

Abstract
VEGFR-1 is a kinase-defective receptor tyrosine kinase (RTK) and negatively modulates angiogenesis by acting as a decoy receptor. The decoy characteristic of VEGFR-1 is required for normal development and angiogenesis. To date, there is no molecular explanation for this unusual characteristic of VEGFR-1. Here we show that the molecular mechanisms underlying the decoy characteristic of VEGFR-1 is linked to the replacement of a highly conserved amino acid residue in the activation loop. This amino acid is highly conserved among all the type III RTKs and corresponds to aspartic acid, but in VEGFR-1 it is substituted to asparagine. Mutation of asparagine (Asn1050) within the activation loop to aspartic acid promoted enhanced ligand-dependent tyrosine autophosphorylation and kinase activation in vivo and in vitro. The mutant VEGFR-1 (Asp1050) promoted endothelial cell proliferation but not tubulogenesis. It also displayed an oncogenic phenotype as its expression in fibroblast cells elicited transformation and colony growth. Furthermore, mutation of the invariable aspartic acid to asparagine in VEGFR-2 lowered the autophosphorylation of activation loop tyrosines 1052 and 1057. We propose that the conserved aspartic acid in the activation loop favors the transphosphorylation of the activation loop tyrosines, and its absence renders RTK to a less potent enzyme by disfavoring transphosphorylation of activation loop tyrosines.
VEGFR-1 (vascular endothelial growth factor receptor-1/FLT-1) is the prototype of a receptor tyrosine kinase gene family encoding structurally related receptors including FLK-1 (VEGFR-2), FLT-3 (FLK-2), and FLT-4 (VEGFR-3) (1). VEGFR-1 is expressed by endothelial cells and acts a high affinity receptor for VEGF-A, VEGF-B, and placenta growth factor (2). Although the precise role of VEGFR-1 in angiogenesis is not known, its presence is required for normal development and angiogenesis. Targeted deletion of VEGFR-1 results in early embryonic lethality due to abnormal overgrowth of endothelial cells (6, 13). Further studies revealed that VEGFR-1 negatively modulates angiogenesis by acting as a decoy receptor by trapping VEGF2 and thus preventing VEGF from binding to and activating VEGFR-2 (5, 7). Consistent with genetic studies, selective activation of VEGFR-1 results in no significant proliferation and migration of endothelial cells in vitro (5, 9). Also stimulation of VEGFR-1 in a defined condition antagonizes VEGFR-2-mediated proliferation of endothelial cells (5, 14).
Tyrosine autophosphorylation represents a crucial event in the activation of RTKs. The basic principles of RTK activation can be summarized as follows: ligand-mediated dimerization of receptor monomers, transphosphorylation by dimerized receptors, and docking of signaling proteins to receptor phosphotyrosines. An increase in the intrinsic catalytic activity and creation of binding sites on the RTKs to recruit cytoplasmic signaling proteins are primary features of RTK activation (11, 33, 39). In this context, VEGFR-1 is considered to be a kinase-impaired RTK (e.g. VEGFR-1 is poorly tyrosine-phosphorylated, and its ability to phosphorylate substrate is negligible).
It could be argued that the decoy characteristic of VEGFR-1 is not an accidental event but rather serves an important biological role in embryonic development and pathological angiogenesis. It is not known to what the decoy characteristic of VEGFR-1 can be attributed. VEGFR-1, like other RTKs, possesses all of the known signatures of RTKs, such as GXGXXG, an ATP binding motif, HRDLA, a motif essential for catalysis, and many potential tyrosine autophosphorylation sites within its kinase domain and carboxyl tail (1–3, 11). Despite having these similarities with other RTKs, ligand stimulation of VEGFR-1 results only in minor tyrosine autophosphorylation of VEGFR-1 in vivo and in vitro (3–5, 9). Thus, it is highly likely that the poor tyrosine autophosphorylation of VEGFR-1 may account for its decoy characteristic. Current literature on VEGFR-1 indicates that the kinase-defective characteristic of VEGFR-1 is not associated with its extracellular domain or juxtamembrane domain. Replacing the extracellular domain of VEGFR-1 with that of CSF-1R or epidermal growth factor receptor (5, 14) or its juxtamembrane region with that of VEGFR-2 does not alter its kinase-impaired characteristic (19). In fact, the extracellular domain of VEGFR-1 displays a higher affinity to VEGF than that of VEGFR-2 (20, 25). A more recent study demonstrated that substitution of carboxyl terminus of VEGFR-2 with that of VEGFR-1 could rescue its kinase-defective activity (9).
In this study, we wished to determine the molecular determinant of decoy characteristic of VEGFR-1. Our search of protein-tyrosine kinase data base revealed an alteration in the activation loop of VEGFR-1. In the activation loop of VEGFR-1, Asp, an amino acid that is highly conserved in all of the type III RTKs is substituted to Asn. We demonstrate that substitution of this invariable amino acid in the activation loop of human VEGFR-1 is responsible for the decoy characteristic of VEGFR-1.
MATERIALS AND METHODS
Reagents and Antibodies
Mouse anti-phosphotyrosine (PY-20) was purchased from Transduction Laboratories (Lexington, KY). Mouse anti-phosphotyrosine (4G10) was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Rabbit anti-VEGFR-2 was made against amino acids corresponding to the carboxyl terminus of VEGFR-2. Rabbit anti-phosphotyrosine 1173 VEGFR-2 was purchased from Cell Signaling Technology, Inc. (Beverly, MA). The following antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA): rabbit and goat polyclonal anti-FLT-1, goat anti-rabbit antibody, goat anti-mouse antibody, and donkey anti-goat antibody antibodies. Recombinant human macrophage colony-stimulating factor was purchased from R&D Systems (Minneapolis, MN). ATP, Protein G-Sepharose, and poly-Glu (4:1 Glu, Tyr) peptide were purchased from Sigma. Anti-phospho-Tyr1057/1059 VEGFR-2 antibody was purchased from BIOSOURCE International.
Site-directed Mutagenesis
The cDNA for human VEGFR-1, chimeric VEGFR-1 (CTR), and chimeric VEGFR-2 (CKR) were used as template to generate the mutants D1050/CTR, D1050/VEGFR-1, and N1054/CKR, respectively. The creation of chimeric VEGFR-1 (CTR) and VEGFR-2 (CKR), in which their extracellular domain is replaced with that of human CSF-1R, was described elsewhere (5). The mutations were made using a PCR-based site-directed mutagenesis method as previously described (5). The reactions were carried out using Accuprime Pfx DNA polymerase (Invitrogen). The resultant mutations were verified by sequencing and were subsequently cloned into pLXSN2 or pLNCX2 retroviral vectors.
