

Abstract
Virtual screening uses computer-based methods to discover new ligands on the basis of biological structures. Although widely heralded in the 1970s and 1980s, the technique has since struggled to meet its initial promise, and drug discovery remains dominated by empirical screening. Recent successes in predicting new ligands and their receptor-bound structures, and better rates of ligand discovery compared to empirical screening, have re-ignited interest in virtual screening, which is now widely used in drug discovery, albeit on a more limited scale than empirical screening.
The dominant technique for the identification of new lead compounds in drug discovery is the physical screening of large libraries of chemicals against a biological target (high-throughput screening). An alternative approach, known as virtual screening, is to computationally screen large libraries of chemicals for compounds that complement targets of known structure, and experimentally test those that are predicted to bind well. Such receptor-based virtual screening faces several fundamental challenges, including sampling the various conformations of flexible molecules and calculating absolute binding energies in an aqueous environment. Nevertheless, the field has recently had important successes: new ligands have been predicted along with their receptor-bound structures — in several cases with hit rates (ligands discovered per molecules tested) significantly greater than with high-throughput screening. Even with its current limitations, virtual screening accesses a large number of possible new ligands, most of which may then be simply purchased and tested. For those who can tolerate its false-positive and false-negative predictions, virtual screening offers a practical route to discovering new reagents and leads for pharmaceutical research.
Problems with virtual screening
A founding idea in molecular biology was that biological function follows from molecular form. If you knew the molecular structure of a receptor — defined here as a biological macromolecule that converts ligand binding into an activity — you could understand and predict its function. This notion has underpinned a 70-year project to determine receptor structures to atomic resolution. From the early X-ray diffraction studies of pepsin and of haemoglobin, to those of macromolecular assemblies like the ribosome and to structural genomics, the taxonomic part of this enterprise (that is, cataloguing receptor structures) has been extraordinarily successful. But still largely unfulfilled is the promise of exploiting receptor structures to discover new ligands that modulate the activities of these molecules and macromolecular assemblies.
As early as the mid-1970s, investigators suggested that computational simulations of receptor structures and the chemical forces that govern their interactions would enable ‘structure-based’ ligand design and discovery1,2. Ligands could be designed on the basis of the receptor structure alone, which would free medicinal chemistry from the tyranny of empirical screening, substrate-based design and incremental modification. Since then, structure-based design has contributed to and even motivated the development of marketed drugs3,4, such as the human immunodeficiency virus (HIV) protease inhibitor Viracept and the anti-influenza drug Relenza, typically through cycles of modification and subsequent experimental structure determination. Computational modelling has been used extensively in these efforts5,6 and indeed in non-receptor-based methods; for example, when searching for new ligands on the basis of their chemical similarity to a known ligand or when matching candidate molecules to a ‘pharmacophore’ that represents the chemical properties of a series of known ligands7. But until recently there have been few instances of completely new ligands (not resembling those previously known) discovered directly from receptor-based computation. Although there are now many more and much better receptor structures than there were in the 1970s and 1980s, and computer speed has grown exponentially, drug discovery and chemical biology remain dominated by empirical screening and substrate-based design.
Three problems have impeded progress in receptor-guided explorations of ligand chemistry. First, chemical space is vast but most of it is biologically uninteresting: blank, lightless galaxies exist within it into which good ideas at their peril wander. Constraining the number of chemical compounds that are searched to biologically relevant and synthetically accessible molecules remains an area of active research. Second, receptor structures are complicated, resembling “tangled knot(s) of viscera”8. They consist of several thousand atoms, each of which is more or less free to move, and they frequently change shape and solvent structure upon binding to a ligand. To predict what molecules might be recognized by a given receptor, energetically accessible receptor and ligand conformations should be calculated. Unfortunately, the number of possible conformations rises exponentially with the number of rotatable bonds, of which there are thousands in a protein–ligand complex, and the full sampling of conformations involves a set of computational problems for which no general solution is known. Third, calculating ligand–receptor binding energies is difficult9. Binding affinity in an aqueous environment is determined by the solvation energies of the individual molecules (high solvation energies typically disfavour binding), and by the interaction energies between them (high interaction energies favour binding). Solvation and interaction energies are both typically much larger in magnitude than the net affinity, making calculation of the latter problematic. Although it has been possible to calculate accurately the differential affinity between two related ligands using thermodynamic integration methods, doing so is time consuming. Calculating the absolute affinities for many thousands of unrelated molecules necessary to encode new chemical functionality remains beyond our reach. So in principle, it could be argued that structure-based computational screens for new ligands do not work at all.
