

Abstract
Transforming growth factor-β1 (TGF-β1) is a multifunctional cytokine involved in differentiation, growth, and survival of mesenchymal cells while inhibiting growth/survival of most other cell types. The mechanism(s) of pro-survival signaling by TGF-β1 in mesenchymal cells is unclear. In this report, we demonstrate that TGF-β1 protects against serum deprivation-induced apoptosis of mesenchymal cells isolated from patients with acute lung injury and of normal human fetal lung fibroblasts (IMR-90). TGF-β receptor(s)-activated signaling in these cells involves rapid activation of the Smad and p38 MAPK pathways within minutes of TGF-β1 treatment followed by a more delayed activation of the pro-survival phosphatidylinositol 3-kinase-protein kinase B (PKB)/Akt pathway. Pharmacological inhibition of p38 MAPK with SB203580 or expression of a p38 kinase-deficient mutant protein inhibits TGF-β1-induced PKB/Akt phosphorylation. Conditioned medium from TGF-β1-treated cells rapidly induces PKB/Akt activation in an SB203580- and suramin-sensitive manner, suggesting p38 MAPK-dependent production of a secreted growth factor that activates this pro-survival pathway by an autocrine/paracrine mechanism. Inhibition of the phosphatidylinositol 3-kinase-PKB/Akt pathway blocks TGF-β1-induced resistance to apoptosis. These results demonstrate the activation of a novel TGF-β1-activated pro-survival/anti-apoptotic signaling pathway in mesenchymal cells/fibroblasts that may explain cell-specific actions of TGF-β1 and provide mechanistic insights into its pro-fibrotic and tumor-promoting effects.
Transforming growth factor-β1 (TGF-β1)1 is a multifunctional cytokine that regulates a number of biological responses including chemotaxis, cell cycle progression, differentiation, and apoptosis of target cells in a context- and cell-specific manner (1, 2). TGF-β1 is critically involved in tissue injury and repair processes (3, 4). Rapid release of TGF-β1 at sites of tissue injury is chemotactic for both inflammatory cells (5) and fibroblasts (6). Pro-angiogenic effects are likely to be important in formation of granulation tissue in the “proliferative” phase of wound healing (7). TGF-β1 exerts multiple effects in the later “maturation” phase of wound repair by inducing extracellular matrix production/remodeling (8, 9) and myofibroblast differentiation (10, 11). Apoptosis of fibroblasts/myofibroblasts is essential for the normal resolution of repair responses and the prevention of scarring/fibrosis (12, 13). The persistence of mesenchymal cells and the up-regulated expression/activation of TGF-β1 at sites of tissue injury and repair are associated with progressive fibrosis with subsequent organ dysfunction in diverse systems including the kidney, liver, and lung (14, 15).
The mechanisms by which TGF-β1 regulates apoptosis/survival signals in mesenchymal cells are not well understood. There is better understanding of the growth-inhibitory/pro-apoptotic effects of TGF-β1 on immune cells (16) and epithelial cells (17), consistent with its anti-inflammatory and tumor-suppressive functions. In contrast to these “suppressive” functions, TGF-β1 generally promotes growth and survival of mesenchymal cells. Growth-promoting effects of TGF-β1 appear to be primarily mediated by indirect effects on the induction of mitogenic growth factor synthesis (18, 19) and/or their receptor(s) up-regulation (20). Relatively few studies have examined direct effects of TGF-β1 on mesenchymal cell/fibroblast apoptosis. Anti-apoptotic effects of TGF-β1 on fibroblasts and myofibroblasts have been reported previously (21–23), although mechanisms are not well understood.
The phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB/Akt) pathway regulates a number of cellular processes including cell cycle progression, glucose metabolism, angiogenesis, cell motility, and apoptosis (24). Multiple targets of PKB/Akt mediate pro-survival/anti-apoptotic effects (reviewed in Ref. 25). Activation of the PI3K/Akt pathway in response to TGF-β1 has been demonstrated in epithelial cells where it mediates epithelial-mesenchymal transition (EMT) (26, 27). Moreover, activation of the PKB/Akt pathway has been demonstrated to “rescue” Hep3B cells from TGF-β1-induced apoptosis (28). There is limited information, however, on the role and regulation of this pathway by TGF-β1 in mesenchymal cells.
TGF-β family members signal via heteromeric transmembrane complexes of type II and type I serine-threonine receptor kinases (1, 2). The best known direct effectors of TGF-β receptor(s) signaling are the Smad proteins that, when activated, function as transcriptional regulators (1). More recently, early post-receptor signaling via Smad-independent pathways have been increasingly recognized (2). The p38 mitogen-activated protein kinase (MAPK) appears to be an important transducer of such responses (29–31). A direct link between activation of the p38 MAPK pathway and TGF-β receptor(s) activation appears to be through binding of X chromosome-linked inhibitor of apoptosis protein (32, 33). Activation of p38 MAPK by TGF-β1 has been shown to induce either EMT (34) or pro-apoptotic effects in epithelial cells (17, 30, 31). The role of the p38 MAPK pathway in the regulation of apoptosis in mesenchymal cells is not well understood.
We hypothesized that activation of PI3K/Akt by TGF-β1 may induce an anti-apoptotic phenotype in mesenchymal cells/fibroblasts and that upstream activation of p38 MAPK may regulate this activity. This study was undertaken to determine effects of TGF-β1 on apoptotic susceptibility of primary cultures of untransformed human lung fibroblasts and the potential role of p38 MAPK-dependent PI3K-Akt activation in the expression of this phenotype.
MATERIALS AND METHODS
Isolation and Culture of Cells
Research protocols involving human subjects received prior approval by the Institutional Review Board at the University of Michigan. Lung mesenchymal cells were isolated by bronchoalveolar lavage from adult patients with respiratory failure due to acute lung injury (ALI) (35). Cells were cultured in medium consisting of Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Sigma), 100 units/ml penicillin/streptomycin (Sigma), and fungizone (Invitrogen); medium was changed every 2 days. Studies were performed on passage 3–5 of a morphologically homogeneous population of spindle-shaped cells that uniformly stained positive for the fibroblast marker, prolyl 4-hydroxylase (36). Normal human fetal lung fibroblasts (IMR-90; Institute for Medical Research, Camden, NJ) were cultured under similar conditions, and studies were performed at passage 5–9. Cells were plated on 60-mm cell culture dishes at a density of 5 × 105 cells/dish or on 96-well ELISA cell culture plates at a density of 15,000 cells per well and incubated in 5% CO2, 95% air. When cells reached 80% confluence, they were growth-arrested for 48 h in DMEM with 0.01% FBS prior to treatment.
