

Abstract
Papillomaviruses (PV) comprise a large family of nonenveloped DNA viruses that include the oncogenic PV types that are the causative agents of human cervical cancer. As is true of many animal DNA viruses, PV are taken into the cell by endocytosis and must escape from the endosomal compartment to the cytoplasm to initiate infection. Here we show that this step depends on the site-specific enzymatic cleavage of the PV minor virion protein L2 at a consensus furin recognition site. Cleavage by furin, a cell-encoded proprotein convertase, is known to be required for endosome escape by many bacterial toxins. However, to our knowledge, furin has not been previously implicated in the viral entry process. This step is potentially a target for PV inhibition.
Keywords: proprotein convertase
Viral capsids have evolved to fulfill numerous roles that are critical to the establishment of viral infection. For nonenveloped viruses, the proteinaceous coat encases and protects the viral nucleic acid and provides the initial interaction of the viral particle with the host cell. After receptor engagement, the virus is internalized and its coat is disassembled to allow the encapsidated genome access to the cellular transcription and replication machinery. For DNA viruses, excluding the poxviruses, this process necessitates the navigation of the genome into the nucleus. Our laboratory has been engaged in the examination of the entry and uncoating mechanisms of papillomaviruses (PV), a family of nonenveloped DNA viruses that includes the oncogenic PV types, the causative agents of human cervical cancer (1).
The PV capsid is composed of two structural proteins: the major capsid protein, L1, which can self-assemble into icosahedral virus-like particles in the absence of the minor capsid protein, L2, which is nonetheless necessary for establishment of infection. Most PV appear to enter the host cell by clathrin-dependent, receptor-mediated endocytosis (2-4). Disassembly of the viral capsid, determined by exposure of the encapsidated genome, occurs within the endosome. Subsequently, L2 and the genome escape into the cytoplasm, enter the nucleus, and colocalize at nuclear domain 10 (5). L2 is mechanistically multifaceted in its contributions to these processes. The localization of the genome to the transcriptionally active nuclear domain 10, which is critical to the efficient establishment of infection, depends on L2. It has also been demonstrated that a C-terminal region of L2 mediates endosomal escape after viral uncoating (6). Additionally, a recent report showed that L2 interacts with syntaxin 18 during entry and possibly uses this resident endoplasmic reticulum protein as a tether for transport toward the nucleus (7).
We recently developed a high-titer PV pseudovirus production system in which the viral L1 and L2 proteins from a given PV encapsidate a target plasmid that encodes a reporter gene. This procedure allows for a straightforward quantification of transduction events (8). Thereby, pseudoviruses can be readily used to screen for compounds that prevent viral infection. We now report that this type of screen has identified an inhibitor of proprotein convertases (PCs) to be a potent inhibitor of infection, as monitored by GFP-encoding PV pseudoviruses. Further investigation has revealed that proteolytic modification of the L2 protein by furin is indispensable for PV infection. Furin is a cell-encoded PC present in the Golgi complex, at the plasma membrane, and within endosomes (9, 10). Furin cleavage is known to be required for endosome escape by several bacterial toxins, including anthrax toxin and Pseudomonas exotoxin A (11). However, this study demonstrates a role for furin in a viral entry process.
Results
Susceptibility of PV Infection to PC Inhibition. In an effort to determine whether specific endosomal proteases can function in the disassembly of PV particles, we evaluated a panel of common protease inhibitors for disruption of infection by using a pseudovirus assay. We found that inhibition of a number of endosomal/lysosomal proteases had a minimal effect on infection by pseudoviruses for HPV16, an oncogenic human PV (HPV) (Fig. 1A, groups A-G). However, the inhibition of furin-like PCs with the specific pharmacological inhibitor decanoyl-RVKR-chloromethylketone (dec.-RVKR-cmk) resulted in a dramatic inhibition of infection (Fig. 1 A, group H). Control chloromethylketone conjugates that do not inhibit furin showed negligible effects on PV infectivity (Fig. 1 A, groups I-K), indicating that the observed inhibition with dec.-RVKR-cmk is not attributable to nonspecific effects of the chloromethylketone moiety. The IC50 of dec.-RVKR-cmk on HPV16 infection was ≈50 nM (Fig. 1B).