In Vitro Kinase Assay and Substrate Phosphorylation
Equal numbers of porcine aortic endothelial (PAE) cells expressing either CTR, D1050/CTR, VEGFR-1, or D1050/VEGFR-1 were serum-starved overnight and lysed without stimulation. CTR and D1050/CTR proteins were immunoprecipitated with anti-VEGFR-1/FLT-1 antibody. The immunoprecipitated proteins were washed once with cold lysis buffer, pH 7.4 (1% Triton X-100, 10 mm Tris-HCl, 5 mm EDTA, 50 mm NaCl, 50 mm NaF, 2 mm sodium orthovanadate, 1 μm leupeptin, 1 mm phenylmethylsulfonyl fluoride, and 20 μg/ml aprotinin) and three times with cold PAN buffer (10 mm PIPES, pH 7.0, 100 mm NaCl, 20 mg/ml aprotinin). An in vitro kinase assay was performed by incubating immunoprecipitated proteins with 20 μl of kinase buffer (10 mm MgCl2, 1 mM dithiothreitol, 100 mm NaCl, 20 mm Tris-HCl, pH 7.4) containing 0.1–1 mm ATP for 15 min at 30 °C. The reaction was stopped by the addition of an equal volume of SDS sample buffer. The samples were denatured and resolved on 7.5% SDS-PAGE and subjected to Western blot analysis using anti-phosphotyrosine antibody. To measure the ability of CTR and mutant D1050/CTR to phosphorylate substrate (poly-Glu peptide), cells were stimulated with ligand for 10 min, lysed, and immunoprecipitated with anti-VEGFR-1 antibody. Substrate phosphorylation was measured as described (27). Briefly, immunoprecipitated proteins were incubated in 10 μCi of [γ-32P]ATP for 15 min at 30 °C in the presence of substrate (5 μg/reaction). The reaction was stopped, and samples were spotted on the p81 paper, and after extensive washing the p81 papers were subjected to a scintillation counter, and their dpm values were measured.
Immunoprecipitation and Western Blot Analysis
PAE cells expressing CTR or D1050/CTR were grown in sparse conditions in 10% FBS and serum-starved overnight. Cells were left either resting or stimulated with 40 ng/ml CSF-1 at 37 °C for appropriate times as indicated in the figure legends. Cells were washed twice with H/S buffer (25 mm HEPES, pH 7.4, 150 mm NaCl, 2 mm Na3VO4) and lysed in lysis (EB) buffer (10 mm Tris-HCl, pH 7.4, 5 mm EDTA, 50 mm NaCl, 50 mm NaF, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, 2 mm Na3VO4, and 20 μg/ml aprotinin). VEGFR-1 proteins were immunoprecipitated with anti-VEGFR-1 antibody, and immune complexes were bound to Protein G-Sepharose. Immunoprecipitates were resolved on a 7.5% SDS-polyacrylamide gel, and the proteins were transferred to polyvinylidene difluoride membrane. For anti-phosphotyrosine Western blot analysis, the membranes were then incubated in Block buffer containing 10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mg/ml bovine serum albumin, 10 mg/ml ovalbumin, 0.05% Tween 20, and 0.005% NaN3 and then incubated with primary antibody diluted in Block. The membranes were then washed and incubated with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit antibodies. Finally, the membranes were washed, and developed with ECL (Amersham Biosciences). In some instances, the membranes were stripped by incubating them in a buffer containing 6.25 mm Tris-HCl, pH 6.8, 2% SDS, and 100 mm β-mercaptoethanol in 50 °C for 30 min and reprobed.
Thymidine Uptake Assay
CSF-1-stimulated cell proliferation was measured by uptake of [3H]thymidine as described before (5). Cells were plated at 5 × 104 cells/ml in DMEM containing 10% FBS in 24-well plates and incubated at 37 °C for 12 h. Cells were then washed twice in phosphate-buffered saline and growth-arrested in DMEM for 24 h at 37 °C. Various amounts of CSF-1 were added and incubated for 18–20 h at 37 °C. Cells were pulsed for 4 h with [3H]thymidine (0.2 μCi/ml) and harvested and measured by a scintillation counter. Quadruplicate samples were performed for each group. Three independent experiments were performed, and essentially the same results were obtained.
Tubulogenesis/in Vitro Angiogenesis Assay
Spheroids of PAE cells expressing CTR, CKR, D1050/CTR, and D1054/CKR were prepared as previously described (9, 10). Cells were suspended in DMEM containing 1% FBS and 0.24% (w/v) carboxymethylcellulose plus 4000 centipoise in nonadherent round bottom 96-well plates at standard cell culture conditions. After 8–12 h, all cells formed one single spheroid per well (750 cells/spheroid). Spheroids were cultured for 2 days before using them in the in vitro angiogenesis assay in the following manner. Before spheroids were embedded in collagen gels, they were centrifuged and suspended in DMEM, containing 0.96% carboxymethylcellulose. Collagen and spheroids were mixed and transferred to prewarmed 24-well plates, and the gels were allowed to polymerize in the incubator. After 30 min, 100 μl of DMEM containing CSF-1 was added on top of the gel. Sprouting and tubulogenesis was observed after 2 days under an inverted phase-contrast microscope (Leica), and pictures were taken using a digital camera system.
Transformation Assays
Colony formation of NIH-3T3 fibroblast cells expressing chimeric VEGFR-1 (CTR) or D1050/CTR were analyzed in a soft agar colony formation assay in the following manner. Briefly, a solution of 1.2% agar was mixed (1:1) with 2× DMEM, supplemented with 7% FBS, and layered onto a 6-well tissue culture plate and allowed to solidify. Cells (103/2.5 ml) were mixed in a 0.36% agar solution prepared in a similar way and layered (2.5 ml/plate) on top of the 0.6% agar. Plates were incubated at 37 °C in 5% CO2 for 18–22 days. Colonies were fixed with 100% methanol, stained with 4% Giemsa blue, and photographed with an inverted phase-contrast microscope (Leica) using a digital camera system. In addition, the focus formation of NIH-3T3 cells expressing chimeric VEGFR-1 (CTR) or D1050/CTR was analyzed. For this purpose, cells were cultured in 10% FBS in the presence of CSF-1 for up to 21 days. Spontaneous focus formation of fibroblast cells was negligible. At the end of the assay, the cells were photographed with an inverted phase-contrast microscope (Leica) using a digital camera system.
RESULTS
A Highly Conserved Aspartic Acid Residue in the Activation Loop of VEGFR-1 Is Substituted to Asparagine
FLT-1 (VEGFR-1) is a member of the gene family receptor tyrosine kinases, encoding structurally related receptors including FLK-1 (VEGFR-2), FLT-3/FLK-2, and FLT-4/VEGFR-3 (1). All of the protein kinases have conserved residues and homologous stretches cored on 12 subdomains within ~300 amino acids. Subdomains VII and VIII represent the activation loop starting with Asp, Phe, and Gly and ending with Ala, Pro, and Glu (21). The activation loop plays a critical role in the enzymatic activation of RTK (1, 21, 25). The amino acid sequence alignment of the activation loop of type III RTKs revealed that in VEGFR-1 a highly conserved amino acid Asp is replaced with Asn (Fig. 1). This substitution is not restricted to human VEGFR-1; it is also conserved among the mouse, rat, and chicken VEGFR-1. All of the other known signatures of RTKs, such HRDLA and DFG sequences, which are essential for catalysis, are preserved in VEGFR-1 (Fig. 1). This uncommon amino acid substitution in a critical position has raised the possibility that this substitution might play a role in a kinase-impaired characteristic of VEGFR-1. To test this notion, we have replaced the asparagine of human VEGFR-1 at position 1050 to aspartic acid and tested its effect on VEGFR-1 activation and function.
FIGURE 1. Amino acid sequence alignment of the activation loop of type III receptor tyrosine kinases.