Successes from virtual screening
However, genuinely novel ligands have been discovered using structure-based computation. Recently, the structures of known ligands in complex with their receptors have been correctly predicted computationally using the structures of the independent receptor and ligand molecules10–12 (Fig. 1). From the standpoint of exploring chemical space, computational screens of chemical databases have identified new ligands for over 50 receptors of known or even, in some cases, computer-modelled structures13,14 (for reviews of recent studies and methods see refs 15 and 16). In these virtual or ‘docking’ screens, large libraries of organic molecules are docked into receptor structures and ranked by the calculated affinity (Fig. 2). Although the energy calculations are crude, the compounds in the library are readily available, making experimental testing easy and false-positives tolerable5.
Figure 1.
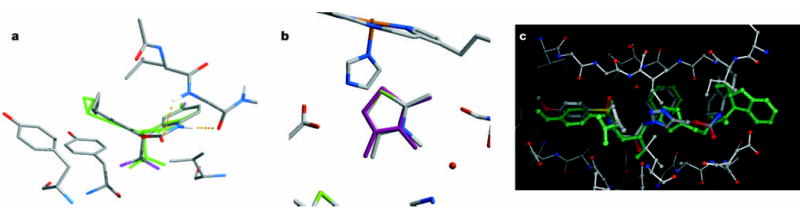
Complexes predicted from virtual screening compared to X-ray crystallographic structures that were subsequently determined. a, Predicted (carbons in grey) and experimental (green) structures for Sustiva in HIV reverse transcriptase10. b, Predicted (magenta) and experimental (carbons in grey) structures of 2,3,4-trimethylthizole in the W191G cavity of cytochrome c peroxidase11. c, Predicted (green)12 and experimental structure (carbons in grey) of an amprenavir mimic in HIV protease (ligands with thick bonds, enzyme residues with thin bonds; structure determined by A. Wlodawer, A. Olson, personal communication).
Figure 2.
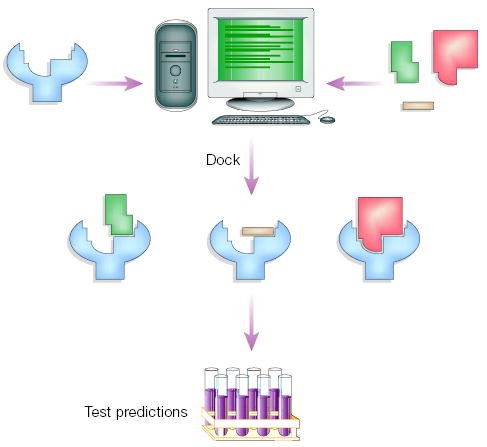
Virtual screening for new ligands. Large libraries of available, often purchasable, compounds are docked into the structure of receptor targets by a docking computer program. Each compound is sampled in thousands to millions of possible configurations and scored on the basis of its complementarity to the receptor. Of the hundreds of thousands of molecules in the library, tens of top-scoring predicted ligands (hits) are subsequently tested for activity in an experimental assay.
Even relatively simple receptor-based constraints can improve the likelihood of finding ligands from among the many possible structures in a library, if only by screening out those that are unlikely to bind the receptor17. In library design, for instance, pre-calculation of possible side chains that would complement a receptor structure resulted in structure-based libraries that were tenfold more likely to contain ligands than random18 or diverse17 libraries constructed at the same time. Similarly, virtual and high-throughput screening have been deployed simultaneously to discover new ligands from libraries of several-hundred-thousand diverse molecules. The virtual screens had ‘hit rates’ (defined as the number of compounds that bind at a particular concentration divided by the number of compounds experimentally tested) that were 100-fold to 1,000-fold higher than those achieved by empirical screens19,20 (Table 1); intriguingly, each technique discovered classes of ligands that the other technique had overlooked19, suggesting that the two screening approaches (virtual and empirical) can be complementary.