Reagents
Porcine-derived TGF-β1 was obtained from R & D Systems, Minneapolis, MN. SB203580, SP600125, wortmannin, and Y-27632 were from Calbiochem. PD98059 and LY294002 were obtained from Cell Signaling Technology, Beverly, MA. Rabbit polyclonal antibodies to phospho-Akt (Ser-473), phospho-ATF-2 (Thr-71), total Akt, and total p38 MAP kinase were from Cell Signaling Technology. Mouse monoclonal antibody to phospho-p38 MAPK (Thr-180/Tyr-182) was from Cell Signaling Technology. Rabbit polyclonal antibody to phospho-Smad2 (Ser-465/467) was from Upstate Biotechnology, Inc., Lake Placid, NY. Goat polyclonal antibody to total Smad2 was from Santa Cruz Biotechnology, Santa Cruz, CA. Mouse monoclonal antibodies to α-smooth muscle actin and β-actin were obtained from Sigma. Mouse monoclonal antibody to single-stranded DNA (ssDNA) and rabbit polyclonal antibody against activated caspase 3 were from Chemicon International, Temecula, CA. Secondary horseradish peroxidase-conjugated anti-goat, anti-mouse, and anti-rabbit antibodies were obtained from Pierce. All other reagents including suramin were from Sigma.
Generation of p38 MAPK Mutant Cells
The mammalian expression plasmids, pcDNA3-HA-p38 (wild type), pcDNA3-p38-KM (kinase mutant), and pcDNA3-MKK3, were provided by Dr. Kun Liang Guan, Department of Biological Chemistry, University of Michigan, Ann Arbor. Plasmid transfections of IMR-90 cells using the cationic lipid reagent LipofectAMINE (Invitrogen) were performed according the manufacturer’s instructions. The optimal ratio of DNA (μg) to LipofectAMINE (μl) was determined to be ~1:5 for IMR-90 cells. Cells were incubated with DNA-lipid complexes in serum-free Opti-MEM I medium (Invitrogen) for 4–5 h prior to introducing 10% FBS for 16–20 h. The next day, transfection medium was replaced by DMEM supplemented with 10% FBS and geneticin (Invitrogen). Geneticin concentrations were 400 μg/ml for selection of stable transfectants and 200 μg/ml as a maintenance dose in cell cultures prior to performing experiments.
Western Immunoblotting
Cell lysates were prepared in RIPA buffer, subjected to SDS-PAGE, and Western blot analyses performed as described previously (37).
In Vitro p38 MAP Kinase Assay
In vitro p38 MAP kinase activity was measured using a nonradioactive assay obtained from Cell Signaling Technology, Beverly, MA. Immobilized antibody to phospho-p38 MAPK was added to 200 μg of whole cell lysates in RIPA buffer and incubated overnight at 4 °C. This was then centrifuged, and the supernatant was removed and the pellet washed three times with lysis buffer and kinase buffer as per the manufacturer’s protocol. The pellet was then suspended in kinase buffer supplemented with ATP and an ATF-2 fusion protein for 30 min at 30 °C. The reaction was terminated with 6× SDS sample buffer, and the samples were boiled, vortexed, and centrifuged. Samples were then subjected to SDS-PAGE followed by immunoblotting with an antibody to phospho-ATF-2.
Assays for Apoptosis
ELISA for ssDNA
Apoptosis was quantitated with the use of an ELISA-based assay for ssDNA (Apoptosis ELISA Kit, Chemicon International, Temecula, CA) according to the manufacturer’s instructions, with minor modifications (38). Cells were seeded directly into 96-well cell culture plates, grown to 80% confluence, growth-arrested for 48 h, and then treated with or without TGF-β1 in the presence of 10% FBS or under serum-deprived conditions for another 120 h. Prior to the assay, the 96-well plate was centrifuged for 5 min. The medium was removed, and cells were fixed to the plate with 80% methanol. The methanol was then removed, and the plate was dried in a 37 °C water bath for 20 min, after which 50 μl of formamide was added to each well for 10 min at room temperature followed by 10 min in a 75 °C water bath. After cooling, the formamide was removed, and 200 μl of 1% bovine serum albumin was added to each well for 1 h to block nonspecific binding. The blocking solution was then removed, and 50 μl of mouse monoclonal anti-ssDNA antibody (1:100) was added to each well for 30 min. After washing, 200 μl of horseradish peroxidase-conjugated anti-mouse secondary antibody was added for 30 min. After repeat washing, 100 μl of 2,2′-azino-bis-(3-benzthiazoline-6-sulfonic) acid solution was added to each well for 30 min followed by “stop solution” supplied by the manufacturer. Absorbance was read with an ELISA plate reader at 405 nm. The “background” absorbance of cells receiving no primary antibody was subtracted, and a relative apoptosis index calculated by dividing the corrected absorbance by cell counts (measured by Coulter counter) was obtained prior to fixing the cells.
Activated Caspase 3/Immunofluorescence Staining
Activated caspase 3 was detected by immunofluorescence staining of fixed cells. Briefly, IMR-90 cells plated on 35 mm of tissue culture were grown to 50% confluence and growth-arrested in medium containing 0.01% serum for 48 h prior to treatments for defined times. Cells were then fixed in 5% formaldehyde and washed three times with cold phosphate-buffered saline prior to permeabilization and after each subsequent step. Permeabilization was performed in buffer consisting of 0.1% Triton in 50 mm PIPES (pH 7.0), 90 mm HEPES (pH 7.0), 0.5 mm MgCl2, 0.5 mm EGTA, and 75 mm KCl. Nonspecific binding sites were blocked with 1% bovine serum albumin for 15 min prior to the addition of antibody to activated caspase 3 (1:25 dilution) for 1 h followed by fluorescein isothiocyanate-conjugated secondary antibody (1:40 dilution) for 1 h. Counterstaining was with DAPI for nuclear staining, and cells were visualized and photographed using a Zeiss fluorescence microscope.
Statistical and Densitometric Analyses
Statistical analysis was performed using Student’s t test and one-way analysis of variance with Bonferonni post test using GraphPad Prism version 3.0 for Windows, GraphPad Software, San Diego (www.graphpad.com). Densitometric analysis of Western blots was performed using the public domain NIH Image program available on the internet at rsb.info.nih.gov/nih-image.
RESULTS
TGF-β1 Protects Human Lung Mesenchymal Cells/Fibroblasts from Serum Deprivation-induced Apoptosis
Adult mesenchymal cells, particularly when isolated from injured tissues, often represent a variably heterogeneous population of cells; moreover, there may be differences in cellular phenotypes and responsiveness between adult and fetal fibroblasts/mesenchymal cells. Therefore, we examined the effects of TGF-β1 on mesenchymal cells isolated both from adult patients with acute lung injury (ALI-MC) and on normal human fetal lung fibroblasts (IMR-90). Spontaneous rates of apoptosis in the presence of serum were slightly higher in ALI-MC than in IMR-90 cells (Fig. 1). Serum deprivation for 5 days increased apoptotic rates in both groups. Treatment of serum-deprived cells with TGF-β1 (2 ng/ml) reduced apoptotic rates in ALI-MC and IMR-90 cells by 50 and 48%, respectively (Fig. 1). Interestingly, TGF-β1 also protected against spontaneous apoptosis (in the presence of serum) in ALI-MC cells (Fig. 1A). These results suggest that TGF-β1 is able to induce cellular resistance against both spontaneous and serum deprivation-induced apoptosis in different fibroblasts/mesenchymal cells, including heterogeneous adult mesenchymal cells (ALI-MC) and more homogeneous cultures of untransformed, human fetal lung fibroblasts (IMR-90).
Fig. 1. TGF-β1 protects human lung mesenchymal cells/fibroblasts from serum deprivation-induced apoptosis.