Fig. 1.

Effect of protease inhibition and expression on PV infectivity. (A) HeLa cells were incubated with various protease inhibitors for the duration of infection with HPV16 pseudovirus containing a GFP expression plasmid. Infection was quantified by flow cytometric analysis of GFP expression. Two concentrations were evaluated for each inhibitor. The lightly shaded bars illustrate the higher concentration, and the darker bars show the lower concentration. The inhibitors are as follows: group A, N-acetyl-Leu-Leu-Met (200 nM, 100 nM); group B, N-acetyl-Leu-Leu-Nle-CHO (200 nM, 100 nM), inhibitors of calpain I and II and cathepsins B and L; group C, Ca-074 (20 μM, 10 μM), an inhibitor of intracellular cathepsin B; group D, calpeptin (1.0 μM, 0.5 μM), a calpain inhibitor; group E, cathepsin-l inhibitor (10 μM, 1 μM); group F, chymostatin (50 μM, 10 μM), an inhibitor of chymotrypsin, papain, and cysteine proteases; group G, pepstatin A (50 μM, 10 μM), an inhibitor of aspartic proteinases; group H, dec.-RVKR-cmk (100 μM, 25 μM), an inhibitor of furin and other PCs; group I, d-VFK-cmk (20 μM, 4 μM), an inhibitor of plasmin; group J, cbz-VNSTLQ-cmk (20 μM, 4 μM), a coronavirus inhibitor; and group K, H-AAF-cmk (20 μM, 4 μM), a tripeptidyl peptidase inhibitor. (B) The IC50 of dec.-RVKR-cmk on PV pseudovirus infection. (C) HPV16 pseudovirus infection of HeLa cells, FD11 cells, and the FD11-plus-furin cells. (D) A comparison of the infectivity of HPV16 pseudoviruses containing either wild-type L2 or the R12S L2 mutation. (E) A comparison of the infectivity of BPV1 pseudoviruses containing either wild-type L2 or the R9S L2 mutation.
Because dec.-RVKR-cmk affects a number of furin-like PCs, we wanted to determine whether the observed inhibition reflected a specific requirement for furin in PV infection. Therefore, we compared the infectivity of a furin-deficient Chinese hamster ovary (CHO) cell line, FD11, and a furin-expressing derivative line (12). FD11 was completely resistant to infection by pseudoviruses for HPV16 (Fig. 1C) and BPV1, a cutaneous bovine PV (BPV) type (data not shown). In contrast, the furin-expressing line was highly infectible by both pseudoviruses. Because CHO cells are known to express most other PCs, it is evident that they cannot substitute for furin in the PV infectious process (13). Additionally, we examined a second furin-deficient line, LoVo, a human colon carcinoma cell line that is defective in proprotein processing because of a point mutation within both alleles of the furin gene (14). We found that these cells are incapable of supporting PV infection until a functional furin gene is provided by transfection (data not shown). LoVo cells have been described to express functional PACE4 protease (15), indicating that PACE4 (paired basic amino acid cleaving enzyme 4) cannot functionally substitute for furin to support PV infection. Thus, in vitro PV infection is a furin-dependent process.
PV L2 Contains a Consensus Furin Cleavage Site. The above results do not distinguish between the possibilities that furin acts directly on the viral capsid during the entry process or that it exerts an indirect effect on PV infection via the processing of a required cellular component. Furin preferentially recognizes the cleavage site sequence R-X-K/R-R (16, 17). Examination of the viral L1 and L2 sequences revealed that a remarkably conserved consensus furin cleavage site close to the N terminus of L2 was present in all PV sequences described in the GenBank database (Table 1), but no potential sites exist in L1, suggesting that furin might be acting directly on L2. Consistent with the conservation of this putative L2 furin cleavage site, infection by all other tested PV pseudotypes (18) and by authentic BPV1 virions was similarly inhibited by dec.-RVKR-cmk (Fig. 1B and data not shown, respectively).