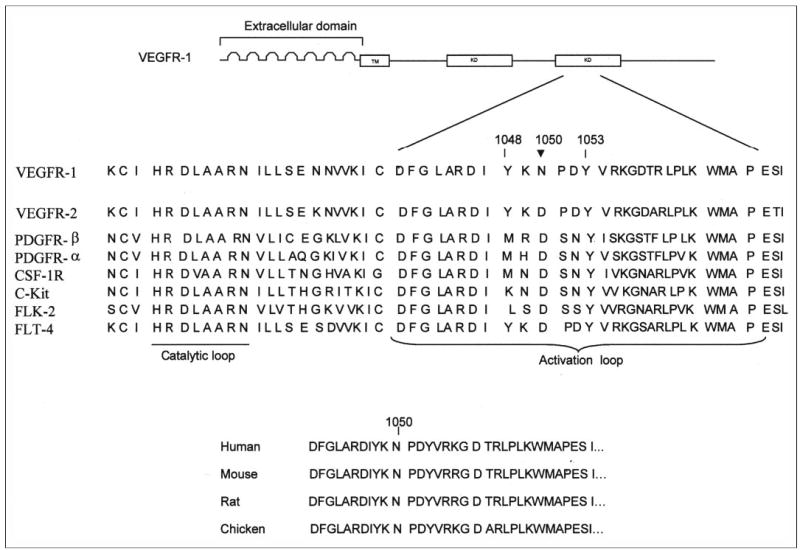
A schematic representation of the VEGFR-1 (FLT-1) structure is shown. The extracellular region contains seven IgG loops, the transmembrane (TM), and the cytoplasmic region containing the kinase domain (KD) indicated. The location of Asn1050 (N1050) in the activation loop of human VEGFR-1 is indicated by an arrow. Tyrosines 1048 and 1053 are putative autophosphorylation sites. Sequence alignment of the catalytic and activation loop regions of human VEGFR-1 and various receptor tyrosine kinases of type III family are shown. The residue corresponding to asparagine 1050 of VEGFR-1 is aspartic acid in type III kinases. This residue is highly conserved and is invariable among the type III family kinases except VEGFR-1. Asparagine 1050 of VEGFR-1 is conserved among humans (GenBank™ accession number NM002019), mice (GenBank™ accession number BAA24498), rats (GenBank™ accession number P53767), and chickens (GenBank™ accession number BAB84690).
The Asp1050 Mutant VEGFR-1 Displays Increased Tyrosine Phosphorylation and Kinase Activation
To determine the effect of the mutation of Asn at position 1050 of human VEGFR-1 to Asp on its ligand-stimulated tyrosine autophosphorylation, we expressed wild type chimeric VEGFR-1 (CTR) and D1050/CTR in PAE cells. We previously have reported the creation and biochemical characterization of a chimeric VEGFR-1 termed CTR in which the extracellular domain of the human CSF-1R was fused to the transmembrane and cytoplasmic domains of human VEGFR-1 (5). We took this unique approach in order to eliminate potential cross-talk between VEGFR-1 and other VEGFR family members.
To measure the ability of CTR and D1050/CTR to undergo in vivo tyrosine autophosphorylation, cells were stimulated with CSF-1, lysed, and immunoprecipitated with an anti-VEGFR-1 antibody and blotted with anti-phosphotyrosine antibody (Fig. 2A). The result showed that the wild type chimeric VEGFR-1 (CTR) after ligand stimulation underwent a low tyrosine phosphorylation. In sharp contrast, ligand stimulation of cells expressing D1050/CTR promoted robust tyrosine phosphorylation of D1050/CTR. The maximal autophosphorylation of D1050/CTR peaked after 10 min and gradually decreased after 30 min of stimulation with ligand (Fig. 2A). For comparative purposes, the ligand-dependent tyrosine phosphorylation of VEGFR-2 chimera (CKR) also is determined. VEGFR-2 is a highly active kinase, and upon ligand stimulation, it undergoes robust tyrosine phosphorylation (5). To further analyze the role of aspartic acid on the catalytic activity of VEGFR-1, we tested the ability of wild type CTR and D1050/CTR to phosphorylate exogenous substrate as described under “Materials and Methods.” As shown in Fig. 2C, the wild type CTR phosphorylated only a trace amount of substrate (less than 0.2-fold). Under identical experimental conditions, a 1.8-fold increase in substrate phosphorylation was observed for D1050/CTR (Fig. 2C). Collectively, the data suggest that mutation of asparagine 1050 to aspartic acid increases the ability of VEGFR-1 to undergo enhanced tyrosine phosphorylation and its capacity to phosphorylate exogenous substrate.
FIGURE 2. Replacement of asparagine 1050 to aspartic acid unleashes the poor tyrosine autophosphorylation and kinase activation of VEGFR-1.
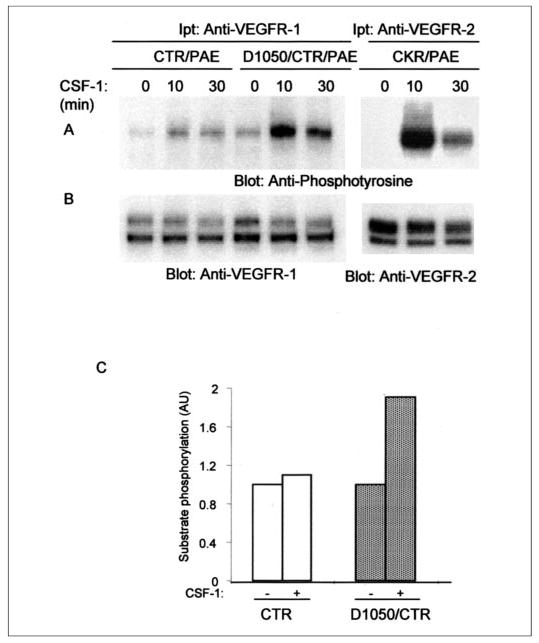
Serum-starved PAE cells individually expressing either wild type chimeric VEGFR-1 (CTR), mutant VEGFR-1 (D1050/CTR), or CKR (chimeric VEGFR-2) were stimulated with CSF-1 for the indicated periods of times. Cell lysates were immunoprecipitated (Ipt) with an anti-VEGFR-1 antibody or anti-VEGFR-2 antibody. The immunoprecipitated proteins were divided to two groups and were subjected to Western blot analysis using either an anti-phosphotyrosine antibody (A) or anti-VEGFR-1 and anti-VEGFR-2 antibodies for protein levels (B). Serum-starved PAE cells expressing CTR and D1050/CTR were left unstimulated or stimulated for 10 min with CSF-1, lysed, immunoprecipitated with anti-VEGFR-1 antibody, and subjected to in vitro kinase assay using poly-Glu peptide as a substrate as described under “Materials and Methods” (C). Results are expressed as arbitrary units (AU) corresponding to the dpm values obtained from counting of the phosphorylation of substrate with a scintillation counter.
To test further the effect of mutation of asparagine 1050 to aspartic acid, we evaluated the ability of D1050/CTR to undergo tyrosine autophosphorylation in vitro in response to ATP stimulation. To this end, serum-starved cells expressing either chimeric VEGFR-1 or Asp1050 mutant chimeric VEGFR-1 were lysed without stimulation with ligand and immunoprecipitated with anti-VEGFR-1 antibody. The immunoprecipitated proteins were incubated with different concentrations of ATP. After 15 min of incubation, the reaction was stopped and was subjected to Western blot analysis using an anti-phosphotyrosine antibody. As presented in Fig. 3A, the wild type chimeric VEGFR-1 (CTR) underwent detectable but not robust tyrosine phosphorylation in response to ATP, and its tyrosine phosphorylation was enhanced with increasing concentrations of ATP. In sharp contrast, the mutant receptor, D1050/CTR, under similar experimental conditions underwent robust tyrosine autophosphorylation. For comparative purposes, PAE cells expressing the VEGFR-2 chimera (CKR) were stimulated with ATP in a similar manner and analyzed for their ability to undergo tyrosine phosphorylation. The quantitation of autophosphorylation of the wild type receptor and the mutant receptor, Asp1050, revealed that the Asp1050 autophosphorylation was at least 2-fold higher than the wild type receptor (Fig. 3C). The basal tyrosine autophosphorylation of Asp1050 receptor also was enhanced in response to both ligand and ATP stimulation (Figs. 2A and 3A).