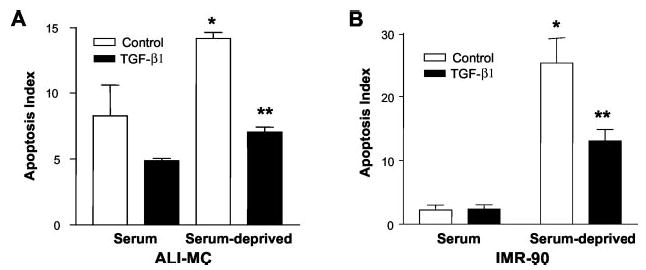
A, mesenchymal cells (ALI-MC) isolated from the lung of a patient with acute lung injury were cultured in 96-well cell culture plates in the presence of 10% FBS and grown to 80% confluence. Cells were then growth-arrested for 48 h and treated with/without TGF-β1 (2 ng/ml) in the presence or absence of serum for an additional 72 h. Apoptosis assays were performed as described under “Materials and Methods.” n = 4 for each group; *, p < 0.001 compared with serum/control; **, p < 0.001 compared with serum-deprived/control. B, normal fetal lung fibroblasts (IMR-90) were cultured, treated with/without TGF-β1, and assayed for apoptosis under identical conditions as described above. n = 4 for each group; *, p < 0.001 compared with serum/control; **, p < 0.01 compared with serum-deprived/control.
TGF-β1 Induces Early Activation of the Smad and p38 MAPK Followed by Delayed Activation of the PI3K-Akt Pathway in Lung Fibroblasts
The PI3K-Akt pathway is known to transduce signals critical for cell survival (24, 25). To determine whether the pro-survival/anti-apoptotic effects of TGF-β1 on fibroblasts may, at least in part, be attributed to this pathway, we examined the effects of TGF-β1 on PKB/Akt activation. In both ALI-MC and IMR-90 cells, phosphorylation of PKB/Akt was observed as early as 3–6 h following TGF-β1 (2 ng/ml) treatment with peak effects at 12 h (Fig. 2). Peak effects at 12 h were completely inhibited by treatment with LY294002 (10 μm) or wortmannin (1 μm) for 2 h prior to cell lysis (data not shown). TGF-β1-induced PKB/Akt phosphorylation was sustained for 24 h and decreased significantly by 48 h following treatment (Fig. 2).
Fig. 2. TGF-β1 induces time-dependent phosphorylation of PKB/Akt in human lung mesenchymal cells/fibroblasts.
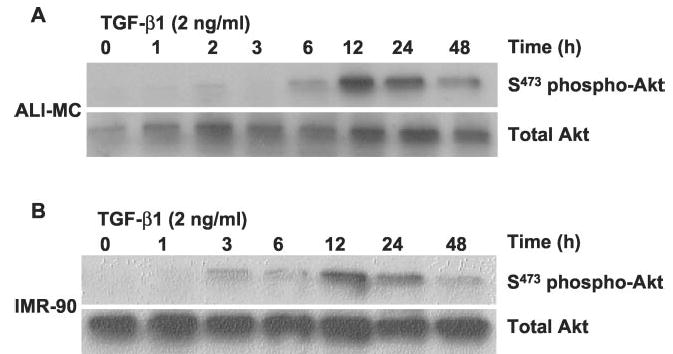
A, mesenchymal cells (ALI-MC) isolated from the lung of a patient with acute lung injury were cultured in the presence of 10% FBS, grown to 80% confluence, and then growth-arrested for 48 h. Cells were then treated with TGF-β1 (2 ng/ml) for the indicated times prior to cell lysis. Cell lysates were then subjected to SDS-PAGE and immunoblotted with phospho-specific antibodies to serine-473 of PKB/Akt (S473 phos-pho-Akt). The same blots were stripped and probed for total Akt. B, normal fetal lung fibroblasts (IMR-90) were similarly analyzed for induction of PKB/Akt phosphorylation in response to TGF-β1 stimulation.
The delayed responses in PKB/Akt phosphorylation suggested that other more proximal TGF-β1 signals mediate this effect. Rapid signaling from TGF-β receptor(s) activation is best recognized to occur by the Smad proteins (1). Recent studies suggest the involvement of Smad-independent pathways involving p38 MAPK (2, 30). Moreover, p38 MAPK appears to mediate PKB/Akt activation in mouse epithelial cells exposed to UV light (39). To determine whether p38 MAPK is rapidly activated in response to TGF-β1 in lung fibroblasts, we assessed the phosphorylated (activated) state of p38 MAPK and compared it with that of Smad2 phosphorylation. Fig. 3 demonstrates that both p38 MAPK and Smad2 are activated within 10 min of TGF-β1 (2 ng/ml) stimulation. Peak activation of these responses to TGF-β1 is observed at 1 h. These results demonstrate that the p38 MAPK pathway is activated rapidly in response to TGF-β1, with similar kinetics to that of Smad2 phosphorylation, and suggested that PKB/Akt phosphorylation might be mediated via p38 MAPK.
Fig. 3. TGF-β1 induces rapid activation of both Smad2 and p38 MAPK pathways in human lung fibroblasts.
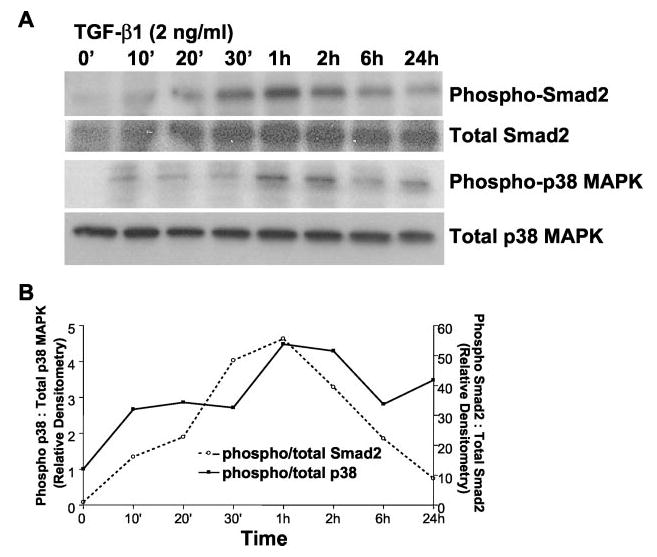
A, cultured human lung fibroblasts (IMR-90) were grown to near-confluence, growth-arrested for 48 h in 0.01% serum, and treated with TGF-β1 (2 ng/ml) for the indicated times prior to cell lysis. Whole cell lysates were subjected to SDS-PAGE and immunoblotted with antibodies to phospho-Smad2 and phospho-p38 MAPK. Each blot was then stripped and probed for total Smad2 or p38 MAPK. B, densitometric analyses of the levels of Smad2 and p38 MAPK phosphorylations represented in A.