Table 1. Alignment of L2 N-terminal sequences from phylogenetically diverse PV types.
| Type | Sequence | Genus/species | Accession no. | FI |
|---|---|---|---|---|
| BPV1 | MSARKRVKRASA... | Delta 4 | CAB57284 | + |
| HPV16 | MRHKRSAKRTKRASA... | Alpha 12 | AAV91690 | + |
| CRPV | MVARSRKRRAAP... | Iota | CAB96120 | + |
| HPV4 | MQSLSRRKRDSV... | Gamma 1 | Q07862 | ND |
| HPV5 | MARAKRVKRDSV... | Beta 1 | D90252 | + |
| HPV6 | MAHSRARRRKRASA... | Alpha 10 | NP_040316 | + |
| HPV18 | MVSHRAARRKRASV... | Alpha 7 | NP_040316 | + |
A representative example from each species was chosen. A consensus furin cleavage site was found in every PV type examined. If available, the pseudovirus was tested for susceptibility of infection to furin inhibition (FI). +, inhibition of infection; ND, not determined because of unavailability; CRPV, cottontail rabbit PV. The P1, P4, and P6 residues are indicated by boldface type.
To explore the possibility that furin cleavage of this L2 consensus site is important for infection, the P1 arginine residue within the putative furin recognition site in HPV16 L2 was mutated to a serine residue (16-R12S), abrogating the consensus cleavage site. This mutation reduced the infectivity of HPV16 to negligible levels (Fig. 1D). The analogous mutation within the BPV1 L2 (B-R9S) produced a similar result (Fig. 1E). The furin-binding pocket does not tolerate well a non-arginine residue at the P1 position. Thus, the infectivity of the L2 mutant pseudoviruses followed their predicted susceptibility to furin cleavage. Both of the L2 mutant pseudovirions incorporated levels of L2 and encapsidated DNA indistinguishable from wild type (data not shown), ruling out these alternate possibilities for their loss of infectivity.
Furin Can Cleave L2 in Vitro. To confirm that L2 is a furin substrate, we performed a series of in vitro proteolysis experiments. Wild-type BPV1 L2 or B-R9S L2 constructs were made that contained the hemagglutinin-derived (HA) epitope tag at either their C or N termini (desginated CHA and NHA, respectively). These L2 proteins were expressed in cells and partially purified. As expected from the location of the cleavage site near the N terminus, furin treatment of the tagged wild-type proteins liberated the NHA tag (Fig. 2) as shown by the lack of detection with an antibody directed against the HA epitope. This construct was still detected easily with an anti-L2 reagent, although the protein migrated slightly faster, indicating the expected small difference in size resulting from cleavage by furin. The C-terminally tagged molecule was also detected as a slightly faster migrating protein after furin treatment. In this instance, the protein could be detected with antibodies directed against either the HA epitope or the L2 protein. By contrast, when the furin cleavage site was abrogated in the L2 mutants, both the proteins' migrations and their detection by anti-HA were unaffected by furin treatment, as observed for the B-R9S-CHA and B-R9S-NHA L2 constructs.
Fig. 2.

In vitro cleavage of BPV L2 proteins. The wild-type BPV1 or BR9S L2 cDNAs fused at either the N- or C-termini with the sequence encoding the HA epitope were transfected into HeLa cells. At 24 h after transfection, the L2 proteins were partially purified and digested with furin or left untreated as indicated. Each sample was divided into two samples and processed for immunoblotting with either an anti-HA antibody (upper gel) or an anti-L2 antibody (lower gel).