FIGURE 3. In vitro kinase activation of CTR and D1050/CTR.
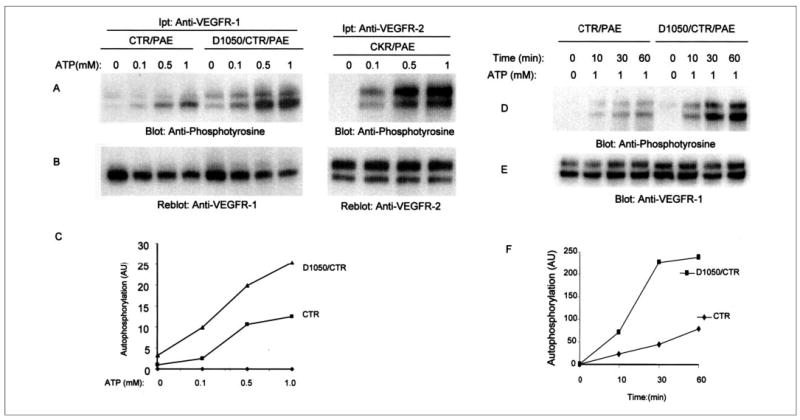
Serum-starved PAE cells expressing wild type chimeric VEGFR-1 (CTR), D1050/CTR, or CKR were lysed without stimulation, and cell lysates were immunoprecipitated (Ipt) with an anti-VEGFR-1 antibody or VEGFR-2 antibody. The immunoprecipitated proteins were subjected to an in vitro kinase assay using ATP at different concentrations as indicated. The reaction stopped after 15 min, and samples were analyzed by Western blot using an anti-phosphotyrosine antibody as described under “Materials and Methods” (A). The same membranes were reprobed with an anti-VEGFR-1 and anti-VEGFR-2 antibodies for protein levels (B). Quantitation of tyrosine phosphorylation of CTR and D1050/CTR in response to ATP stimulation is shown (C). The total protein level was used to normalize the tyrosine phosphorylation values. AU (arbitrary units). Serum-starved PAE cells expressing wild type chimeric VEGFR-1 (CTR), or D1050/CTR were lysed without stimulation, and cell lysates were immunoprecipitated with an anti-VEGFR-1 antibody and divided into two groups. One group was subjected to in vitro kinase assay using cold ATP (1 mm) and incubated for the indicated periods of time. Samples were analyzed by Western blot using an anti-phosphotyrosine antibody as described under “Materials and Methods” (D). The second group was blotted for protein level using anti-VEGFR-1 antibody (E). Quantitation of tyrosine phosphorylation of CTR and D1050/CTR in response to ATP stimulation is shown (F). The Eastman Kodak Co. ID Image analysis program was used to quantify the data. The total protein level was used to normalize the tyrosine phosphorylation values. AU, arbitrary units.
The Asp1050 Mutation Increases the Threshold of Tyrosine Autophosphorylation of VEGFR-1
To further analyze the effect of Asp1050 mutation on VEGFR-1 autophosphorylation, we analyzed the kinetics of autophosphorylation of wild type chimeric VEGFR-1 (CTR) and D1050/CTR in response to 1 mm ATP stimulation. As presented in Fig. 3, Asp1050 VEGFR-1 underwent tyrosine autophosphorylation at a higher rate, and its maximal autophosphorylation was reached after a 30-min incubation with ATP. The tyrosine autophosphorylation of wild type chimeric VEGFR-1 in response to ATP was slow. The magnitude and intensity of its tyrosine phosphorylation never reached that of D1050/CTR (Fig. 3D). Indeed, the maximum tyrosine phosphorylation of wild type VEGFR-1 in response to ATP was reached after 60 min of incubation. In contrast, the tyrosine phosphorylation of D1050/CTR was much enhanced; its maximum tyrosine phosphorylation reached after 30 min of incubation with ATP and remained high after 60 min of incubation (Fig. 3, D and F). Altogether, these results suggest that the decoy characteristic of VEGFR-1 is associated with its inability to induce a threshold of its kinase activation. The lack of maximal threshold activation of VEGFR-1 appears to be directly associated with the presence of asparagine 1050.
D1050 Mutant VEGFR-1 Promotes Proliferation but Not Tubulogenesis of Endothelial Cells
Proliferation and tubulogenesis of endothelial cells are critical steps in angiogenesis. To test the biological impact of mutation of asparagine 1050 to aspartic acid, we tested the ability of the wild type and the mutant Asp1050 mutant receptor to stimulate proliferation of endothelial cells. The results showed that the activation of wild type chimeric VEGFR-1 as previously reported (4, 9) is not associated with the proliferation of endothelial cells (Fig. 4A). In contrast, stimulation of the D1050/CTR induced proliferation of endothelial cells in a dose-dependent manner (Fig. 4A). For comparative purposes, we subjected PAE cells expressing VEGFR-2 chimera (CKR) to a proliferation assay in a similar manner. As shown in Fig. 4A, D1050/CTR promoted proliferation of PAE cells slightly better than the CKR. We next evaluated the capacity of the Asp1050 mutant receptor to promote tubulogenesis of endothelial cells. Stimulation of neither CTR (chimeric VEGFR-1) nor the Asp1050 mutant receptor (D1050/CTR) induced tubulogenesis of PAE cells (Fig. 4B). However, the VEGFR-2 chimera (CKR) as we previously reported promoted sprouting and tubulogenesis (10). Activation of the wild type chimeric VEGFR-1 induces no significant PLCγ1 stimulation (9). Surprisingly, the mutant VEGFR-1, although highly phosphorylated, is unable to elevate phosphorylation of PLCγ1 (Fig. 4C). Altogether, this suggests that tubulogenesis of endothelial cells is uniquely associated with VEGFR-2 activation and that VEGFR-1 activity is not directly involved in tubulogenesis even when VEGFR-1 is able to undergo tyrosine autophosphorylation.
FIGURE 4. Replacement of asparagine 1050 to aspartic acid allows VEGFR-1 to stimulate cell proliferation but not tubulogenesis.