Pharmacological Inhibition of p38 MAPK Blocks TGF-β1-induced PKB/Akt Phosphorylation
To determine whether early p38 MAPK activation is necessary for the more delayed activation of PKB/Akt, we examined the effect of various protein kinase inhibitors on TGF-β1-mediated PKB/Akt phosphorylation. In both ALI-MC and IMR-90 cells, blockade of the ERK-1/2 MAPK pathway with the MEK1 inhibitor, PD98059 (20 μm), had little effect on PKB/Akt phosphorylation (Fig. 4). In contrast, the p38 MAPK inhibitor, SB203580 (6 μm), completely inhibited TGF-β1-induced PKB/Akt phosphorylation to below base-line levels in both ALI-MC and IMR-90 cells (Fig. 4). Similar results were obtained when the effects of TGF-β1 were examined separately at 12 and 24 h after treatment (data not shown). An inhibitor of c-Jun N-terminal kinase (SP600125, 100 nm) more modestly attenuated the induction of PKB/Akt phosphorylation by 48% in ALI-MC and 27% in IMR-90 cells (Fig. 4). In the presence of the PI3K inhibitor, LY294002 (10 μm), PKB/Akt phosphorylation was virtually undetectable in IMR-90 cells (Fig. 4, C and D). Similarly, wortmannin (1 μm) reduced PKB/Akt phosphorylation to about 50% of control levels in ALI-MC (Fig. 4, A and B). Interestingly, the Rho kinase inhibitor, Y-27632 (15 nm), inhibited PKB/Akt phosphorylation to a greater degree in ALI-MC than in IMR-90 cells. Overall, these results suggest that pharmacological inhibition of the p38 MAPK pathway reliably and consistently blocks TGF-β1-induced PKB/Akt phosphorylation; more variable and apparently cell-specific responses are observed with an inhibitor of Rho kinase.
Fig. 4. Pharmacological inhibition of p38 MAP kinase inhibits TGF-β1-induced PKB/Akt phosphorylation in human lung fibro-blasts.
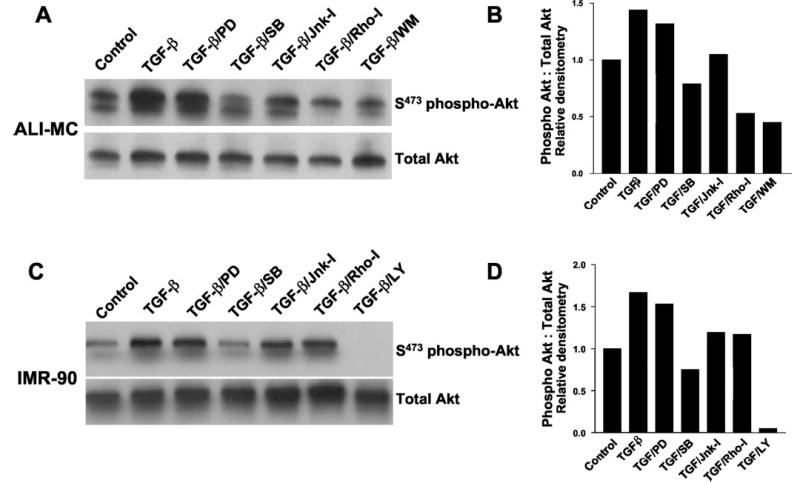
A, mesenchymal cells isolated from the lung of a patient with acute lung injury (ALI-MC) were grown to near-confluence and growth-arrested for 48 h prior to treatment with/without TGF-β1 (2 ng/ml for 12 h) in the presence/absence of the indicated protein kinase inhibitors. Cell lysates were then obtained and subjected to SDS-PAGE followed by immunoblotting against phosphorylated (serine 473) PKB/Akt (S473 phospho-Akt). The blot was stripped and probed for total Akt. B, densitometric analyses of the Western blots in A. C, human lung fibroblasts (IMR-90) were similarly cultured and treated ± TGF-β1 (2 ng/ml) ± protein kinase inhibitors as described above. Western blot analyses for Ser-473-phospho-Akt and total Akt are shown. D, densitometric analyses of the Western blots in C. PD, PD98059 (20 μm); SB, SB203580 (6 μm); Jnk-I, SP600125 (100 nm); Rho-I, Y27632 (15 nm); LY, LY294002 (10 μ m); and WM, wortmannin (50 nm).
Protein kinase inhibitors may exert non-specific effects, particularly at higher concentrations (40). SB203580 may directly inhibit PKB/Akt, although the IC50 values are 100–500-fold higher than for p38 MAPK (40). In vitro assays indicate that at a concentration of 10 μm, SB203580 almost completely inhibits p38 MAPK activity (98%), whereas PKB/Akt is inhibited by about 38% (40). To define better effective concentrations at which SB203580 inhibits TGF-β1-induced PKB/Akt phosphorylation, we performed a dose-response study by co-treating cells with increasing doses (0.6, 3, 6, and 18 μm) of SB203580 and TGF-β1 (2 ng/ml for 12 h). Fig. 5 demonstrates dose-dependent inhibition of TGF-β1-induced PKB/Akt phosphorylation. An ~50% inhibition was noted at 3 μm, and complete inhibition was achieved at 6 μm; at 18 μm, there was additional inhibition of the base-line levels of PKB/Akt phosphorylation (Fig. 5). Moreover, 6 μm SB203580 alone (in the absence of TGF-β1) had no effect on base-line PKB/Akt phosphorylation in these cells (data not shown). These findings suggest that the observed blockade of TGF-β1 stimulated PKB/Akt phosphorylation is more likely related to inhibitory effects of SB203580 on upstream p38 MAPK activation than to more direct effects of this compound on PKB/Akt.
Fig. 5. The p38 MAPK inhibitor, SB203580, blocks TGF-β1-induced phosphorylation of Akt in a dose-dependent manner.
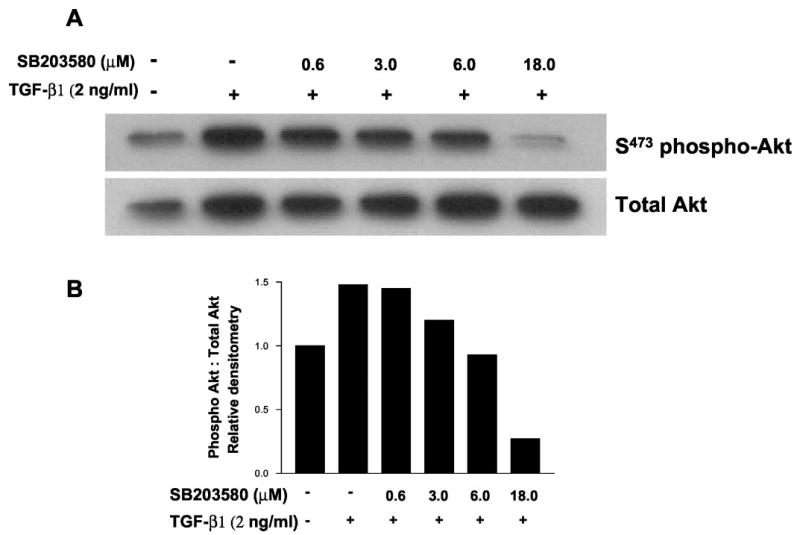
A, cultured IMR-90 cells were grown to near-confluence, growth-arrested for 48 h, and then stimulated with/without TGF-β1 (2 ng/ml) in the presence of the indicated concentrations of the p38 MAPK inhibitor, SB203580. Cell lysates were obtained 12 h after treatment and subjected to SDS-PAGE followed by immunoblotting with an antibody to phospho(serine 473)-specific PKB/Akt. The blot was stripped and probed with antibody to total Akt. B, densitometric analyses of phospho-Akt:total Akt Western blots represented in A.