Furin Cleavage of L2 Is Necessary for Endosome Escape. We next sought evidence for in vivo digestion of the N-terminal furin cleavage site during infection. The low number of L2 proteins per virion and the lack of synchronicity during the viral entry process prevented us from performing in vivo biochemical analysis. However, it was possible to follow the trafficking of L2 proteins microscopically during infection by using an assay we recently developed to detect uncoated pseudovirus (5). In this assay, the pseudoviral genome is labeled with 5-bromodeoxyuridine (BrdUrd) during pseudovirus production. When viral entry is studied with these BrdUrd-labeled pseudovirions, the encapsidated genomes become detectable with anti-BrdUrd antibodies only after disassembly of the viral particles in the endosome. The CHA also becomes detectable only after uncoating (5). To detect L2 cleavage in vivo, we also examined the NHA, whose tag would be predicted to be lost during infection. First, we verified that expression of de novo-synthesized NHA and CHA L2 proteins resulted in their intranuclear detection. As expected, both constructs show a punctate intranuclear distribution indicating association with nuclear domain 10 as described in ref. 19. This pattern was indistinguishable between the NHA- and CHA-tagged proteins and was detectable both with anti-L2 antibodies (data not shown) and anti-HA antibodies (Fig. 5, which is published as supporting information on the PNAS web site).
The PV entry process has extremely slow kinetics, with disassembly being initially detected 6-8 h after entry (5). By 24 h after entry, the C-terminally tagged L2 protein was predominantly detected in the nucleus, along with the pseudoviral genome (Fig. 3A), as described (5). However, the N-terminally tagged L2 protein was not detectable in the nucleus (Fig. 3B), although it was seen in cytoplasmic vesicles. The pseudoviral genome was detected in the nucleus at equivalent levels for the CHA- and NHA-containing pseudoviruses (Fig. 3 C and D), and both pseudoviruses were equally infectious (data not shown). We conclude that the N-terminal tag is removed from the L2 protein in vivo before its entry into the nucleus during the infectious process.
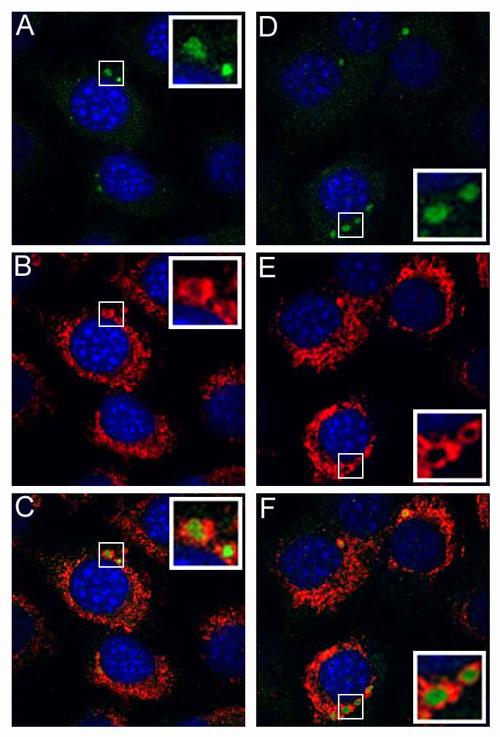
Fig. 3.

Uncoating and nuclear trafficking of HA-epitope-tagged wild-type L2 BPV1 pseudovirions or B-R9S-L2 pseudovirions. HeLa cells were allowed to internalize pseudovirions assembled in the presence of 20 μM BrdUrd. At 24 h after entry, the cells were fixed and processed for either L2-HA detection or BrdUrd detection. (A and B) The detection of L2 proteins with an anti-HA antibody. (A) CHA-L2. (B) NHA-L2. (C-F) The detection of the encapsidated pseudogenome with anti-BrdUrd antibody. (C) CHA-L2. (D) NHA-L2. (E) CHA-B-R9S-L2. (F) NHA-B-R9S-L2.
