
Glial Fibrillary Acidic Protein
- KA:
Kainic acid
- KO:
knockout
- NMDA:
N-methyl-D-aspartate
- PPS:
perforant path stimulation
- SE:
status epilepticus
- SEM:
standard error of mean
- TUNEL:
terminal deoxynucleotidyl transferase-mediated UTP nick end labeling
- WT:
wild type
References
- Baraban SC, Hollopeter G, Erickson JC, Schwartzkroin PA, Palmiter RD. Knockout mice reveal a critical antiepileptic role for neuropeptide Y. J. Neurosci. 1997;17:8927–8936. doi: 10.1523/JNEUROSCI.17-23-08927.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartfai T, Lu T, Badie-Mahdavi H, M. Barr AM, Mazarati A, Hua X, Yaksh T, Haberhauer G, Trembleau L, Conde Ceide S, Somogyi L, Rebek J., Jr.Galmic, a non peptide galanin receptor agonist, affects behaviors in seizure, pain and forced swim tests Proc. Natl. Acad. Sci. USA 2004. In Press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchek TA, Smith KE, Gerald C, Walker MW. Galanin receptor subtypes. Trends Pharmacol. Sci. 2000;21:109–117. doi: 10.1016/s0165-6147(00)01446-2. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GL, Henley GM. Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature. 1996;379:78–81. doi: 10.1038/379078a0. [DOI] [PubMed] [Google Scholar]
- Consolo S, Baldi G, Russi G, Civenni G, Bartfai T, Vezzani A. Impulse flow dependency of galanin release in vivo in the rat ventral hippocampus. Proc Natl Acad Sci U S A. 1994;91:8047–8051. doi: 10.1073/pnas.91.17.8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consolo S, Bertorelli R, Girotti P, La Porta C, Bartfai T, Parenti M, Zambelli M. Pertussis toxin-sensitive G-protein mediates galanin's inhibition of scopolamine-evoked acetylcholine release in vivo and carbachol-stimulated phosphoinositide turnover in rat ventral hippocampus. Neurosci. Lett. 1991;126:29–32. doi: 10.1016/0304-3940(91)90363-x. [DOI] [PubMed] [Google Scholar]
- Depczynski B, Nichol K, Fathi Z, Iismaa T, Shine J, Cunningham A. Distribution and Characterization of the Cell Types Expressing GALR2 mRNA in Brain and Pituitary Gland. Ann N Y Acad Sci. 1998;863:120–128. doi: 10.1111/j.1749-6632.1998.tb10689.x. [DOI] [PubMed] [Google Scholar]
- Dutar P, Lamour Y, Nicol R. Galanin blocks the slow cholinergic EPSP in CA1 pyramidal neurons from ventral hippocampus. Eur. J. Pharmacol. 1989;164:355–360. doi: 10.1016/0014-2999(89)90477-9. [DOI] [PubMed] [Google Scholar]
- Elliott-Hunt CR, Marsh B, Bacon A, Pope R, Vanderplank P, Wynick D. Galanin acts as a neuroprotective factor to the hippocampus. Proc. Natl. Acad. Sci. USA. 2004;101:5105–5110. doi: 10.1073/pnas.0304823101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetissov SO, Jacoby AS, Brumovsky PR, Shine J, Iismaa TP, Hökfelt T. Altered Hippocampal Expression of Neuropeptides in Seizure-prone GALR1 Knockout Mice. Epilepsia. 2003;44:1022–1033. doi: 10.1046/j.1528-1157.2003.51402.x. [DOI] [PubMed] [Google Scholar]
- Fisone G, Wu CF, Consolo S, Nordstrom O, Brynne N, Bartfai T, Melander T, Hokfelt T. Galanin inhibits acetylcholine release in the ventral hippocampus of the rat: histochemical, autoradiographic, in vivo, and in vitro studies. Proc Natl Acad Sci U S A. 1987;84:7339–7343. doi: 10.1073/pnas.84.20.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B. Lithium-pilocarpine-induced status epilepticus produces necrotic neurons with internucleosomal DNA fragmentation in adult rats. Eur J Neurosci. 1999;11:1605–1614. doi: 10.1046/j.1460-9568.1999.00573.x. [DOI] [PubMed] [Google Scholar]
- Gould E, Tanapat P. Lesion-induced proliferation of neuronal progenitors in the dentate gyrus of the adult rat. Neuroscience. 1997;80:427–436. doi: 10.1016/s0306-4522(97)00127-9. [DOI] [PubMed] [Google Scholar]
- Gustafson EL, Smith KE, Durkin MM, Gerald C, Branchek TA. Distribution of a rat galanin receptor mRNA in rat brain. Neuroreport. 1996;7:953–957. doi: 10.1097/00001756-199603220-00025. [DOI] [PubMed] [Google Scholar]