PKB/Akt Phosphorylation by TGF-β1 Is Dependent on Functional p38 MAPK Activation
To address further the role of p38 MAPK activation in TGF-β1-induced PKB/Akt phosphorylation, we generated stable cell lines of IMR-90 cells expressing a kinase-deficient/mutant p38 MAPK (p38-KM), a constitutively active MKK3 (MKK3), and control (pcDNA, empty vector) plasmid constructs. TGF-β1 (2 ng/ml for 1 h) induced phosphorylation of p38 MAPK in all cell lines (Fig. 6A, top panel). However, p38 MAPK activity measured by in vitro phosphorylation of ATF-2 was not induced in p38-KM cells at the same time point (Fig. 6A, upper middle panel), consistent with functionally inactive p38 MAPK in these cells. Overexpression of MKK3 did not appear to augment further the ATF-2 phosphorylation in response to TGF-β1, and basal levels appeared to be lower than control pcDNA cells. The pattern of ATF-2 phosphorylation/p38 activity closely correlated with PKB/Akt phosphorylation induced by TGF-β1 in all cell lines. Importantly, TGF-β1-induced PKB/Akt phosphorylation was completely inhibited in p38-KM cells (Fig. 6A, lower middle panel, and B). Moreover, the activation of this pathway appears to be less important for TGF-β1-induced α-smooth muscle cell actin, a marker of myofibroblast differentiation, because this response is preserved in p38-KM cells (Fig. 6A, lower panel). These results indicate that activation of p38 MAPK by TGF-β1 is required for the induction of PKB/Akt and suggest that activation of this pathway may be relatively more specific for pro-survival signaling than for myofibroblast differentiation-inducing effects of TGF-β1.
Fig. 6. Functional p38 MAPK activity is required for TGF-β1-induced PKB/Akt phosphorylation.
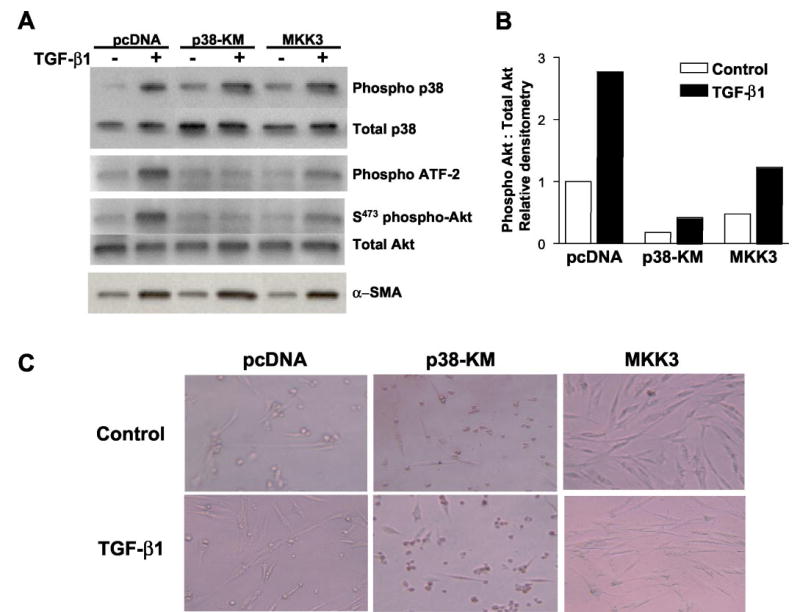
A, IMR-90 cells stably transfected with control plasmid (pcDNA), a kinase-deficient p38 MAPK construct (p38-KM), or wild-type MKK3 (MKK3) were grown to near-confluence, growth-arrested for 48 h prior to treatment with or without TGF-β1 (2 ng/ml), and cell lysates obtained at 1, 12, and 16 h. Phosphorylation of p38 MAPK was assessed at 1 h following TGF-β1 treatment by Western immunoblotting with antibodies to phospho- and total p38 MAPK (top panels). p38 MAP kinase activity was assessed at 1 h following TGF-β1 treatment by in vitro phosphorylation of the p38 MAPK substrate, ATF-2, as described under “Materials and Methods” (middle top panel). Phosphorylation of PKB/Akt was assessed at 12 h following TGF-β1 treatment by Western immunoblotting with antibodies to phospho- and total Akt (middle bottom panels). Cell lysates obtained 16 h after treatment with TGF-β1 were subjected to Western immunoblotting for α-smooth muscle actin (α-SMA), a marker of myofibroblast differentiation (bottom panel). B, densitometric analyses of the phospho-Akt:total Akt blots represented in A. C, stably transfected cells were cultured to 80% confluence, growth-arrested in DMEM with 0.01% FBS for 48 h, treated with/without TGF-β1 (2 ng/ml) for 2 days. Representative photographs demonstrate cellular morphology under these serum-deprived conditions.
The morphology of these stable cell lines in the presence and absence of TGF-β1 after 48 h of serum deprivation was examined (Fig. 6C). Stable p38-KM cells demonstrated reduced viability with large numbers of rounded cells, whereas cells overexpressing MKK3 were more spread/fibroblast-like in appearance. These observations support the notion that activation of the p38 MAPK pathway is important for the pro-survival effects of TGF-β1 in serum-deprived conditions.
TGF-β1-induced PKB/Akt Activation Is Mediated by p38 MAPK dependent Autocrine Growth Factor Stimulation
The delayed phosphorylation of PKB/Akt relative to the early and rapid activation of p38 MAPK in response to TGF-β1 suggested that production of an intermediary mediator/growth factor may activate this pro-survival pathway in an autocrine manner. To test this possibility, we examined the effects of conditioned medium from control and TGF-β1-treated cells on the activation of PKB/Akt in untreated IMR-90 cells. PKB/Akt phosphorylation is rapidly induced within 15 min when conditioned medium collected from cells treated with TGF-β1 for 16 h is added to untreated, serum-deprived IMR-90 cells; less pronounced stimulation is seen with conditioned medium from control/untreated cells (Fig. 7, A and B). This suggests that the secreted factor(s) responsible for activation of PKB/Akt may be produced at basal levels in control cells and that this production may be up-regulated by TGF-β1. To determine whether this factor(s) was functioning by ligand-receptor interactions at the cell surface, cells were pre-treated with suramin (300 μm), a nonspecific inhibitor of growth factor receptor activation that functions by blocking the binding of growth factor ligands to their extracellular membrane receptors (41). Suramin blocked the activation of PKB/Akt by conditioned medium from both control and TGF-β1-treated cells (Fig. 7, A and B), suggesting the involvement of a secreted factor(s) that mediates this action via a cell-surface receptor(s)-mediated mechanism. To determine whether the production of this factor was dependent on the early activation of p38 MAPK by TGF-β1, conditioned medium from cells treated with TGF-β1 in the presence/absence of SB203580 (10 μm) was added to IMR-90 cells, and early responses on PKB/Akt phosphorylation were determined. TGF-β1 conditioned medium-induced PKB/Akt phosphorylation was blocked when this medium was generated in the presence of the p38 MAPK inhibitor, SB203580, but not by the MEK1 (ERK1/2 MAPK) inhibitor, PD98059. Cumulatively, these results suggest that activation of p38 MAPK is required for the production/secretion of a putative growth factor(s) that mediates rapid activation of the PI3K-Akt pathway in an autocrine/paracrine manner.