To identify the step during infection that required furin cleavage of L2, we followed the localization of particles assembled with the L2 furin-cleavage mutants. There were no gross differences in the entry and trafficking kinetics, as visualized with anti-L1 antibodies (data not shown). Moreover, furin cleavage was not necessary for uncoating of the viral capsid, because the BrdUrd-labeled pseudoviral genome was readily detected. However, the genome appeared to be retained in the endosomal compartment (Fig. 3 E and F), in contrast to the wild-type virus, for which genome was detectable both in endosomes and the nucleus (Fig. 3 C and D). A similar retention of the genome and of the L2 protein was observed when the entry of wild-type virus was performed in the presence of dec.-RVKR-cmk (Fig. 6, which is published as supporting information on the PNAS web site). It has been shown that infectious PV traverses the endocytic pathway (2), and we confirmed that the genome- and L2-containing vesicles localize within vesicles limited by Lamp-1 staining, indicating retention in the late endosomal/lysosomal compartment (Fig. 6). We have not observed the delivery of L2 or genome to the nucleus either in the presence of dec.-RVKR-cmk or with the furin cleavage-L2 mutants. We conclude that furin cleavage of L2 is essential for the correct trafficking of the genome and L2 out of the endocytic compartment before their transit into the nucleus.
Precleavage of L2 Can Bypass the Requirement for Cellular Furin. The above studies indicated that furin cleavage of L2 is necessary for infection. To test whether this proteolysis is the sole furin activity required for infection, we sought a condition in which in vitro furin treatment of a wild-type pseudovirus before infection might overcome the ability of the furin inhibitor to prevent infection. We have recently determined that when pseudoviruses are produced, they initially form as immature particles with a loose conformation and then undergo a maturation process, resulting in particles with a more compact conformation (20). Furin treatment of mature pseudovirions did not render them infectious in cells treated with furin inhibitor (Table 2). This negative result is consistent with published data demonstrating that antibodies directed to the N terminus of L2 are not able to bind intact viral particles (21). However, furin treatment of immature particles did enable them, when added to cells treated with furin inhibitor, to reach ≈30% of the infectivity of pseudovirus added to untreated cells (Table 2). This result strongly suggests that L2 cleavage by furin is the only furin-dependent effect required for pseudovirus infection and that furin apparently acts during PV entry after the capsid has undergone an initial conformational change.
Table 2. In vitro furin cleavage of immature pseudovirus bypasses the requirement for cellular furin.
| Pseudovirus added, μl
|
||||
|---|---|---|---|---|
| FI | 1.25 | 0.63 | 0.32 | 0.16 |
| untx. iPV | ||||
| − | 30.7 | 17.9 | 14.1 | 7.4 |
| + | 0.4 | 0.2 | 0.1 | 0.1 |
| furin-tx. iPV | ||||
| − | 29.9 | 20.2 | 10.7 | 8.1 |
| + | 11.6 | 4.6 | 2.2 | 0.8 |
| untx. mPV | ||||
| − | 28.7 | 17.2 | 14.9 | 6.3 |
| + | 1.2 | 0.2 | 0.1 | 0.2 |
| furin-tx. mPV | ||||
| − | 29.2 | 17.4 | 14.9 | 6.0 |
| + | 1.6 | 0.6 | 0.4 | 0.3 |
Immature (iPV) or mature (mPV) BPV1 pseudoviruses were either treated with furin (furin-tx.) or left untreated (untx.). The infectivity of these preparations was evaluated on untreated HeLa cells (−) or HeLa cells treated with furin inhibitor (+). The percent of GFP-positive cells is shown. FI, furin inhibition.
Furin Does Not Affect Production of Infectious PV. The requirement for furin during PV entry contrasts sharply with the previously described role of furin in the production of a variety of infectious enveloped viruses. For these viruses, furin cleavage of an envelope protein must occur within the Golgi complex to yield the mature, fusogenic form of the protein, which is expressed on the cell surface before particle budding (22, 23). Because PV are nonenveloped viruses, they would not be expected to use furin during virus production. Indeed, furin inhibition during pseudovirus production, in contrast to pseudovirus entry, did not affect the titer of the resultant virus (Fig. 4).
Fig. 4.

Furin inhibition during pseudovirus production has no effect on PV infectivity. The titer of pseudovirus that was produced either by normal methodology, as previously described, or in the presence of 10 μM dec.-RVKR-cmk was compared. HeLa cells were infected with pseudoviruses for 72 h, and GFP-expressing cells were quantified by flow cytometry.