Table 1.
Hit rates and drug-like properties for inhibitors discovered with high-throughput and virtual screening against the enzyme PTP-1B (ref.19)
| Technique | Compounds tested | Hits with IC50 < 100μM | Hits with IC50 < 10μM | Lipinski compliant hits* | Hit rate† |
|---|---|---|---|---|---|
| HTS | 400,000 | 85 | 6 | 23 | 0.021% |
| Docking | 365‡ | 127 | 18 | 57 | 34.8% |
Number of 100 μM or better inhibitors that passed all four of the drug-like criteria identified in Lipinski’s ‘rule of five’25;
The number of compounds experimentally tested divided by the number of compounds with IC50 values of 100 μM or less;
The number of top-scoring docking hits that were experimentally tested; IC50, The concentration of inhibitor at which the enzyme is 50% inhibited.
In a few cases the structures of the new ligands in complex with the receptors have been subsequently determined experimentally — typically by X-ray crystallography. Although the docking-derived hits are very different from natural ligands for a given receptor, they often bind at the active site, interacting with conserved receptor groups, as predicted by the docking program21–24 (Fig. 3). From a molecular recognition perspective, this suggests that the structural ‘code’ for binding is plastic in that multiple ligand scaffolds can be recognized by the same receptor site. Methodologically, these structures suggest that although virtual screens are plagued by false-positives, in favourable circumstances they can predict genuinely novel ligands and do so for the right reasons.
Figure 3.
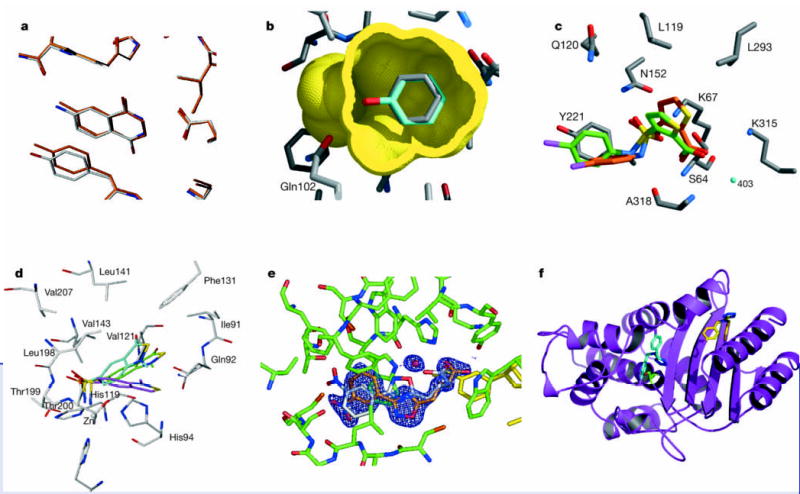
Comparing the structures of new ligands predicted from virtual screening to the structures subsequently determined experimentally. a, The docked (carbons in orange) versus the crystallographic structure (carbons in grey) of the 8.3 μM inhibitor 4-aminophthalhydrazide bound to transfer RNA guanine transglycosylase (ligand in the centre surrounded by enzyme residues)21. b, The docked (carbons in cyan) versus the crystallographic structure (carbons in grey) of the 100 μM ligand phenol bound to a cavity site in T4 lysozyme (ligand in the centre surrounded by the molecular surface of the surrounding protein residues)24. c, The docked (carbons in green) versus the crystallographic structure (carbons in red) of the 26 μM inhibitor 3-((4-chloroanilino)-sulphonyl)-thiophene-2-carboxylate bound to AmpC β-lactamase (enzyme carbons in grey)22. d, The docked (carbons in magenta), re-scored (carbons in cyan) and crystallographic (carbons in grey) structures of a 0.25 μM inhibitor bound to carbonic anhydrase (enzyme carbons in grey)23. Oxygen atoms in red, sulphurs in yellow, nitrogens in blue. e, The docked (ligand carbons in grey) versus the crystallographic structure (ligand carbons in orange) for a new inhibitor of aldose reductase (enzyme carbons in green). Electron density maps for the ligand are shown in blue. The ordered water (red sphere) observed in the experimental structure was not considered in the docking28 (H. Steuber and G. Klebe, unpublished work). f, The docked (carbons in cyan) versus the crystallographic structure (carbons in yellow) of the new inhibitor of TEM-1 β-lactamase (enzyme in magenta)29. The experimentally observed binding mode — 16 Å from the active site targeted in the docking calculations — occurs in a cryptic site absent from the native structure.