Fig. 7. TGF-β1 induces p38 MAPK-dependent production/secretion of a growth factor that mediates PKB/Akt activation in an autocrine manner.
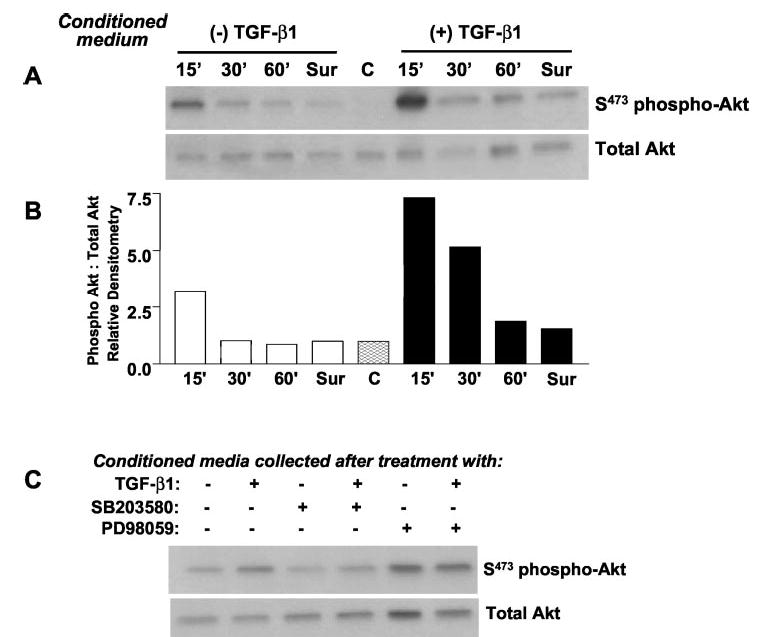
A, IMR-90 cells were treated with/without TGF-β1, and the conditioned medium was collected at 16 h. Conditioned media were added to a different set of “reporter” IMR-90 cell cultures for the shorter periods indicated. Cell lysates were then subjected to SDS-PAGE and immunoblotted with antibodies against phosphorylated (serine 473) PKB/Akt (S473 phospho-Akt); blots were stripped and re-probed for total Akt. Suramin (300 μ m, Sur) was pre-added to reporter cells 30 min prior to incubation with conditioned medium for 15 min. Control cells (C) that received no conditioned medium are also represented. B, densitometric analyses of the ratio of phospho-Akt to total Akt determined from immunoblots in A. C, conditioned media were collected from IMR-90 cells treated with TGF-β1 in the presence/absence of inhibitors of p38 MAPK (SB203580, 10 μ m) or ERK1/2 MAPK (PD98049, 20 μ m) for 16 h. Conditioned medium was then added to reporter IMR-90 cells that had been serum-deprived for 24 h. Cells were then harvested at 15 min and lysates subjected to Western blot analysis for phosphorylated (serine 473) PKB/Akt (S473 phospho-Akt); blots were stripped and probed for total Akt.
A Functional PI3K-Akt Pathway Is Required to Confer Apoptosis Resistance to Serum Deprivation in Lung Fibroblasts by TGF-β1
The PI3K-Akt pathway has been linked to pro-survival/anti-apoptotic signaling (25). To determine whether activation of this pathway is essential for anti-apoptotic effects of TGF-β1, we examined the effect of the PI3K inhibitor, LY294002 (10 μm), on serum-induced apoptosis. Inhibition of PI3K completely reverses the protective effect of TGF-β1 on serum deprivation-induced apoptosis (Fig. 8A). This effect is also observed when apoptosis is assayed by counting the number of cells that stain positively for activated caspase 3 (Fig. 8, B and C). These results suggest that activation of the PI3K-Akt pathway is required for induction of the apoptosis-resistant phenotype in mesenchymal cells/fibroblasts exposed to TGF-β1.
Fig. 8. TGF-β1-induced protection from serum deprivation-induced apoptosis of lung fibroblasts is dependent on the PI3K/Akt pathway.
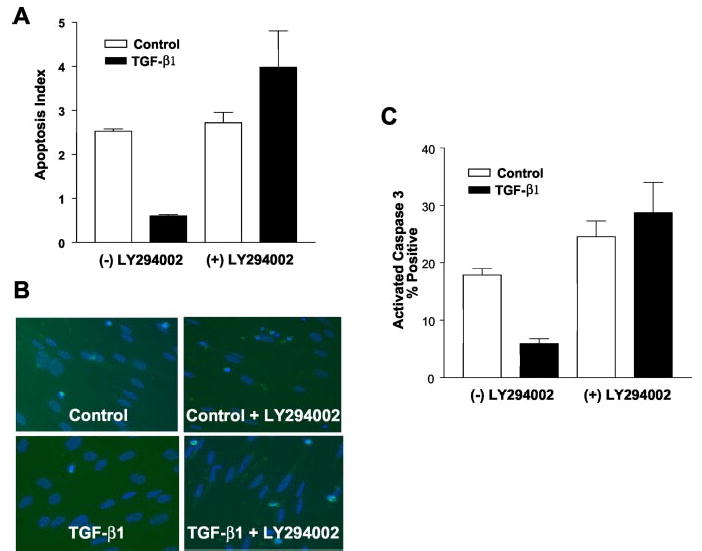
A, IMR-90 cells were grown to near-confluence in a 96-well ELISA culture plate and growth-arrested for 48 h in 0.01% serum prior to treatment with TGF-β1 ± LY294002 (10 μ m) as indicated for 5 days. Apoptosis rates were measured using an ELISA for single-stranded DNA as described under “Materials and Methods.” Findings represent one of three independent experiments. Values are mean ± S.E., n = 3. *, p < 0.05 compared with untreated controls. B, cultured IMR-90 cells were grown to 50% confluence, growth-arrested in 0.01% serum for 48 h, and then treated with/without TGF-β1 (2 ng/ml) in the presence/absence of the PI3K inhibitor, LY294002 (10 μ m), as indicated. Immunofluorescence staining for activated caspase 3 was performed 5 days after treatment. Counter staining with DAPI (blue) was used to identify nuclear morphology. C, quantitative assessment of apoptosis by activated caspase 3-positivity for the experimental conditions shown in B. Three random 20× fields containing 50–60 cells per field were selected from each group. % apoptosis was measured by determining dividing the total number of activated caspase 3-positive cells (fluorescein isothiocyanate/green) by the total number of cells (DAPI/blue). Findings are representative of three independent experiments. Values are mean ± S.E., n = 3. *, p < 0.05 compared with untreated controls.