Discussion
In this report, we used multiple approaches to implicate furin as an L2-processing protease that is critical for the establishment of PV infection. Based on genetic, biochemical, and cytological evidence, we conclude that furin removes the N terminus of the L2 protein early in the infectious entry pathway and that this cleavage is essential for processes leading to L2-mediated endosome escape. L2 that is not cleaved by furin does not leave the endocytic compartment, and the accompanying genome is likewise withheld.
These findings represent an example of a viral entry pathway that is clearly dependent on a specific single proteolytic cleavage event. Reoviruses and Ebola virus use the lysosomal cathepsin proteases to initiate their uncoating processes (24, 25). For most substrates, cathepsins act as relatively nonspecific, processive proteases (26), and for these viruses the cathepsins mediate a stepwise proteolysis of the viral structural proteins. By contrast, cathepsin inhibitors had no effect on PV infection, and furin inhibition did not affect capsid uncoating, which appears to be an L2-independent event (6). It would be interesting to determine whether other nonenveloped viruses employ PCs to activate incoming virions. We have found that BK and JC viruses, two polyomaviruses with a virion structure that is similar to PV, are not sensitive to furin inhibition, although they both contain consensus cleavage sites at the C termini of their minor capsid proteins, VP2 and VP3 (data not shown).
Furin is a type I membrane protein localized predominantly in the transGolgi network (TGN). However, furin has also been demonstrated to be present in an active form both on the cell surface and within the endosomal compartment and, thus, is present in cellular locations where it could intersect with incoming viral capsids (10, 27, 28). This protease plays a role in the endoproteolytic processing of cell-encoded precursor proteins in mammalian cells within the TGN. During the production of infectious viruses, many viral envelope proteins, including those of avian influenza virus, HIV-1 and measles virues, are also processed by furin or other PCs during their exocytic transit through the TGN (reviewed in ref. 10). This processing yields the mature, fusogenic form of the envelope protein and disruption of the processing results in the production of noninfectious particles. However, no PC, including furin, has previously been implicated as playing a role in virus entry.
The activation of bacterial exotoxins is frequently a furin-dependent process, with furin cleavage occurring either at the cell surface or within the endosomal compartment, as with anthrax toxin and Pseudomonas exotoxin, respectively (10, 12, 29-31). With PV, endosome escape and subsequent trafficking were furin-dependent, although the actual furin cleavage may be occurring at an earlier stage. Thus, viruses and bacterial toxins use the same protease to promote endosomal translocation, representing an interesting example of convergent evolution. By analogy with toxins, the cleavage of L2 could occur on the cell surface or within the early endosomal compartment. It has been demonstrated that HPV33 capsids undergo a subtle conformational change after cell attachment (32), raising the possibility of cell-surface cleavage due to exposure of the N terminus of L2. Alternatively, cleavage might occur in an endocytic compartment, because the capsids clearly undergo further conformational changes, as shown by the exposure of the genome and to antibodies (5).
Furin digestion of immature capsids before their incubation with cells overcame the inhibition of cellular furin with dec.-RVKR-cmk. This direct evidence that furin cleavage of L2 is its essential role in PV infection suggests that (i) a conformational change in the capsid must occur before cleavage, because mature capsids were not susceptible to furin cleavage, (ii) cleavage in a particular subcellular compartment or at a precise time in the infectious process is not required, and (iii) the short L2 cleavage product does not have an indispensable role in infection.