How can these successes be reconciled with the field’s methodological weaknesses? Virtual screening avoids the problem of broad searches of chemical space by restricting itself to libraries of specific, accessible compounds (often those that can simply be purchased). This avoids costly syntheses and restricts the search to compounds that are interesting enough biologically to have been previously made, albeit for another reason. Filters may be applied to ensure that the library meets some standard of biological relevance or ‘drug-likeness’25,26. Progress in both the number and quality of molecules in docking libraries has contributed to the increasingly drug-like character of docking hits in recent studies19. Although the problems of sampling molecular conformations and of calculating affinities remain acute, progress has been made both algorithmically16 and in the computer resources available for these calculations. Moreover, we can define success in virtual screening as ‘finding some interesting new ligands’, and not as ‘correctly ranking all the molecules in the library’ or ‘finding all the possible ligands in a library’. Virtual screening thus adopts the same logic as high-throughput screening: as long as some interesting ligands are found, false-negatives are tolerated. Indeed, the two techniques, because of their emphasis on large libraries, share other similarities: both accept limited accuracy in return for screening on a large scale; both look to enrich a list of likely-but-not-certain candidates for further quantitative study; and both are dogged by curious false-positive hits27. Although high-throughput screening remains the dominant technique, virtual screening is now commonly used in pharmaceutical research.
Finally, it must be admitted that these successes retain an episodic character. Even expert practitioners are frequently surprised and sometimes disappointed. Geometries of true ligands may be slightly (Fig. 3e)28 or conspicuously (Fig. 3f)29 mis-predicted and hit rates can vary greatly. We have had hit rates as high as 35% (ref. 19)against an enzyme, protein tyrosine phosphatase 1B (PTP1B), with which we had little experience, and as low as 5% (ref. 22) against an enzyme, AmpC β-lactamase, that we had studied intensely. For many medicinal chemists and structural biologists, such unpredictability lends a whiff of sulphur to an enterprise that has been advertised as ‘rational drug design’.
Prospects
Notwithstanding these caveats, virtual screening will be an evermore important tool for exploring biologically relevant chemical space. Large high-throughput screens have liabilities of their own, and are inaccessible to many investigators (although this will begin to change with the advent of screening resource centres30). In contrast, virtual screening processes large libraries (in principle, libraries that are larger than any library used by empirical screening) and any receptor for which there is a structure at little cost. What advances might be anticipated to make virtual screening reliable and accessible enough to be widely used?
Improved sampling and ‘scoring functions’ (calculations of ligand–receptor energetics) will undoubtedly help. The good news is that the fundamentals of molecular interactions are well understood, and so the field has a clear way forward. But the challenge, as always, will be to implement good physical models for hundreds of thousands of possible ligands, each one sampled in many thousands of possible receptor complexes. Indeed, accurate calculation of absolute binding affinity in screens of large, diverse libraries will remain beyond us for the foreseeable future; even predicting the rank order of affinity for disparate ligands in a hit list will be difficult. What we may anticipate are improved explorations of conformational states for ligand and receptor, and scoring functions that use more sophisticated models of solvation and a better balance of electrostatic and non-polar terms. An interesting strategy will be the use of higher-level, typically much slower methods to re-score initial hits from virtual screening, using the screening calculation as a fast first filter31. From these we can hope for better hit rates and better predictions of geometries23 (Fig. 3d), which are the first and most important goals of virtual screening.