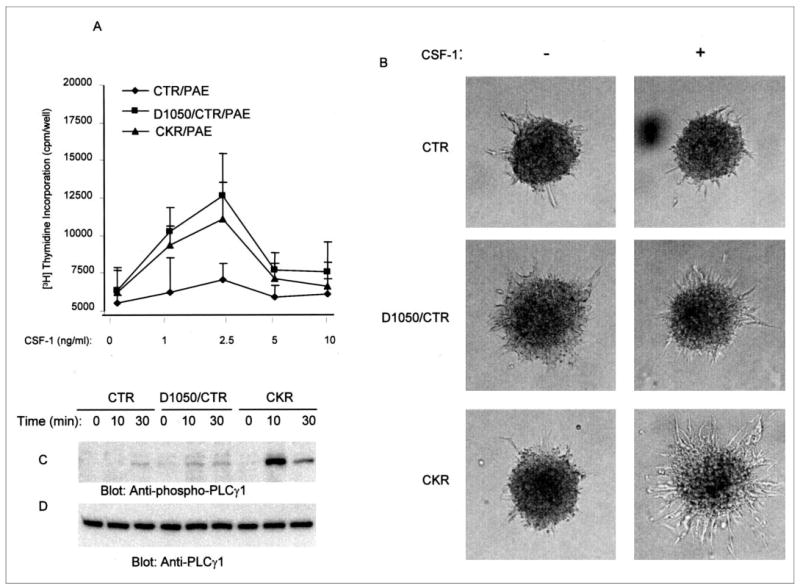
Serum-starved PAE cells expressing wild type chimeric VEGFR-1 (CTR), mutant chimeric VEGFR-1 (D1050/CTR), or chimeric VEGFR-2 (CKR) were stimulated with different concentrations of CSF-1, and DNA synthesis was measured by [3H]thymidine uptake method as described under “Materials and Methods.” The results are expressed as the mean (cpm/well) ±S.D. of quadruplicates (A). PAE cells expressing wild type CTR or D1050/CTR were prepared as spheroids and subjected to in vitro angiogenesis with or without CSF-1. In addition, PAE cells expressing chimeric VEGFR-2 (CKR) were prepared and used in a similar manner as a positive control. Sprouting and tubulogenesis was observed after 24 h under an inverted phase-contrast microscope (Leica), and pictures were taken using a Leica digital camera (B). Serum-starved PAE cells individually expressing either wild type chimeric VEGFR-1 (CTR), mutant VEGFR-1 (D1050/CTR), or CKR (chimeric VEGFR-2) were stimulated with CSF-1 for the indicated periods of time. Total cell lysates were subjected to Western blot analysis using an anti-pospho-PLCγ1 antibody (C) or an anti-PLCγ1 antibody (D).
Asp1050 Mutant VEGFR-1 Promotes Transformation of Fibroblast Cells
To further understand the biological importance of substitution of aspartic acid on the VEGFR-1 function, we expressed the wild type chimeric VEGFR-1 and Asp1050 mutant chimeric VEGFR-1 in NIH-3T3 fibroblast cells and analyzed for their transforming potentials in focus-forming and soft agar growth assays. To this end, an equal number of NIH-3T3 cells expressing either chimeric VEGFR-1 or D1050/CTR were plated in a 6-well plate and stimulated with CSF-1. After 20 days in culture, the transformed morphologies of cells were viewed under microscope and photographed. As shown in Fig. 5A, the control wild type NIH-3T3 cells displayed no apparent transformed morphological changes. NIH-3T3 cells expressing chimeric VEGFR-1 showed a negligible morphological and transformation changes. In contrast, NIH-3T3 cells expressing D1050/CTR displayed a significant morphological change focus formation, the phenotype that is associated with transformed cells. In further analysis, when these cells were subjected to a soft agar colony formation assay, cells expressing D1050/CTR exhibited a highly transformed phenotype and formed large colonies (Fig. 5B). NIH-3T3 cells and NIH-3T3 cells expressing CTR formed only small colonies (Fig. 5B). In sum, these results demonstrate that the oncogenic potential of VEGFR-1 is severely suppressed by the presence of asparagine 1050, and its replacement with aspartic acid converts VEGFR-1 from a decoy and nononcogenic RTK to a harmful and potent oncogenic RTK.
FIGURE 5. D1050 mutant VEGFR-1 promotes transformation of fibroblast cells.
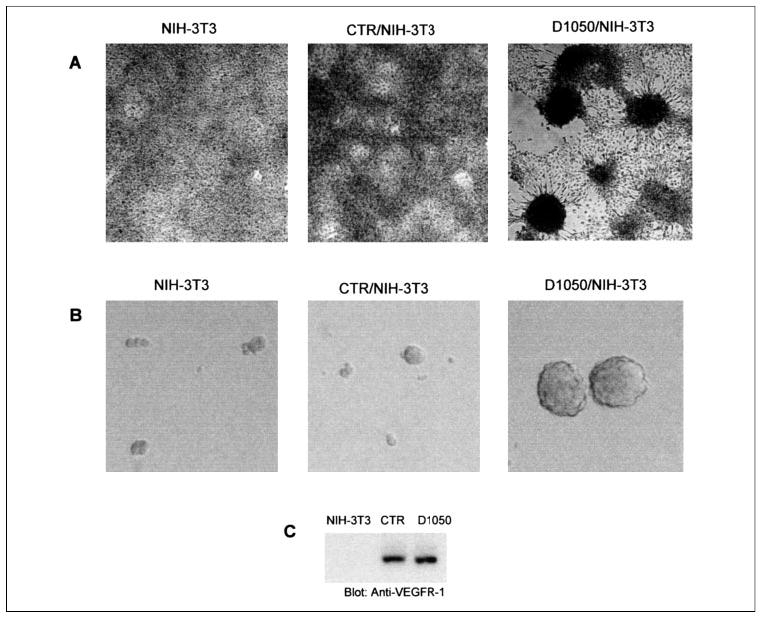
NIH-3T3 cells expressing CTR and D1050/CTR were plated in 10% FBS plus CSF-1 (20 μg/ml) and maintained for 3 weeks. Additional CSF-1 was added into medium every 2 days, and the foci formation of cells was evaluated. The foci formation of cells was viewed under an inverted phase-contrast microscope (Leica) and pictures were taken using a Leica digital camera (A). NIH-3T3 cells expressing CTR and D1050/CTR were subjected to soft agar colony formation assay as described under “Materials and Methods.” After 22 days, colonies were counted. NIH-3T3 cells and NIH-3T3 cells expressing CTR did not form large colonies. NIH-3T3 cells expressing D1050/CTR formed 145 large colonies. The representative colony formations of NIH-3T3, NIH-3T3 cells expressing CTR, and NIH-3T3 cells expressing D1050/CTR are shown (B). Expression of CTR and D0150/CTR in NIH-3T3 is shown (C).
Replacement of Asparagine 1050 to Aspartic Acid in Nonchimeric VEGFR-1 Enhances Kinase Activation of VEGFR-1
Although it is highly unlikely that the significance of mutation of asparagine 1050 in the chimeric VEGFR-1 (CTR) is different from that of the VEGFR-1, nevertheless it is important to validate the effect of mutation of asparagine 1050 to aspartic acid in a nonchimeric system. For this reason, we have created an aspartic acid 1050 mutation on VEGFR-1 (D1050/VEGFR-1) and expressed it in PAE cells. Endothelial cells in general express both VEGFR-1 and VEGFR-2. For this very reason, in our study we have employed a chimeric system to study selective activation of VEGFR-1 and VEGFR-2 and their signaling properties in endothelial cells (5, 9, 10). VEGFR-2 is not expressed in PAE cells, but VEGFR-1 although at a low level, is expressed in these cells (5). To this end, analysis of the ectopic expression of VEGFR-1 and D1050/VEGFR-1 in PAE cells demonstrates that mutation of asparagine 1050 to aspartic acid in VEGFR-1 also increases the kinase activation of VEGFR-1 (Fig. 6A). Indeed, autophosphorylation of D1050/VEGFR-1 is almost identical to that of D1050/CTR (Fig. 3A, Fig. 6C). Also, as shown in Fig. 6A, the wild type VEGFR-1, like the chimeric receptor (CTR), displayed low kinase activation. Altogether, the results obtained from analysis of kinase activation of asparagine 1050 mutation to aspartic acid in both chimeric and nonchimeric VEGFR-1 reaffirm the conclusion that the presence of asparagine 1050 hinders the ability of VEGFR-1 to act as a strong RTK.
FIGURE 6. Replacement of asparagine 1050 to aspartic acid in nonchimeric VEGFR-1 enhances tyrosine autophosphorylation of VEGFR-1.