Although our in vitro and in vivo results support the conclusion that cleavage occurs at the consensus furin recognition site, the asynchronous nature of PV infection and the low amount of L2 incorporated into the virion made it infeasible to biochemically address the in vivo cleavage of L2. One question that this limitation leaves unanswered is the number of L2 molecules per virion that must be cleaved for infection to proceed. However, biochemical approaches would, at best, give an average estimate of L2 cleavage but not a per virion estimate, which would be the essential information. Our data indicate that furin has cleaved most or all of the L2 molecules that accompany the viral genome into the nucleus because we cannot detect the NHA tag in the nucleus. What percentage of L2 needs to be processed by furin for efficient infection remains unanswered. The appearance of some uncleaved NHA-L2 in the late endosomes may indicate that not all L2 molecules per virion need to be cleaved for the virus to establish infection. Alternatively, the uncleaved L2 may represent capsids that have been routed down a nonproductive pathway.
Furin cleavage of L2 could serve several functions: First, furin cleavage of L2 could enable the release of the L2-genome complex from L1, because L1 does not appear to exit from the endosomal compartment (5); second, it could promote L2 binding to a specific receptor. Interestingly, a syntaxin 18 binding site on L2 has been mapped to a peptide immediately downstream of the cleavage site (7). It would be interesting to determine whether the furin-cleaved form of L2 preferentially interacts with syntaxin during the entry process. Third, furin cleavage of L2 could prevent exposure of L2-specific neutralizing B cell epitopes until the capsids are internalized. Consistent with this latter conjecture, the broadly cross-type neutralization epitopes of L2 map near the furin cleavage site (33). L2 immunogens exposing this cleavage site might be particularly effective at inducing broadly cross-neutralizing PV antibodies.
Based on the above considerations, we have developed a speculative model for the viral entry process, which is initiated by the binding of viral particles to the cell surface. Virion binding leads to a conformational change in the capsid that in turn permits furin to cleave the N terminus of L2, which may occur at the cell surface or within an early endosomal compartment. The capsid undergoes uncoating in a late endosomal compartment, leading to furin-cleaved L2 and the associated genome to escape from the endosome into the cytoplasm via a mechanism that also involves the C terminus of L2 (6). Furin inhibition by dec.-RVKR-cmk prevents the escape of L2 and the genome from the endosome but does not appear to interfere with virion trafficking or uncoating.
It was previously reported that an N-terminally truncated BPV L2 could not produce infectious virions (34). Like our results with the R9S mutation, truncated L2 was incorporated into the virion, and the viral genome was encapsidated at wild-type levels. The truncated protein was initiated just after the furin cleavage site. A priori, we would expect this mutant to behave like the in vitro digested pseudovirus and effectively lead to infection. A possible explanation for the discrepancy is that initiation at the ninth amino acid results in a modified residue that cannot participate in downstream events. Either the additional N-terminal methionine residue is not cleaved or the next residue, an alanine, is modified. N-terminal alanine residues are often acetylated. An alternative explanation is that the truncated L2 protein does not correctly participate in a necessary precleavage conformational change, which is mimicked in the immature capsids.
Furin belongs to the family of PCs, which are subtilisin-like eukaryotic endoproteases (reviewed in ref. 35). These enzymes are differentially expressed in various cells and tissues and display similar specificity for basic motives, as typified by R-X-R/K-R. The data obtained with the furin-deficient CHO and LoVo cell lines indicate that the endogenous PCs expressed in these two lines cannot substitute functionally for furin. However, it remains formally possible that a PC family member not expressed in these cells might be able to do so, because dec.-RVKR-cmk inhibits substrate binding to furin and related PCs (36). Keratinocytes, which are the natural host cell for PV, are known to express furin, PACE4, PC5/6, and PC7/8 (37). CHO cells express PC7/8 but not PACE4 (13), whereas LoVo express PACE4 (15, 23). Thus, PC5/6 has not been ruled out as a putative physiologic PC for cleavage of the L2 sequence.
The discovery that furin activity is required for PV infection raises the possibility that furin (or furin/PC5/6) inhibitors might be effective as topical microbicides to prevent genital infection by HPV, and they might also inhibit viruses, such as HIV, whose production depends on furin (38, 39). Furin inhibitors that prevent the activation of bacterial toxins, such as anthrax toxin (40), are under development. Results from mice protected against anthrax toxemia by systemic treatment with furin inhibitors suggest that topical application might have acceptable side effects (41).