To bring virtual screening to a wide community it will be important to democratize the resources on which it depends. Receptor structures are already available through the Protein Data Bank or PDB (for experimental structures), and through databases such as MODBASE (for a much larger number of structures from computer-based modelling32). Several groups provide docking programs without charge to the academic community, although these programs often require some effort to learn. Programs less demanding of expert knowledge, perhaps as a web-accessible resource, would bring docking to many interested non-specialists. Finally, community-accessible chemical libraries are needed. The National Cancer Institute (NCI) provides calculated structures for about 140,000 of its compounds, and will provide at least some of these for experimental testing (http://cactus.nci.nih.gov/). MDL Inc. sells the Available Chemicals Directory (ACD; http://www.mdl.com/products/experiment/available_chem_dir/index.jsp) of commercially available compounds and the ACD-SC for screening collections. To use these libraries in docking screens, molecular properties such as protonation, charge, stereochemistry, accessible conformations and solvation must be calculated. Even details such as stereochemistry, tautomerization and protonation, which we frequently take for granted, are often ambiguous, or can change on binding to a receptor. Recently, about one million commercially accessible molecules have become available through the ZINC database (http://blaster.docking.org/zinc/). ZINC is a free, web-accessible database constructed with docking, substructure searching and compound purchasing in mind.
In the immediate future, virtual screening is mature enough to benefit from an aggressive programme of experimental testing. As more docking predictions are evaluated, and sometimes falsified, the methods will improve, especially if care is taken to remove the false-positives that have plagued both high-throughput and virtual screening27. Subsequent solution of receptor–ligand complex structures will be particularly informative; so far, too few of these have been determined. For those who can tolerate its false-positives, structure-based virtual screening is reliable enough to justify its use in active ligand discovery projects, providing an important complementary approach to empirical screening. For some projects, especially those centred in academic laboratories, virtual screening will be the best way to access a large chemical space without the commitment in time, material and infrastructure that an empirical screen demands.
Acknowledgments
I thank G. Klebe, A. Olson, and W. Jorgensen for contributing figures and comments, and I. D. Kuntz, M. Jacobson, A. Sali, K. Dill and J. Irwin for many insightful conversations. My laboratory’s research in docking is supported by NIGMS.
Footnotes
Competing interests statement The author declares competing financial interests: details accompany the paper on www.nature.com/nature.
References
- 1.Beddell CR, Goodford PJ, Norrington FE, Wilkinson S, Wootton R. Compounds designed to fit a site of known structure in human haemoglobin. Br J Pharmacol. 1976;57:201–209. doi: 10.1111/j.1476-5381.1976.tb07468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen SS. A strategy for the chemotherapy of infectious disease. Science. 1977;197:431–432. doi: 10.1126/science.195340. [DOI] [PubMed] [Google Scholar]
- 3.Itzstein MV, et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;36:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 4.Varney MD, et al. Crystal-structure-based design and synthesis of Benz[cd]indole-containing inhibitors of thymidylate synthase. J. Med. Chem. 1992;35:663–676. doi: 10.1021/jm00082a006. [DOI] [PubMed] [Google Scholar]
- 5.Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 6.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 7.Stahura FL, Bajorath J. Virtual screening methods that complement HTS. Comb Chem High Throughput Screen. 2004;7:259–269. doi: 10.2174/1386207043328706. [DOI] [PubMed] [Google Scholar]
- 8.Perutz MF. The hemaglobin molecule. Sci Am. 1964;211:64–76. doi: 10.1038/scientificamerican1164-64. [DOI] [PubMed] [Google Scholar]
- 9.van Gunsteren WF, Berendsen HJC. Computer simulation of molecular dynamics: methodology, applications, and perspectives in chemistry. Angew Chem Int Ed Engl. 1990;29:992–1023. [Google Scholar]