DISCUSSION
TGF-β1 is a prototypical, multifunctional cytokine that mediates varied and contextual responses in different phases of tissue repair/fibrosis following injury as well as in other complex disease processes such as carcinogenesis. TGF-β1 actions are also highly cell-specific. Growth/survival of epithelial cells is suppressed whereas that of fibroblasts (and myofibroblasts) is enhanced. These are key pathophysiological features of the dysregulated repair process in chronic, progressive fibrotic diseases. The results of this study indicate that the pro-survival/anti-apoptotic phenotype of human lung fibroblasts/mesenchymal cells induced by TGF-β1 is mediated, at least in part, by activation of the PI3K-Akt pathway. Our data demonstrate that activation of PI3K-Akt is mediated by a putative secreted growth factor(s) whose production is dependent on early activation of p38 MAPK by TGF-β receptor(s) signaling.
Cell-specific differences between epithelial and mesenchymal cells in the response to TGF-β1 have been described, but the mechanisms are unclear (1). The results of our study suggest that differences in the activation and integration of post-receptor signaling pathways may account, in large part, for the observed differences in cell-specific responses. There is better understanding of TGF-β1 signaling by Smad-independent pathways in epithelial cells than in mesenchymal cells. There is substantial evidence that TGF-β1-induced p38 MAPK activation in epithelial cells can lead to enhanced apoptotic responses (17, 30, 31), a finding consistent with current models of fibrotic disease (42). Interestingly, when the PI3K-Akt pathway is activated in epithelial cells by TGF-β1, a different biological response, that of EMT, may result (26). In other studies in which TGF-β1-induced EMT has been observed, and p38 MAPK implicated in the response, specific roles for PI3K-Akt were not examined (34, 43). Moreover, the activation of the PI3K/Akt pathway in epithelial cells may be important not only for EMT but to confer resistance to apoptosis (44). This suggests that the ability to activate the PI3K-Akt may determine very different cell fates downstream of the early activation of p38 MAPK by TGF-β1 in epithelial cells, i.e. EMT/apoptosis resistance versus apoptosis. However, the relationship and integration between these TGF-β1-activated pathways are unclear.
Our studies demonstrate for the first time, in any cell type, that TGF-β1-induced activation of PI3K-Akt pathway is dependent on the early and rapid activation of p38 MAPK. Early p38 MAPK activation is essential for the production/secretion of a secreted factor(s) that is then able to rapidly activate PI3K-Akt by an autocrine mechanism (Fig. 9). Candidate molecules for this effect likely involve receptor tyrosine kinase(s)-linked growth factors such as FGF-2 (19) or IGF-1 (45). The stability of the soluble secreted factor in conditioned medium and the observed inhibition by suramin argue against non-receptor-ligand mechanisms, such as oxidant generation, in mediating PKB/Akt activation (46, 47). Our previous studies also show that TGF-β1 may activate integrin-dependent focal adhesion kinase (11), which may associate with and activate PI3K at focal adhesions (48, 49). However, integrin-extracellular matrix signaling is unlikely to fully account for this response based on the soluble nature of the secreted factor(s).
Fig. 9. Schematic representation of pro-survival signaling in mesenchymal cells by TGF-β1.
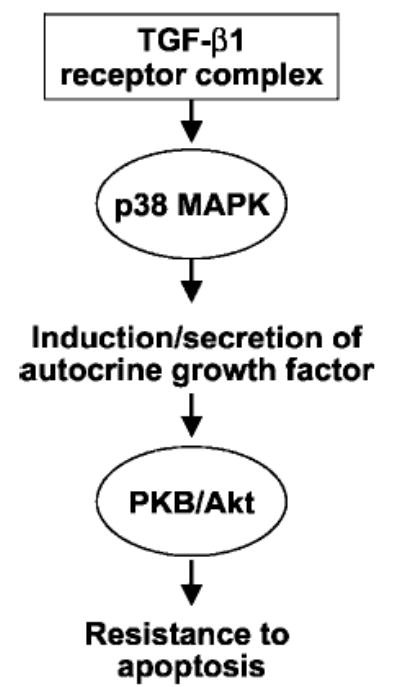
In the current study, we have demonstrated that TGF-β1 induces rapid activation of p38 MAPK followed by a more delayed activation of the PKB/Akt pathway in lung mesenchymal cells/fibroblasts. Activation of the PI3K/Akt pathway is mediated by p38 MAPK-dependent production of a putative growth factor(s) that functions in an autocrine/paracrine manner. Activation of the PI3K/Akt pathway is sufficient to confer a pro-survival/apoptosis-resistant phenotype to human mesenchymal cells/fibroblasts.
An important focus of the current study was to identify Smad-independent pathways that are activated in mesenchymal cells by TGF-β1. Although our data clearly suggest a role for p38 MAPK in pro-survival/anti-apoptotic signaling in these cells, it is possible that Smads and p38 MAPK play co-operative roles in modulating this cellular phenotype (29). However, the cell-specific and/or contextual induction of autocrine PI3K-Akt activation may also function to suppress Smad-dependent pathways and its associated tumor-suppressive effects (50). Additionally, overexpression of MKK3 in our studies did not result in augmentation of p38 MAPK activation in response to TGF-β1 or in the downstream activation of PKB/Akt. This may be related to the recently described MAPKK-independent mechanisms of p38 MAPK activation by TGF-β1 (32, 51).
Previous studies from our laboratory demonstrated a critical role for focal adhesion kinase in myofibroblast differentiation induced by TGF-β1 (11); the current study suggests that activation of PI3K/Akt is not essential for TGF-β1-induced myofibroblast differentiation but is perhaps more important in protecting these cells from apoptosis. Thus, dual activation of these pathways by TGF-β1 may serve to mediate both myofibroblast differentiation and prolonged survival of myofibroblasts in injured tissues undergoing active repair/remodeling. The activation of the PI3K/Akt pathway in mesenchymal cells/fibroblasts may be a key regulatory event in a number of human fibrotic diseases in which persistent mesenchymal cell activation/survival and overexpression of TGF-β1 are hallmarks of disease pathogenesis (3, 15). In addition, activation of such autocrine pathways may play an important role in epithelial-mesenchymal transition/cross-talk in late stages of carcinogenesis, where TGF-β1 may function as a “tumor promoter” rather than as a “tumor suppressor” (52).
Acknowledgments
We thank Dr. Kun Liang Guan (Department of Biological Chemistry, University of Michigan) for providing the p38 kinase-deficient, MKK3, and control plasmid constructs used in these studies.
Footnotes
This work was supported by Grants R01 HL-67967 and P50 HL-74021 from the National Institutes of Health.
The abbreviations used are: TGF-β, transforming growth factor-β1; MAP, mitogen-activated protein; MAPK, MAP kinase; PI3K, phosphatidylinositol 3-kinase; EMT, epithelial-mesenchymal transition; DAPI, 4,6-diamidino-2-phenylindole; PIPES, 1,4-piperazinediethanesulfonic acid; DMEM, Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; ssDNA, single-stranded DNA; ELISA, enzyme-linked immunosorbent assay; PKB, protein kinase B; ALI, acute lung injury; MC, mesenchymal cells; KM, kinase mutant.