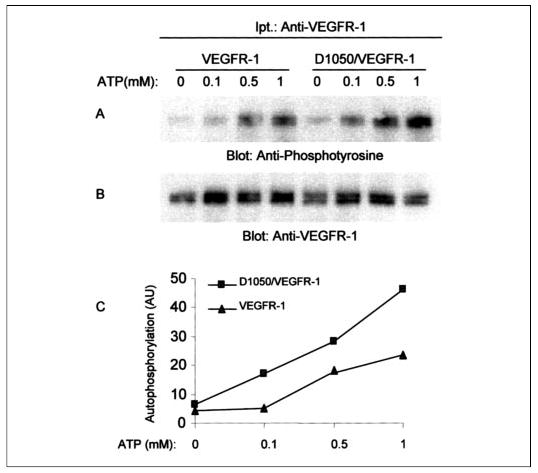
Serum-starved PAE cells expressing wild type VEGFR-1 or D1050/VEGFR-1 were lysed without stimulation, and cell lysates were immunoprecipitated with an anti-VEGFR-1 antibody and divided into two groups. One group was subjected to an in vitro kinase assay using cold ATP with different concentrations of ATP as indicated. Samples were analyzed by Western blot using an anti-phosphotyrosine antibody as described under “Materials and Methods” (A). The second group was blotted for protein level using anti-VEGFR-1 antibody (B). The graph was generated by quantitation of tyrosine phosphorylation of VEGFR-1 and D1050/VEGFR-1 in response to ATP stimulation using the Kodak ID Image analysis program (C). The total protein levels were used to normalize the tyrosine phosphorylation values. AU, arbitrary units.
Mutation of Conserved Aspartic Acid to Asparagine in VEGFR-2/FLK-1 Alters Autophosphorylation of Activation Loop Tyrosines
To address how replacement of asparagine with a conserved aspartic acid alters the autophosphorylation and kinase activation of VEGFR-1, we hypothesized that the presence of asparagine at position 1050 of VEGFR-1 disfavors trans-autophosphorylation of tyrosine autophosphorylation sites in the activation loop of VEGFR-1. However, the presence of aspartic acid in this location favors ligand-induced trans-autophosphorylation of activation loop tyrosines. To test this possibility, we utilized a well characterized VEGFR-2/FLK-1 system. In VEGFR-2, tyrosines 1052 and 1057 are phosphorylated upon ligand stimulation (17, 21, 22), and the antibody that recognizes the phosphorylated tyrosines 1052 and 1057 is also available (see “Materials and Methods”). To this end, we mutated the analogous site, aspartic acid 1054, to asparagine in chimeric VEGFR-2 (CKR) and herein referred to it as N1054/CKR. The wild type CKR and N1054/CKR were expressed in PAE cells and comparatively were analyzed for their capacity to undergo autophosphorylation at the tyrosine 1052 and 1057 sites. As shown in Fig. 7A, ligand stimulation of the wild type CKR promoted phosphorylation of tyrosines 1052 and 1057. In contrast, phosphorylation of these sites on N1054/CKR was significantly less than what we observed in wild type CKR. Quantitation of tyrosine phosphorylation of 1052 and 1057 revealed that phosphorylation of these sites are reduced by 39.6% in N1054/CKR compared with that of wild type CKR (Fig. 7C). To test whether the presence of aspartic acid affects only phosphorylation of tyrosines 1052 and 1057 in mouse VEGFR-2 or also affects phosphorylation of other tyrosines, we evaluated phosphorylation of another major autophosphorylation site, tyrosine 1173, located in the carboxyl terminus of VEGFR-2, which binds to p85 of phosphatidylinositol 3-kinase and PLCγ1 (10, 40). The results show that phosphorylation of tyrosine 1173 in N1054/CKR was not affected by this mutation (Fig. 7D). In addition, we have evaluated the ability of N1054/CKR to induce tubulogenesis of endothelial cells. The results demonstrate that the ability of N1054/CKR to promote tubulogenesis of PAE cells is severely impaired, suggesting that the presence of aspartic acid is highly critical for normal function of VEGFR-2 (Fig. 7F). Altogether, the data favor the idea that the presence of aspartic acid at position 1054 creates a bias for autophosphorylation of tyrosine sites within the activation loop, whereas asparagine hinders efficient autophosphorylation of these sites.
FIGURE 7. Mutation of conserved aspartic acid to asparagine in VEGFR-2/FLK-1 alters auto-phosphorylation of activation loop tyrosines.
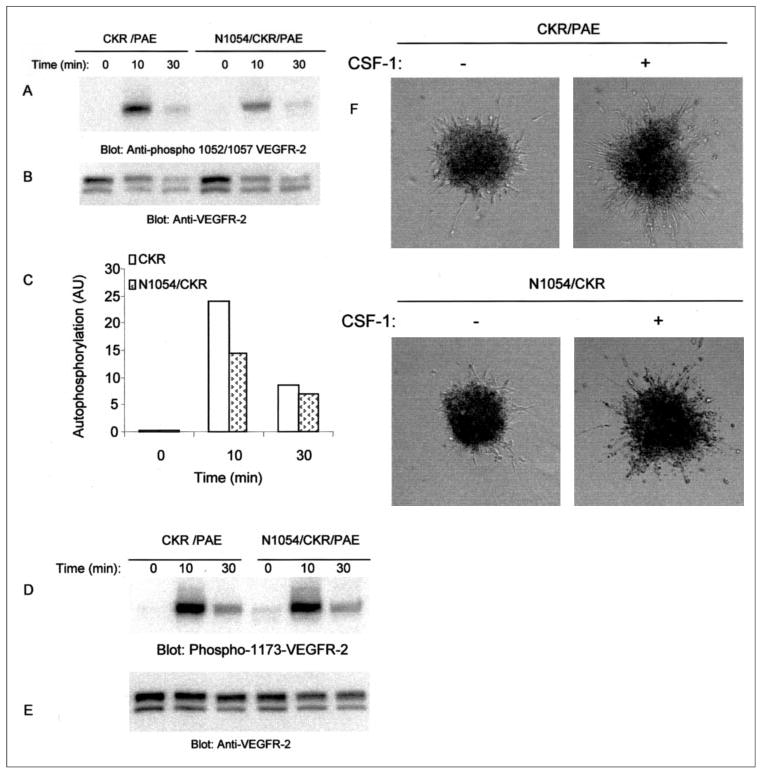
Equal numbers of serum-starved PAE cells expressing chimeric VEGFR-2 (CKR) and N1054/CKR were stimulated with CSF-1 for the indicated periods of time and lysed, and total cell lysates were subjected to Western blot analysis using anti-phospho-Tyr1052/1057 VEGFR-2 antibody (A). The same membrane was reprobed for protein levels (B). Phosphorylation of the activation loop tyrosines (Tyr1052 and Tyr1057) was quantitated, and results are expressed as arbitrary units (AU). The total protein levels were used to normalize the tyrosine phosphorylation values (C). The same cell lysates were subjected to Western blot analysis using anti-phospho-Tyr1173 VEGFR-2 antibody (D). The same membrane was reprobed for protein level using anti-VEGFR-2 antibody (E). PAE cells expressing wild type CKR or D1050/CKR were prepared as spheroid and subjected to in vitro angiogenesis with or without CSF-1 as described under “Materials and Methods.” Sprouting and tubulogenesis was observed after 24 h under an inverted phase-contrast microscope (Leica), and pictures were taken using a Leica digital camera (F).
