

Abstract
We have developed an improved method for preparing cell aggregates for in vitro chondrogenesis studies. This method is a modification of a previously developed conical tube-based culture system that replaces the original 15-mL polypropylene tubes with 96-well plates. These modifications allow a high-throughput approach to chondrogenic cultures, which reduces both the cost and time to produce chondrogenic aggregates, with no detrimental effects on the histological and histochemical qualities of the aggregates. We prepared aggregates in both systems with human bone marrow-derived mesenchymal stem cells (hMSC). The aggregates were harvested after 2 and 3 weeks in chondrogenic culture and analyzed for their ability to differentiate along the chondrogenic pathway in a defined in vitro environment. Chondrogenic differentiation was assessed biochemically by DNA and glycosaminoglycan (GAG) quantification assays and by histological and immunohistologic assessment. The chondrogenic cultures produced in the 96-well plates appear to be slightly larger in size and contain more DNA and GAG than the aggregates made in tubes. When analyzed histologically, both systems demonstrate morphological characteristics that are consistent with chondrogenic differentiation and cartilaginous extracellular matrix production.
INTRODUCTION
Three-dimensional (3-D) cell aggregate culture has been used to study in vitro chondrogenesis. This system, which dates back to the 1960s when it was used by Holtzer et al. (1) to study organ-cultured chondrocyte aggregates on nutrient agar, has evolved through the years. Terminal differentiation studies of rat (2,3) and rabbit chondrocytes (4) used a modification of this culture system using 15-mL polypropylene centrifuge tubes to establish the aggregates. More recently, the chondrogenic potential of rabbit (5), human bone marrow-derived mesenchymal progenitor cells (6), and rat periosteum-derived progenitor cells (7) have also been studied, using 15-mL polypropylene centrifuge tubes to produce and maintain the aggregates. The unusual use of polypropylene in a cell culture application is a critical component of this technique, because the pelleted cells do not adhere to the tube walls, but rather to one another to form free-floating cell aggregates, in which cell-cell, rather than cell-substrate, interactions take place. One of our research goals is the development of tissue-engineered cartilage implants for the repair of articular cartilage defects. In this context, our group is investigating factors that contribute to the chondrogenic differentiation of human bone marrow-derived mesenchymal stem cells (hMSCs) in an in vitro bioreactor environment (8). We have shown that chondrogenic preconditioning of hMSCs in aggregate culture results in improved chondrogenesis in these tissue engineered constructs (9). This chondrogenic preconditioning paradigm requires the preparation of large numbers of aggregates, but doing so using 15-mL centrifuge tubes is both time-consuming and expensive. We have developed a novel high-throughput method for the preparation and maintenance of cell aggregates that significantly decreases the time required for the preparation of the aggregates and lowers the cost, while maintaining reproducible chondrogenic differentiation. As a result, a more efficient culture protocol to promote chondrogenic differentiation was developed. This method can now be used to efficiently study the effects of growth factors and cytokines on the chondrogenic potential of hMSCs. We have used this system to study the effects of the fibroblast growth factor-2 on the mitogenic and chondrogenic potential of hMSCs (10) and in other experiments that require large quantities of individual aggregates. This paper describes the development of this method in detail and presents a protocol for high-throughput production of chondrogenic cell aggregates.
MATERIALS AND METHODS
hMSC Isolation
Bone marrow aspirates obtained from the MSC core facility at Case Western Reserve University (CWRU; Cleveland, OH, USA) were used in this study. The cells were derived from bone marrow that was harvested from the posterior iliac crest of healthy volunteer donors. The marrow aspiration procedure was approved by an Institutional Review Board, and informed consent was obtained from all volunteers. The procedure for harvesting hMSCs followed the method described by Haynesworth et al. (11). Briefly, the bone marrow samples were washed with low glucose Dulbecco’s modified Eagle medium (DMEM-LG; Invitrogen, Carlsbad, CA, USA) supplemented with 10% of a selected lot of fetal bovine serum (FBS; Invitrogen) (12). To isolate the mononucleated cells, the sample was subjected to a preformed Percoll™ (Sigma, St. Louis, MO, USA) density gradient (1.073 g/mL) centrifugation step. The hMSC primary cultures were seeded at a density of 1.8 × 105 cells/cm2 in serum-supplemented medium in a 10-cm plate. The hMSCs were cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2. Nonadherent cells were removed from the plate after 4 days by changing the medium. The medium was changed every 3 days thereafter. The primary cultures were subcultured after approximately 2 weeks and seeded at 5 × 103 cells/cm2.