- Haberman RP, Samulski RJ, McCown TJ. Attenuation of seizures and neuronal death by adeno-associated virus vector galanin expression and secretion. Nature Med. 2003;9:1076–1080. doi: 10.1038/nm901. [DOI] [PubMed] [Google Scholar]
- Hampson DR, Manalo JL. The activation of glutamate receptors by kainic acid and domoic acid. Nat Toxins. 1998;6:153–158. doi: 10.1002/(sici)1522-7189(199805/08)6:3/4<153::aid-nt16>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Hua XY, Hayes CS, Hofer A, Fitzsimmons B, Kilk K, Langel U, Bartfai T, Yaksh TL. Galanin Acts at GalR1 Receptors in Spinal Antinociception: Synergy with Morphine and AP-5. J Pharmacol Exp Ther. 2004;308:574–582. doi: 10.1124/jpet.103.058289. [DOI] [PubMed] [Google Scholar]
- Jacoby AS, Holmes F, Hort YJ, Shine J, Iismaa T. Phenotypic analysis of Galr1 knockout mice reveals a role for GALR1 galanin receptor in modulating seizure activity but not nerve regeneration. Lett. Pept. Sci. 2002a;8:139–146. [Google Scholar]
- Jacoby AS, Hort YJ, Constantinescu G, Shine J, Iismaa TP. Critical role for GALR1 galanin receptor in galanin regulation of neuroendocrine function and seizure activity. Mol. Brain Res. 2002b;107:195–200. doi: 10.1016/s0169-328x(02)00451-5. [DOI] [PubMed] [Google Scholar]
- Jope RS, Simonato M, Lally K. Acetylcholine content in rat brain is elevated by status epilepticus induced by lithium and pilocarpine. J. Neurochem. 1987;49:944–951. doi: 10.1111/j.1471-4159.1987.tb00985.x. [DOI] [PubMed] [Google Scholar]
- Jureus A, Langel U, Bartfai T. l-Ala-substituted rat galanin analogs distinguish between hypothalamic and jejunal galanin receptor subtypes. Peptide Res. 1997;49:195–200. doi: 10.1111/j.1399-3011.1997.tb00878.x. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S. Kainate receptor-mediated inhibition of presynaptic Ca2+ influx and EPSP in area CA1 of the rat hippocampus. J. Physiol. (London) 1998;509:833–845. doi: 10.1111/j.1469-7793.1998.833bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerekes N, Mennicken F, O'Donnell D, Hokfelt T, Hill R. Galanin increases membrane excitability and enhances Ca2+ currents in adult, acutely dissociated dorsal root ganglion neurons. Eur J Neurosci. 2003;11:2957–2966. doi: 10.1111/j.1460-9568.2003.03057.x. [DOI] [PubMed] [Google Scholar]
- Kokaia M, Holmberg K, Nanobashvili A, Xu Z D, Kokaia Z, Lendahl U, Hilke S, Theodorsson E, Kahli U, Bartfai T, Lindvall O, Hokfelt T. Suppressed kindling epileptogenesis in mice with ectopic overexpression of galanin. Proc Natl Acad Sci U S A. 2001;98:14006–14011. doi: 10.1073/pnas.231496298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J, Paternain AV, Rodríguez-Moreno A, López-García J. Molecular physiology of kainate receptors. Physiol. Rev. 2001;81:971–998. doi: 10.1152/physrev.2001.81.3.971. [DOI] [PubMed] [Google Scholar]
- Lin EJ, Richichi C, Young D, Baer K, Vezzani A, During MJ. Recombinant AAV-mediated expression of galanin in rat hippocampus suppresses seizure development. Eur J Neurosci. 2003;18:2087–2092. doi: 10.1046/j.1460-9568.2003.02926.x. [DOI] [PubMed] [Google Scholar]
- Liu HX, Brumovsky P, Schmidt R, Brown W, Payza K, Hodzic L, Pou C, Godbout C, Hökfelt T. Receptor subtype-specific pronociceptive and analgesic actions of galanin in the spinal cord: selective actions via GalR1 and GalR2 receptors. Proc. Natl.Acad. Sci. USA. 2001;98:9960–9964. doi: 10.1073/pnas.161293598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh DJ, Baraban SC, Hollopeter G, Palmiter RD. Role of the Y5 neuropeptide Y receptor in limbic seizures. Proc Natl Acad Sci U S A. 1999;96:13518–13523. doi: 10.1073/pnas.96.23.13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati AM, Halaszi E, Telegdy G. Anticonvulsive effects of galanin administered into the central nervous system upon the picrotoxin-kindled seizure syndrome in rats. Brain Res. 1992;589:164–166. doi: 10.1016/0006-8993(92)91179-i. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Hohmann JG, Bacon A, Liu H, Sankar R, Steiner RA, Wynick D, Wasterlain CG. Modulation of hippocampal excitability and seizures by galanin. J Neurosci. 