DISCUSSION
VEGFR-1/FLT-1 is devoid of ligand-dependent autophosphorylation, and its activation is inadequate to promote endothelial cell proliferation and angiogenesis (2–4, 10). There is no molecular explanation for this characteristic of VEGFR-1. The results reported here demonstrate that in the kinase domain of VEGFR-1, a highly conserved aspartic acid residue is substituted to asparagine. Converting asparagine back to aspartic acid enables VEGFR-1 to undergo robust tyrosine autophosphorylation and kinase activation and to stimulate proliferation of endothelial cells.
All known RTKs contain an evolutionary conserved kinase domain, and the available crystal structure of the kinase domain of RTKs also shows that its conformation is highly conserved (1, 11). Among all of the type III receptor tyrosine kinases, including VEGFR-2, platelet-derived growth factor receptor, CSF-1R, c-Kit, FLK-2, and FLT-4, the aspartic acid residue is conserved (1) (Fig. 1). In contrast, in VEGFR-1, this aspartic acid residue is replaced with asparagine, and this unique surrogate residue is conserved among the human, mouse, rat, and chicken VEGFR-1 (Fig. 1). The fact that VEGFR-1 proteins from different species are all deficient in kinase activity may hint that substitution of a conserved aspartic acid to asparagine is not an accidental event and has a functional role. Such a role for other RTKs, such as ErbB3, which has no kinase activity but plays a key biological role, has been demonstrated (23, 24). The kinase-impaired activity of ErbB3 is thought to be associated with alteration of a residue in the HRDL motif, where Asp is substituted to Asn (29, 30). Similarly, amino acid substitutions in the activation loop, in particular in GXGXXG, HRDL, and DFG motifs, are identified in kinase-impaired RTKs such as CCK-4, KLG, Ro1, and Ror2 (31, 32). In addition, mutations in the kinase domain of RTKs have been linked to a number of inherited human diseases, such as cancer and venous malformation (34–36). Both gain-of-function mutations and loss-of-function mutations have been identified (34, 37, 38).
It could be argued that due to evolutionary pressure, VEGFR-1, like other kinase-defective RTKs, has lost its kinase activity due to replacement of the conserved aspartic acid to asparagine. Nevertheless, VEGFR-1 has retained its ability to bind VEGF at high affinity (12) and form heterodimers with VEGFR-2 (8). The exact mechanism by which asparagine 1050 prevents full VEGFR-1 activation needs further analysis, including resolving the crystal structure of VEGFR-1. To date, the structure of the cytoplasmic domain of VEGFR-1 is not resolved. Also, the crystal structure of a highly related RTK, namely FLK-1/VEGFR-2, is determined, but due to the mobile nature of the activation loop, electron density was not obtained (17). Based on data presented here, we propose that the presence of asparagine, which contains an uncharged polar side chain (unlike aspartic acid, which contains a negatively charged side chain), averts phosphorylation of tyrosines in the so-called autophosphorylation sites of the activation loop. Activation loop tyrosines of RTKs, such as insulin receptor kinase, are thought to prevent both ATP and substrate binding. In the activated form, phosphorylated activation loop tyrosines stabilize the molecule and allow kinase activity (25). In fibroblast growth factor receptor, activation loop tyrosines are suggested to interfere with substrate binding but not ATP binding (26). Activation loop tyrosines in VEGFR-1 correspond to tyrosines 1048 and 1053 (Fig. 1). The data presented in this work indicate that replacement of asparagine to aspartic acid increases autophosphorylation of VEGFR-1 and its ability to phosphorylate substrate.
The idea that aspartic acid but not asparagine at position 1050 may favor transphosphorylation of activation loop tyrosines is further supported by our finding that in VEGFR-2, substitution of aspartic acid to asparagine reduced the transphosphorylation of activation loop tyrosines 1052 and 1057 without affecting phosphorylation of tyrosine 1173, a residue that is located outside the kinase domain of VEGFR-2. Although activation loop tyrosine autophosphorylation is generally thought to regulate tyrosine autophosphorylation and kinase activation of RTKs (11, 25), activation loop tyrosines may play a different role, depending on the individual RTK. For example, phosphorylation of activation loop tyrosines may selectively abolish phosphorylation of certain tyrosines and thus certain biological responses while sparing others. In support of this view, mutation of activation loop tyrosines in Trk/NGF receptor selectively impairs phosphorylation of Tyr785, a PLCγ1 binding site, without affecting tyrosine sites involved in binding with SHC and SNT/FRS-2 (41). Similar findings were reported for insulin-like growth factor-1 receptor (42) and Met/hepatocyte growth factor receptor (43). More importantly, phosphorylation of tyrosine 1057 in human VEGFR-2 (Tyr1057 in human VEGFR-2 corresponds to Tyr1059) also was reported to impair proliferation without affecting VEGFR-2-mediated permeability (44).
VEGFR-1 also can gain an oncogenic potential when a certain mutation is introduced to its kinase domain, as recently demonstrated using random site-directed mutagenesis (28). Data presented here suggest that the presence of asparagine at position 1050 imposes a hindrance to autophosphorylation of the receptor itself and to substrate phosphorylation. Consequently, this makes VEGFR-1 an RTK with a latent kinase activity, capable of basal autophosphorylation without reaching to an activation, a threshold that is required for the initiation of signal transduction and biological responses. At low ATP concentrations (0.1 mm), the mutant VEGFR-1 undergoes rapid autophosphorylation, whereas the autophosphorylation of wild type VEGFR-1 is barely detectable (Fig. 2). This observation further argues that replacement of asparagine 1050 to aspartic acid predisposes VEGFR-1 to transphosphorylation and kinase activation. We propose that the presence of an acidic amino acid residue, such as aspartic acid, at this location makes activation loop tyrosines a better substrate for transphosphorylation. It is also possible that aspartic acid 1050 via hydrogen bonding makes the activation loop in a less inhibitory conformation, creating a better environment for tyrosine phosphorylation of VEGFR-1.
Regardless of how asparagine contributes to poor activation of VEGFR-1, the presence of this residue is critical for this unique characteristic of VEGFR-1. One fascinating aspect of VEGFR-1 is that although replacement of asparagine to aspartic acid promotes its ability to undergo tyrosine autophosphorylation and kinase activation, the mutant receptor selectively promotes endothelial cell proliferation but not tubulogenesis. It has been shown that tubulogenesis of endothelial cells is associated with activation of PLCγ1 (10). Stimulation of the wild type chimeric VEGFR-1 and Asp1050 mutant VEGFR-1 resulted only in a trivial amount of activation of PLCγ1 (Fig. 4), suggesting that tubulogenesis of endothelial cells is mainly regulated by VEGFR-2 via activation of PLCγ1 but not VEGFR-1. Furthermore, the inability of D1050/CTR to phosphorylate PLCγ1 suggests that either VEGFR-1 possesses no PLCγ1 binding site or the putative tyrosine site is not phosphorylated in order to recruit PLCγ1. Some RTKs, such as CSF-1 receptor, however, are highly tyrosine-phosphorylated and activate many signaling proteins but do not activate PLCγ1 (18). Thus, in this regard, VEGFR-1 (D1050/CTR) acts like CSF-1 receptor.
Coordinated endothelial cell migration, growth, and differentiation are essential requirements for normal vessel development. A recurring scenario in this process is that the complexity of blood vessel development necessitated more stringent control over VEGFR-1 signaling in development. Thus, VEGFR-1 evolved to have higher affinity to bind VEGF but lost its potency in enzymatic activity, creating a unique mechanism to control its function. It appears that its poor tyrosine phosphorylation in part is established by replacement of a highly conserved aspartic residue to asparagine. In short, the current study gives us a better understanding of how VEGFR-1 acts as a decoy receptor.