Chondrogenic Assay
Culture-expanded hMSCs are typically used at the end of first passage to prepare the cell aggregates. The hMSCs are subcultured at approximately 80% confluence to prevent contact inhibition of growth and spontaneous differentiation (13). The cultures are rinsed with sterile Tyrode’s salt solution to remove residual FBS present in the growth medium. The Tyrode’s solution is then aspirated, 0.05% trypsin-EDTA (Invitrogen) is added, and the cultures are returned to the incubator for 5-10 min. When the cells have detached, bovine calf serum (Hyclone, Logan, UT, USA) is added to inhibit the trypsin. The cells are transferred to a 50-mL conical tube (BD Biosciences, Bedford, MA, USA) and centrifuged for 5 min at 200× g. The supernatant is discarded, and the cells are resuspended in a defined chondrogenic medium [e.g., that described by Johnstone et al. (5)]. This defined medium is DMEM-HG (DMEM with 4 g/L glucose; Biowhittaker, Walkersville, MD, USA) supplemented with 1% BD™ ITS+ Premix (BD Biosciences; consisting of 6.25 μg/mL insulin, 6.25 μg/mL transferrin, 6.25 ng/mL selenious acid, 1.25 mg/mL serum albumin, and 5.35 μg/mL linoleic acid), 37.5 μg/mL ascorbate-2-phosphate (WAKO, Richmond, VA, USA), and 10-7 M dexamethasone (Sigma). Additional supplements such as L-glutamine, antibiotic antimycotic (10,000 U/mL penicillin G sodium, 10,000 μg/mL streptomycin sulfate, and 25 μg/mL amphotericin B in 0.85% saline), nonessential amino acids, and sodium pyruvate (all from Invitrogen) are also added to the medium at 1%. The cells are counted using a hemacytometer, and the suspension volume is adjusted to a final cell density of 1.25 × 106 cells/mL. Transforming growth factor β1 (TGF-β1; Peprotech, Rocky Hill, NJ, USA) is added to the cell suspension to a final concentration of 10 ng/mL. The cell suspension is mixed by gentle pipeting to ensure homogeneity. Two hundred-microliter aliquots are then dispensed into the wells of an autoclave-sterilized 96-well, V Bottom, 300-μL polypropylene microplate (Phenix, Hayward, CA, USA) using a repeater pipet (Eppendorf, Westbury, NY, USA) to which a large-orifice tip (Fisher Scientific, Hampton, NH, USA) has been affixed to allow smooth delivery of the aliquots into the wells. A polypropylene lid (Phenix) is placed on the plate, and the plate is centrifuged for 5 min at 500× g and incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2. In the following 12-16 h, the cells coalesce and form a high-density cell aggregate (5). Twenty-four hours after seeding, we ensure that the aggregates can float freely by releasing them from the bottom of the wells. To this end, we aspirate 100 μL of the medium and gently release it back into the wells using an 8-channel pipet (Fisher Scientific). The medium is changed every other day thereafter using DMEM-HG plus supplements plus 10 ng/mL TGF-β1. To avoid possible acidification problems, strict adherence to the medium change regimen is critical with cultures of this size.
DNA Assay
The cellularity of five replicate aggregates was determined by measuring the DNA content. Aggregates were digested with 200 μL papain (25 μg/mL papain, Sigma; consisting of 2 mM cysteine, 50 mM NaH2PO4, 2 mM EDTA, pH 6.5) (14) at 65°C. The papain-digested extracts were then combined with 400 μL 0.1 M NaOH and incubated at room temperature for 30 min. The extracts were neutralized with 400 μL 0.1 M HCl in 5 M NaCl, 100 mM NaH2PO4. In a 96-well microplate, 100 μL of the extracts were combined with 100 μL of 0.7 μg/mL Hoechst 33258 (Sigma) in water. Fluorescence was read in a GENios Pro™ microplate reader (Tecan, Durham, NC, USA) at an excitation wavelength of 340 nm and an emission wavelength of 465 nm and compared to that of a certified calf thymus DNA standard (Amersham Biosciences, Piscataway, NJ, USA).
Glycosaminoglycan Quantification
Replicate aggregates were papain-digested as described above. A nitro-cellulose membrane (0.45 μM pore size) prewetted with distilled water was placed into a dot-blot apparatus (BioRad Laboratories, Hercules, CA, USA). Two hundred fifty-microliter aliquots of 0.02% Safranin O in 50 mM sodium acetate, pH 4.8, were pipeted into each well, and 25-μL aliquots of the papain-digested extracts were then pipeted into the Safranin O. Vacuum was applied until the samples filtered through. The wells were rinsed with distilled water, and the filter was removed from the apparatus. The individual dots were cut out, transferred to microcentrifuge tubes, and incubated for 20 min in 10% cetylpyridinium chloride at 37°C. The absorbance of these extracts was read at 530 nm in a GENios Pro microplate reader and compared to that of chondroitin sulfate C (shark cartilage) standards (Seikagaku America, East Falmouth, MA, USA) (15).