2000;20:6276–6281. doi: 10.1523/JNEUROSCI.20-16-06276.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati AM, Liu H, Soomets U, Sankar R, Shin D, Katsumori H, Langel U, Wasterlain CG. Galanin modulation of seizures and seizure modulation of hippocampal galanin in animal models of status epilepticus. J Neurosci. 1998;18:10070–10077. doi: 10.1523/JNEUROSCI.18-23-10070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati AM, Lu X, Kilk K, Langel U, Wasterlain CG, Bartfai T. Galanin type 2 receptors regulate neuronal survival, susceptibility to seizures and seizure-induced neurogenesis in the dentate gyrus. Europ. J. Neurosci. 2004;19:3235–3244. doi: 10.1111/j.0953-816X.2004.03449.x. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Wasterlain CG. N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci Lett. 1999;265:187–90. doi: 10.1016/s0304-3940(99)00238-4. [DOI] [PubMed] [Google Scholar]
- McKhann GMn, Wenzel HJ, Robbins CA, Sosunov AA, PA. S. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122:551–561. doi: 10.1016/s0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- Melander T, Staines WA, Rokaeus A. Galanin-like immunoreactivity in cholinergic neurons of the septum-basal forebrain complex projecting to the hippocampus of the rat. Neuroscience. 1986;19:223–240. doi: 10.1016/0006-8993(85)91228-4. [DOI] [PubMed] [Google Scholar]
- Morrisett RA, Jope RS, Snead OC. Effects of drugs on the initiation and maintenance of status epilepticus induced by administration of pilocarpine to lithium-pretreated rats. Exp. Neurol. 1987;97:193–200. doi: 10.1016/0014-4886(87)90293-7. [DOI] [PubMed] [Google Scholar]
- Niquet JF, Ben-Ari Y, Represa A. Glial reaction after seizure induced hippocampal lesion: immunohistochemical characterization of proliferating glial cells. J Neurocytol. 1994;23:641–656. doi: 10.1007/BF01191558. [DOI] [PubMed] [Google Scholar]
- O’Donnel D, Sultan A, Wahlestedt C, Walker P. Expression of the novel galanin receptor subtype GALR2 in the adult rat CNS: distinct distribution from GALR1. J. Comp. Neurol. 1999;409:469–481. [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate Granule Cell Neurogenesis Is Increased by Seizures and Contributes to Aberrant Network Reorganization in the Adult Rat Hippocampus. J. Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd Academic Press, Academic Press; 2001. [Google Scholar]
- Saar K, Mazarati AM, Mahlapuu R, Hallnemo G, Soomets U, Kilk K, Hellberg S, Pooga M, Tolf BR, Shi TS, Hokfelt T, Wasterlain C, Bartfai T, Langel U. Anticonvulsant activity of a nonpeptide galanin receptor agonist. Proc Natl Acad Sci U S A. 2002;99:7136–7141. doi: 10.1073/pnas.102163499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued L, Hopkins K. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Shilbey H, Smith BN. Pilocarpine-induced status epilepticus results in mossy fiber sprouting and spontaneous seizures in C57BL/6 and CD-1 mice. Epilepsy Research. 2002;49:109–120. doi: 10.1016/s0920-1211(02)00012-8. [DOI] [PubMed] [Google Scholar]
- Zhao M, Momma S, Delfani K, Carlen M, Cassidy RM, Johansson CB, Brismar H, Shupliakov O, Frisen J, Janson AM. Evidence for neurogenesis in the adult mammalian substantia nigra. Proc. Natl. Acad. Sci. USA. 2003;100:7925–7930. doi: 10.1073/pnas.1131955100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zini S, Roisin M, Langel U, Bartfai T, Ben-Ari Y. Galanin reduces release of endogenous excitatory amino acids in the rat hippocampus. Eur. J. Pharmacol. 1993a;245:1–7. doi: 10.1016/0922-4106(93)90162-3. [DOI] [PubMed] [Google Scholar]
- Zini S, Roisin MP, Armengaud C, Ben-Ari Y. Effects of potassium channels modulators on the release of glutamate induced by ischaemic-like conditions in rat hippocampal slices. Neurosci. Lett. 1993b;153:202–205. doi: 10.1016/0304-3940(93)90322-c. [DOI] [PubMed] [Google Scholar]