- 10.Rizzo R, Wang D, Tirado-Rives J, Jorgensen W. Validation of a model for the complex of HIV-1 reverse transcriptase with sustiva through computation of resistance profiles. J Am Chem Soc. 2000;122:12898–12900. [Google Scholar]
- 11.Rosenfeld RJ, et al. Automated docking of ligands to an artificial active site: augmenting crystallographic analysis with computer modeling. J. Comput. Aided Mol. Des. 2003;17:525–536. doi: 10.1023/b:jcam.0000004604.87558.02. [DOI] [PubMed] [Google Scholar]
- 12.Brik A, et al. Rapid diversity-oriented synthesis in microtiter plates for in situ screening of HIV protease inhibitors. Chembiochem. 2003;4:1246–1248. doi: 10.1002/cbic.200300724. [DOI] [PubMed] [Google Scholar]
- 13.Schapira M, et al. Discovery of diverse thyroid hormone receptor antagonists by high-throughput docking. Proc. Natl Acad. Sci. USA. 2003;100:7354–7359. doi: 10.1073/pnas.1131854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evers A, Klebe G. Ligand-supported homology modeling of G-protein-coupled receptor sites: models sufficient for successful virtual screening. Angew Chem Int Ed Engl. 2004;43:248–251. doi: 10.1002/anie.200352776. [DOI] [PubMed] [Google Scholar]
- 15.Shoichet BK, McGovern SL, Wei BI, Irwin JJ. Lead discovery using molecular docking. Curr Opin Chem Biol. 2002;6:439–446. doi: 10.1016/s1367-5931(02)00339-3. [DOI] [PubMed] [Google Scholar]
- 16.Schneidman-Duhovny D, Nussinov R, Wolfson HJ. Predicting molecular interactions in silico: II. Protein-protein and protein-drug docking. Curr Med Chem. 2004;11:91–107. doi: 10.2174/0929867043456223. [DOI] [PubMed] [Google Scholar]
- 17.Wyss PC, et al. Novel dihydrofolate reductase inhibitors. Structure-based versus diversity-based library design and high-throughput synthesis and screening. J. Med. Chem. 2003;46:2304–2312. doi: 10.1021/jm020495y. [DOI] [PubMed] [Google Scholar]
- 18.Kick EK, et al. Structure-based design and combinatorial chemistry yield low nanomolar inhibitors of cathepsin D. Chem. Biol. 1997;4:297–307. doi: 10.1016/s1074-5521(97)90073-9. [DOI] [PubMed] [Google Scholar]
- 19.Doman TN, et al. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 20.Paiva AM, et al. Inhibitors of dihydrodipicolinate reductase, a key enzyme of the diaminopimelate pathway of Mycobacterium tuberculosis. Biochim. Biophys. Acta . 2001;1545:67–77. doi: 10.1016/s0167-4838(00)00262-4. [DOI] [PubMed] [Google Scholar]
- 21.Gradler U, et al. A new target for shigellosis: rational design and crystallographic studies of inhibitors of tRNA-guanine transglycosylase. J. Mol. Biol. 2001;306:455–467. doi: 10.1006/jmbi.2000.4256. [DOI] [PubMed] [Google Scholar]
- 22.Powers RA, Morandi F, Shoichet BK. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure (Camb) 2002;10:1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 23.Gruneberg S, Stubbs MT, Klebe G. Successful virtual screening for novel inhibitors of human carbonic anhydrase: strategy and experimental confirmation. J Med Chem. 2002;45:3588–3602. doi: 10.1021/jm011112j. [DOI] [PubMed] [Google Scholar]
- 24.Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 25.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 26.Oprea TI. Current trends in lead discovery: are we looking for the appropriate properties? Mol Divers. 2002;5:199–208. doi: 10.1023/a:1021368007777. [DOI] [PubMed] [Google Scholar]
- 27.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 28.Krämer O, Hazemann I, Podjarny AD, Klebe G. Virtual screening for inhibitors of human aldose reductase. Proteins. 2004;55:814–823. doi: 10.1002/prot.20057. [DOI] [PubMed] [Google Scholar]
- 29.Horn JR, Shoichet BK. Allosteric inhibition through core disruption. J Mol Biol. 2004;336:1283–1291. doi: 10.1016/j.jmb.2003.12.068. [DOI] [PubMed] [Google Scholar]
- 30.Kaiser J. NIH Gears up for chemical genomics. Science. 2004;304:1728. doi: 10.1126/science.304.5678.1728a. [DOI] [PubMed] [Google Scholar]
- 31.Kalyanaraman, C., Bernacki, K., & Jacobson, M. P. Virtual screening against highly charged active sites: Identifying substrates of alpha-beta barrel enzymes. Biochemistry in the press. [DOI] [PubMed]
- 32.Pieper U, Eswar N, Stuart AC, Ilyin VA, Sali A. MODBASE, a database of annotated comparative protein structure models. Nucleic Acids Res. 2002;30:255–259. doi: 10.1093/nar/30.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]