Histology/Collagen Immunohistochemistry
Replicate aggregates were fixed with 10% neutral-buffered formalin, side-by-side paraffin-embedded, and sectioned for histological and immunohistochemical evaluation. Adjacent sections were either stained with toluidine blue to evaluate proteoglycan content or were stained for collagen type I using monoclonal anti-collagen type I (clone col-1; Sigma), anti-collagen type II (Developmental Studies Hybridoma bank, University of Iowa), or anti-collagen type X (Gary J. Gibson, Ph.D., Breech Research Laboratory, Bone & Joint Center, Henry Ford Hospital & Medical Centers, Detroit, MI). Negative controls, which used either mouse pre-immune immunoglobulin G (IgG; Vector Laboratories, Burlingame, CA, USA) or no primary antibody, were run in parallel. For immunohistochemistry, sections were deparaffinized in xylene in three 5-min washes, rehydrated in decreasing ethanol concentrations (in duplicate) for 2 min each, and washed with distilled water for 3 min. The sections were digested in a humidified chamber, with a 1 mg/mL pronase (Sigma) in phosphate-buffered saline (PBS) solution for 15 min, and then blocked with 5% bovine serum albumin (BSA) in PBS for 30 min. The primary antibody, diluted 1:200 in 1% BSA in PBS, was applied to the sections for 1 h. Fluorescein isothyocianate (FITC)-conjugated goat anti-mouse IgG (MP Biomedicals, Irvine, CA, USA) was used as the secondary antibody, which was also diluted 1:200 in 1% BSA in PBS and applied for 45 min. After each step in the protocol, the sections were washed with PBS for 5 min. The slides were then mounted using 5% N-propyl gallate in glycerol to slow photobleaching and were photographed with a SPOT-RT™ digital camera (Diagnostic Instruments, Sterling Heights, MI, USA) using a fluorescence microscope.
RESULTS AND DISCUSSION
Human bone marrow-derived MSC aggregates produced in the 96-well plates are larger in comparison to those produced using previous systems such as that described by Kato et al. (4) (Figure 1, A and B). Chondrogenic assessment by means of immunohistochemistry indicated no type I collagen in aggregates of 15-mL tubes or 96-well plates (Figure 1, C and D). Conversely, type II collagen immunoreactivity was present throughout the extracellular matrix in both the tube and plate aggregates, except for a small centralized region in the tube aggregates (Figure 1, E and F). Type X collagen immunoreactivity was also present throughout the extracellular matrix in both aggregate systems, with the exception of the outermost cell layers (Figure1, G and H). All negative controls remained unstained (not shown), indicating that the type I, II, and X collagen immunoreactivity was specific to the antigens of interest. There was no central necrosis detected in the plate or tube aggregates. However, this can become an issue, due to mass transport limitations, if the size of the aggregates is increased significantly beyond what is shown here. In this regard, however, we also did not notice any differences between plate and tube-based aggregates.
Figure 1.
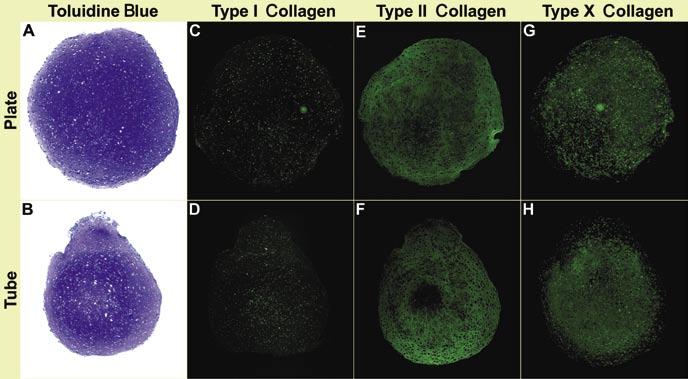
Microscopic characteristics of aggregates. (A and B) Toluidine blue-stained human bone marrow-derived mesenchymal stem cell (hMSC) aggregates prepared in 15-mL polypropylene tubes or 96-well plates cultured in chondrogenic medium for 3 weeks. (C-H) hMSC aggregates prepared in 15-mL polypropylene tubes or 96-well plates cultured in chondrogenic medium for 3 weeks and immunostained for collagen types I, II, and X.