Footnotes
This work was supported in part by National Institutes of Health Grants EY0137061 and EY012997 (to N. R.).
The abbreviations used are: VEGF, vascular endothelial growth factor; RTK, receptor tyrosine kinase; FBS, fetal bovine serum; DMEM, Dulbecco’s modified Eagle’s medium; PLCγ1, phospholipase Cγ1.
References
- 1.Hanks S, Quinn AM. Methods Enzymol. 1999;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- 2.Tjwa M, Luttun A, Autiero M, Carmeliet P. Cell Tissue Res. 2003;314:5–14. doi: 10.1007/s00441-003-0776-3. [DOI] [PubMed] [Google Scholar]
- 3.de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- 4.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. J Biol Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 5.Rahimi N, Dayanir V, Lashkari K. J Biol Chem. 2000;275:16986–16992. doi: 10.1074/jbc.M000528200. [DOI] [PubMed] [Google Scholar]
- 6.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 7.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Proc Natl Acad Sci U S A. 1998;95:9349–9354. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, Katayama S, Awata T, Leigh N, Lang-Lazdunski L, Dewerchin M, Shaw C, Moons L, Vlietinck R, Morrison KE, Robberecht W, Van Broeckhoven C, Collen D, Andersen PM, Carmeliet P. Nat Genet. 2003;34:383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- 9.Meyer RD, Singh A, Majnoun F, Latz C, Lashkari K, Rahimi N. Oncogene. 2004;23:5523–5531. doi: 10.1038/sj.onc.1207712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer RD, Latz C, Rahimi N. J Biol Chem. 2003;278:16347–16355. doi: 10.1074/jbc.M300259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hubbard SR, Till JH. Annu Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 12.Shibuya M. Cell Struct Funct. 2001;26:25–35. doi: 10.1247/csf.26.25. [DOI] [PubMed] [Google Scholar]
- 13.Fong GH, Zhang L, Bryce DM, Peng J. Development. 1999;126:3015–3025. doi: 10.1242/dev.126.13.3015. [DOI] [PubMed] [Google Scholar]
- 14.Zeng H, Dvorak HF, Mukhopadhyay D. J Biol Chem. 2001;276:26969–26979. doi: 10.1074/jbc.M103213200. [DOI] [PubMed] [Google Scholar]
- 15.Ohno-Matsui K, Yoshida T, Uetama T, Mochizuki M, Morita I. Biochem Biophys Res Commun. 2003;303:962–967. doi: 10.1016/s0006-291x(03)00446-7. [DOI] [PubMed] [Google Scholar]
- 16.LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, Hillan KJ, Ferrara N. Science. 2003;299:890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- 17.McTigue MA, Wickersham JA, Pinko C, Showalter RE, Parast CV, Tempc-zyk-Russell A, Gehring MR, Mroczkowski B, Kan CC, Villafranca JE, Appelt K. Struct Fold Des. 1999;7:319–330. doi: 10.1016/s0969-2126(99)80042-2. [DOI] [PubMed] [Google Scholar]
- 18.Downing JR, Margolis BL, Zilberstein A, Ashmun RA, Ullrich A, Sherr CJ, Schlessinger J. EMBO J. 1989;11:3345–3350. doi: 10.1002/j.1460-2075.1989.tb08496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gille H, Kowalski J, Yu L, Chen H, Pisabarro MT, Davis-Smyth T, Ferrara N. EMBO J. 2000;19:4064–4073. doi: 10.1093/emboj/19.15.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanks SK, Quinn AM, Hunter T. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 21.Dougher M, Terman BI. Oncogene. 1999;18:1619–1627. doi: 10.1038/sj.onc.1202478. [DOI] [PubMed] [Google Scholar]
- 22.Singh AJ, Meyer RD, Band H, Rahimi N. Mol Biol Cell. 2005;4:2106–2118. doi: 10.1091/mbc.E04-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA, Di Fiore PP, Kraus MH. Oncogene. 1995;10:1813–1821. [PubMed] [Google Scholar]
- 24.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, III, Hynes NE. Proc Natl Acad Sci U S A. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammadi M, Schlessinger J, Hubbard SR. Cell. 1996;86:577–587. doi: 10.1016/s0092-8674(00)80131-2. [DOI] [PubMed] [Google Scholar]
- 26.Hubbard SR. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahimi N, Hung W, Tremblay E, Saulnier R, Elliott B. J Biol Chem. 1998;273:33714–33721. doi: 10.1074/jbc.273.50.33714. [DOI] [PubMed] [Google Scholar]
- 28.Maru Y, Yamaguchi S, Shibuya M. Oncogene. 1998;16:2585–2595. doi: 10.1038/sj.onc.1201786. [DOI] [PubMed] [Google Scholar]
- 29.Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL., III Proc Natl Acad Sci U S A. 1994;91:8132–8136. doi: 10.1073/pnas.91.17.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knighton DR, Cadena DL, Zheng J, Ten Eyck LF, Taylor SS, Sowadski JM, Gill GN. Proc Natl Acad Sci U S A. 1993;90:5001–5005. doi: 10.1073/pnas.90.11.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mossie K, Jallal B, Alves F, Sures I, Plowman GD, Ullrich A. Oncogene. 1995;11:2179–2184. [PubMed] [Google Scholar]
- 32.Chou YH, Hayman MJ. Proc Natl Acad Sci U S A. 1991;88:4897–4901. doi: 10.1073/pnas.88.11.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huse M, Kuriyan J. Cell. 2002;109:275–283. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 34.Blume-Jensen P, Hunter T. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 35.Robertson SC, Tynan JA, Donoghhue DJ. Trends Genet. 2000;16:265–271. doi: 10.1016/s0168-9525(00)02021-7. [DOI] [PubMed] [Google Scholar]
- 36.Dibb NJ, Dilworth SM, Mol CD. Nat Rev Cancer. 2004;9:718–727. doi: 10.1038/nrc1434. [DOI] [PubMed] [Google Scholar]
- 37.Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, Alitalo K, Finegold DN. Nat Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- 38.Zwick E, Bange J, Ullrich A. Trends Mol Med. 2002;8:17–23. doi: 10.1016/s1471-4914(01)02217-1. [DOI] [PubMed] [Google Scholar]
- 39.Pawson T, Raina M, Nash P. FEBS Lett. 2002;513:2–10. doi: 10.1016/s0014-5793(01)03292-6. [DOI] [PubMed] [Google Scholar]
- 40.Dayanir V, Meyer RD, Lashkari K, Rahimi N. J Biol Chem. 2001;276:17686–17692. doi: 10.1074/jbc.M009128200. [DOI] [PubMed] [Google Scholar]
- 41.Cunningham ME, Stephens RM, Kaplan DR, Greene LA. J Biol Chem. 1997;272:10957–10967. doi: 10.1074/jbc.272.16.10957. [DOI] [PubMed] [Google Scholar]
- 42.Li S, Ferber A, Miura M, Baserga R. J Biol Chem. 1994;269:32558–32564. [PubMed] [Google Scholar]
- 43.Longati P, Bardelli A, Ponzetto C, Naldini L, Comoglio PM. Oncogene. 1994;9:49–57. [PubMed] [Google Scholar]
- 44.Zeng H, Sanyal S, Mukhopadhyay D. J Biol Chem. 2001;276:32714–32719. doi: 10.1074/jbc.M103130200. [DOI] [PubMed] [Google Scholar]