References
- 1.Massague J. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 2.Roberts, A. B., and Derynck, R. (2001) Science’s STKE http:/www.stke.org/cgi/content/full/OC_sigtrans;2001/PE43 [DOI] [PubMed]
- 3.Border WA, Ruoslahti E. J Clin Investig. 1992;90:1–7. doi: 10.1172/JCI115821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grande JP. Proc Soc Exp Biol Med. 1997;214:27–40. doi: 10.3181/00379727-214-44066. [DOI] [PubMed] [Google Scholar]
- 5.Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, Sporn MB. Proc Natl Acad Sci U S A. 1987;84:5788–5792. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postlethwaite AE, Keski-Oja J, Moses HL, Kang AH. J Exp Med. 1987;165:251–256. doi: 10.1084/jem.165.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, Fauci AS. Proc Natl Acad Sci U S A. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ignotz RA, Massague J. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 9.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 10.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. J Cell Biol. 1993;122:103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 12.Desmouliere A, Redard M, Darby I, Gabbiani G. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 13.Grinnell F, Zhu M, Carlson MA, Abrams JM. Exp Cell Res. 1999;248:608–619. doi: 10.1006/excr.1999.4440. [DOI] [PubMed] [Google Scholar]
- 14.Blobe GC, Schiemann WP, Lodish HF. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 15.Border WA, Noble NA. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 16.Brown TL, Patil S, Cianci CD, Morrow JS, Howe PH. J Biol Chem. 1999;274:23256–23262. doi: 10.1074/jbc.274.33.23256. [DOI] [PubMed] [Google Scholar]
- 17.Dai C, Yang J, Liu Y. J Biol Chem. 2003;278:12537–12545. doi: 10.1074/jbc.M300777200. [DOI] [PubMed] [Google Scholar]
- 18.Leof EB, Proper JA, Goustin AS, Shipley GD, DiCorleto PE, Moses HL. Proc Natl Acad Sci U S A. 1986;83:2453–2457. doi: 10.1073/pnas.83.8.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finlay GA, Thannickal VJ, Fanburg BL, Paulson KE. J Biol Chem. 2000;275:27650–27656. doi: 10.1074/jbc.M000893200. [DOI] [PubMed] [Google Scholar]
- 20.Thannickal VJ, Aldweib KD, Rajan T, Fanburg BL. Biochem Biophys Res Commun. 1998;251:437–441. doi: 10.1006/bbrc.1998.9443. [DOI] [PubMed] [Google Scholar]
- 21.Jelaska A, Korn JH. Arthritis Rheum. 2000;43:2230–2239. doi: 10.1002/1529-0131(200010)43:10<2230::AID-ANR10>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 22.Zhang HY, Phan SH. Am J Respir Cell Mol Biol. 1999;21:658–665. doi: 10.1165/ajrcmb.21.6.3720. [DOI] [PubMed] [Google Scholar]
- 23.Kim G, Jun JB, Elkon KB. Arthritis Rheum. 2002;46:1504–1511. doi: 10.1002/art.10314. [DOI] [PubMed] [Google Scholar]
- 24.Brazil DP, Park J, Hemmings BA. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- 25.Datta SR, Brunet A, Greenberg ME. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 26.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 27.Nicolas FJ, Lehmann K, Warne PH, Hill CS, Downward J. J Biol Chem. 2003;278:3251–3256. doi: 10.1074/jbc.M209019200. [DOI] [PubMed] [Google Scholar]
- 28.Chen RH, Su YH, Chuang RL, Chang TY. Oncogene. 1998;17:1959–1968. doi: 10.1038/sj.onc.1202111. [DOI] [PubMed] [Google Scholar]
- 29.Hanafusa H, Ninomiya-Tsuji J, Masuyama N, Nishita M, Fujisawa J, Shibuya H, Matsumoto K, Nishida E. J Biol Chem. 1999;274:27161–27167. doi: 10.1074/jbc.274.38.27161. [DOI] [PubMed] [Google Scholar]
- 30.Yu L, Hebert MC, Zhang YE. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edlund S, Bu S, Schuster N, Aspenstrom P, Heuchel R, Heldin NE, Ten Dijke P, Heldin CH, Landstrom M. Mol Biol Cell. 2003;14:529–544. doi: 10.1091/mbc.02-03-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi K, Nagai S, Ninomiya-Tsuji J, Nishita M, Tamai K, Irie K, Ueno N, Nishida E, Shibuya H, Matsumoto K. EMBO J. 1999;18:179–187. doi: 10.1093/emboj/18.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K. Science. 1996;272:1179–1182. doi: 10.1126/science.272.5265.1179. [DOI] [PubMed] [Google Scholar]
- 34.Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. J Biol Chem. 2001;276:46707–46713. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- 35.Ware LB, Matthay MA. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 36.Janin A, Konttinen YT, Gronblad M, Karhunen P, Gosset D, Malmstrom M. Clin Exp Rheumatol. 1990;8:237–242. [PubMed] [Google Scholar]
- 37.Thannickal VJ, Aldweib KD, Fanburg BL. J Biol Chem. 1998;273:23611–23615. doi: 10.1074/jbc.273.36.23611. [DOI] [PubMed] [Google Scholar]
- 38.Frankfurt OS, Krishan A. J Immunol Methods. 2001;253:133–144. doi: 10.1016/s0022-1759(01)00387-8. [DOI] [PubMed] [Google Scholar]
- 39.Nomura M, Kaji A, He Z, Ma WY, Miyamoto K, Yang CS, Dong Z. J Biol Chem. 2001;276:46624–46631. doi: 10.1074/jbc.M107897200. [DOI] [PubMed] [Google Scholar]
- 40.Davies SP, Reddy H, Caivano M, Cohen P. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sachsenmaier C, Radler-Pohl A, Zinck R, Nordheim A, Herrlich P, Rahmsdorf HJ. Cell. 1994;78:963–972. doi: 10.1016/0092-8674(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 42.White ES, Lazar MH, Thannickal VJ. J Pathol. 2003;201:343–354. doi: 10.1002/path.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. J Cell Sci. 2002;115:3193–3206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- 44.Valdes F, Alvarez AM, Locascio A, Vega S, Herrera B, Fernandez M, Benito M, Nieto MA, Fabregat I. Mol Cancer Res. 2002;1:68–78. [PubMed] [Google Scholar]
- 45.Gooch JL, Tang Y, Ricono JM, Abboud HE. J Biol Chem. 2001;276:42492–42500. doi: 10.1074/jbc.M102994200. [DOI] [PubMed] [Google Scholar]
- 46.Thannickal VJ, Fanburg BL. J Biol Chem. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 47.Esposito F, Chirico G, Gesualdi NM, Posadas I, Ammendola R, Russo T, Cirino G, Cimino F. J Biol Chem. 2003;278:20828–20834. doi: 10.1074/jbc.M211841200. [DOI] [PubMed] [Google Scholar]
- 48.Chen HC, Guan JL. Proc Natl Acad Sci U S A. 1994;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM. J Biol Chem. 1999;274:20693–20703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- 50.Song K, Cornelius SC, Reiss M, Danielpour D. J Biol Chem. 2003;278:38342–38351. doi: 10.1074/jbc.M304583200. [DOI] [PubMed] [Google Scholar]
- 51.Ge B, Xiong X, Jing Q, Mosley JL, Filose A, Bian D, Huang S, Han J. J Biol Chem. 2003;278:2286–2293. doi: 10.1074/jbc.M210918200. [DOI] [PubMed] [Google Scholar]
- 52.Massague J, Blain SW, Lo RS. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]