DNA analysis demonstrated higher DNA content in the plate aggregates compared to the tubes (Figure 2A). Similarly, glycosaminoglycan (GAG) analysis of the plate aggregates indicates higher GAG content per aggregate (Figure 2B) and normalized to DNA content (Figure 2C). We have also successfully extracted RNA from 96-well plate aggregates. The typical RNA yields from aggregates grown in 96-well plates were in the range of 3-7 μg RNA per aggregate.
Figure 2.
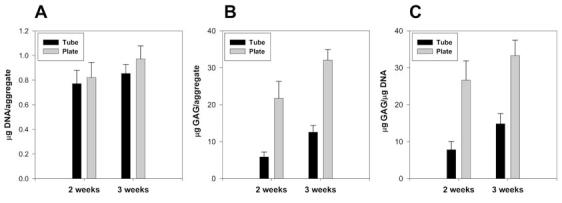
DNA and GAG content for hMSC aggregates made in 15-mL polypropylene tubes (black) or 96-well plates (gray). (A) DNA content for five replicate aggregates. (B) GAG content for five replicate aggregates. (C) GAG content for five replicate aggregates normalized to DNA. hMSC, human bone marrow-derived mesenchymal stem cells; GAG, glycosaminoglycan.
The typical time course for chondrogenesis in aggregates has been described previously (6). Briefly, however, metachromatic toluidine blue staining for GAG usually first appears around day 5, with type I collagen present from day 1, and decreasing through day 14. Type II collagen typically appears at day 5, while type X collagen is generally detected in the matrix by day 7, with both being detected throughout the aggregate by day 14. When cultures in tubes and plates were compared, there were no detectable differences between these markers at 1, 2, or 3 weeks, suggesting that chondrogenesis proceeds in parallel under both culture methods.
Biochemical analyses indicate that the larger size of the plate aggregates are a result of an increase in the number of cells as well as higher matrix production, which suggests that the 96-well plates are a seemingly better choice for the preparation of aggregates. Although our group has successfully used the Kato et al. (4) protocol to promote chondrogenesis, the method is cumbersome, time-consuming, and expensive for experiments requiring large numbers of replicate cultures or study variables. We present here a protocol that will result in an overall cost and time reduction. A typical aggregate culture experiment for our group frequently consists of 400 aggregates or more, which would require a substantial amount of incubator space to contain the aggregates if using 15-mL tubes. At typical plastic-ware prices, replacing the tubes necessary for the production of 400 aggregates with 96-well plates results in a 90% cost savings. Another nontrivial improvement with our system is a decrease in the amount of time required for the preparation and maintenance of the aggregates. Using our method, 400 aggregates can be prepared and maintained in about one-fourth of the time needed when using the tubes. This time savings is due to the time required to uncap/recap and rack the tubes.
To adapt this assay for use with microplates, we tested a variety of 96-well polypropylene microplates (Fisherbrand®; Fisher Scientific) with different-shaped bottoms to select those which produce consistent aggregate formation and chondrogenic differentiation of bone marrow-derived MSCs. Microplates that had wells with wide-angle bottoms did not allow the cell-cell interactions that are necessary for the coalescence of the cells, while those with narrow angles limited the circumferential growth of the aggregates as new extracellular matrix was fabricated by the differentiating chondrocytes. The 96-well, V Bottom, 300-μL polypropylene plates from Phenix (cat. no. MPU-8355) were found to be the most effective for the production of our aggregates. Although polypropylene plates are normally intended for use in PCR applications and are supplied nonsterile, there are no apparent adverse effects from autoclave sterilization.
To summarize, we have successfully produced consistent and reproducible chondrogenic aggregates using this high-throughput culture system. This protocol can be a practical approach to examine multiple variables to study the chondrogenic potential of hMSCs.
ACKNOWLEDGMENTS
The authors wish to thank Dr. Arnold Caplan’s Skeletal Research Center Mesenchymal Stem Cell facility headed by Dr. Donald Lennon for supplying the hMSCs used in this study and Ms. Margie Harris for her dedication in the preparation of the primary cell cultures. We would also like to acknowledge Ms. Teresa Pizzuto and Amad Awadallah for the histology. This work was supported, in part, by grants from the Arthritis Foundation (J.F.W.) and the Presidential Research Initiative (J.F.W. and L.A.S.), sponsored by the Ohio Board of Regents and Case Western Reserve University.
Footnotes
The authors declare no competing interests.
To purchase reprints of this article, contact Reprints@BioTechniques.com
REFERENCES
- Holtzer H, Abbott J, Lash J, Holtzer S. The loss of phenotypic traits by differentiated cells in vitro, I. dedifferentiation of cartilage cells. Proc. Natl. Acad. Sci. USA. 1960;46:1533–1542. doi: 10.1073/pnas.46.12.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballock RT, Heydemann A, Wakefield LM, Flanders KC, Roberts AB, Sporn MB. TGF-beta 1 prevents hypertrophy of epiphyseal chondrocytes: regulation of gene expression for cartilage matrix proteins and metalloproteases. Dev. Biol. 1993;158:414–429. doi: 10.1006/dbio.1993.1200. [DOI] [PubMed] [Google Scholar]
- Ballock RT, Reddi AH. Thyroxine is the serum factor that regulates morphogenesis of columnar cartilage from isolated chondrocytes in chemically defined medium. J. Cell Biol. 1994;126:1311–1318. doi: 10.1083/jcb.126.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Iwamoto M, Koike T, Suzuki F, Takano Y. Terminal differentiation and calcification in rabbit chondrocyte cultures grown in centrifuge tubes: regulation by transforming growth factor beta and serum factors. Proc. Natl. Acad. Sci. USA. 1988;85:9552–9556. doi: 10.1073/pnas.85.24.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone B, Hering TM, Caplan AI, Goldberg VM, Yoo JU. In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp. Cell Res. 1998;238:265–272. doi: 10.1006/excr.1997.3858. [DOI] [PubMed] [Google Scholar]
- Yoo JU, Barthel TS, Nishimura K, Solchaga L, Caplan AI, Goldberg VM, Johnstone B. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J. Bone Joint Surg. Am. 1998;80:1745–1757. doi: 10.2106/00004623-199812000-00004. [DOI] [PubMed] [Google Scholar]
- Hanada K, Solchaga LA, Caplan AI, Hering TM, Goldberg VM, Yoo JU, Johnstone B. BMP-2 induction and TGF-beta 1 modulation of rat periosteal cell chondrogenesis. J. Cell Biochem. 2001;81:284–294. doi: 10.1002/1097-4644(20010501)81:2<284::aid-jcb1043>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Solchaga LA, Goldberg VM, Mansour JM, Caplan AI, Welter JF. Cartilage tissue engineering: In vitro generation of large cartilaginous constructs. Trans. Orthop. Res. Soc. 2003;28:944. [Google Scholar]
- Welter JF, Baskaran H, Berilla J, Penick K, Goldberg VM, Mansour J, Caplan AI, Solchaga LA. Modulation of growth and differentiation conditions enhances chondrogenesis in large tissue engineered constructs. Trans. Orthop. Res. Soc. 2004;29:719. [Google Scholar]
- Solchaga LA, Penick K, Porter JD, Goldberg VM, Caplan AI, Welter JF. FGF-2 enhances the mitotic and chondrogenic potentials of human adult bone marrow-derived mesenchymal stem cells. J. Cell Physiol. 2005;203:398–409. doi: 10.1002/jcp.20238. [DOI] [PubMed] [Google Scholar]
- Haynesworth SE, Goshima J, Goldberg VM, Caplan AI. Characterization of cells with osteogenic potential from human marrow. Bone. 1992;13:81–88. doi: 10.1016/8756-3282(92)90364-3. [DOI] [PubMed] [Google Scholar]
- Lennon DP, Haynesworth SE, Bruder SP, Jaiswal N, Caplan AI. Human and animal mesenchymal progenitor cells from bone marrow: Identification of serum for optimal selection and proliferation. In Vitro Cell. Dev. Biol. 1996;32:602–611. [Google Scholar]
- Solchaga LA, Welter JF, Lennon DP, Caplan AI. Generation of pluripotent stem cells and their differentiation to the chondrocytic phenotype. Methods Mol. Med. 2004;100:53–68. doi: 10.1385/1-59259-810-2:053. [DOI] [PubMed] [Google Scholar]
- Ponticiello MS, Schinagl RM, Kadiyala S, Barry FP. Gelatin-based resorbable sponge as a carrier matrix for human mesenchymal stem cells in cartilage regeneration therapy. J. Biomed. Mater. Res. 2000;52:246–255. doi: 10.1002/1097-4636(200011)52:2<246::aid-jbm2>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Carrino DA, Arias JL, Caplan AI. A spectrophotometri modification of a sensitive densitometric Safranin O assay for glycosaminoglycans. Biochem. Int. 1991;24:485–495. [PubMed] [Google Scholar]