Materials and Methods
Pseudovirus Production. Pseudoviruses were made as described in ref. 8. To create the mutations within the L2 sequences, the QuikChange II site-directed mutagenesis kit (Stratagene) was used according to manufacturer's directions. The oligonucleotides used for mutagenesis were as follows (in both cases the reverse complement was also used but is not listed): HPV16 R12S, 5′-GCGCCAAGAGGACCAAGAGCG-CCAGCGCCACCCAGC-3′; BPV R9S, 5′-GCCCGCAAGAGAGTGAAGAGCGCCAGCGCCTACGACCTG-3′. Construction of CHA-L2 is described in ref. 5. To create NHA-L2, the previously described vector PMLH containing the BPV codon-modified L2 sequence was digested with the restriction endonucleases KpnI and BssHI and ligated with the annealed and likewise digested primers (5′-CACGCGTGGTACCGCCATGTACCCATACGATGTTCCAGATTACGCTAGCGCCCGCAAGAGAGTGAAGCGCGCCAGCGCC-3′ and its reverse complement). Sequences for all constructs were confirmed on both strands.
Cell Lines. HeLa cells and C127 cells were grown in DMEM supplemented with 10% FBS and antibiotics. FD11 cells and FD11-plus-furin cells were the kind gifts from Stephen Leppla (National Institute of Allergy and Infectious Disease, National Institutes of Health) (12). The cells were cultured in DMEM supplemented with 10% FBS, antibiotics, and proline.
Pseudovirus Infection. Cells were plated in a 24-well plate at a density of 1 × 105 per well. After adherence, pseudoviruses were added at the concentrations indicated in the figure legends. When included, inhibitors were added in unison with the pseudovirus. Infection was monitored at 72 h by flow cytometric analysis of GFP expression as described in ref. 8. Ca-074 was obtained from Sigma, pepstatin A, cathepsin-l inhibitor, chymostatin, calpeptin, ALLN, ALLM, dec.-RVKR-cmk, d-VFK-cmk, cbz-VNSTLQ-cmk, and H-AAF-cmk were all purchased from Calbiochem.
In Vitro Furin Cleavage of L2. HeLa cells were transfected with the cDNA for either the wild-type CHA-L2 or CHA-BPV R9S-L2 or the wild-type NHA-L2 or NHA-BPV R9S-L2. At 24 h after transfection, nuclei were isolated by standard methodology and disrupted by sonication. L2 proteins were immunoprecipitated with a polyclonal anti-L2 antiserum (serum 17/28) conjugated to protein A Sepharose (Pierce). The Sepharose-antibody-L2 complexes were resuspended in furin digestion buffer (100 mM Hepes/1 mM CaCl2/0.5% Triton X-100), and each sample was divided into two equal portions, one of which was treated with 5 units of furin (Alexis Biochemical, San Diego). All samples were incubated at 37°C for 16 h. Each sample was again divided in two equal aliquots. One aliquot was immunopurified with anti-HA magnetic beads according to the manufacturer's directions (Miltenyi Biotec, Auburn, CA). The eluates from this purification were separated by SDS/PAGE, transferred to a polyvinylidene fluoride membrane, and probed with a mouse anti-HA antibody (Babco, Richmond, CA). The second aliquot was subjected to Western blotting with a mouse anti-BPV L2 antibody, C6 (21).
Immunofluorescence. Cells were seeded onto glass coverslips in a 24-well plate at a density of 1 × 105 per well. Cells were incubated with 50 ng of pseudovirus for 24 h. The detection of L2-HA and the BrdUrd-labeled pseudogenome is described in ref. 5. This method utilizes a mild, nondenaturing procedure for BrdUrd detection. All images were acquired with a Zeiss LSM 510 confocal system interfaced with a Zeiss Axiovert 100M microscope. Images were collated with the photoshop software (Adobe Systems, San Jose, CA).
In Vitro Furin Cleavage of Pseudovirus. Immature and mature pseudoviruses were prepared as described in ref. 20. Pseudovirus preparations were clarified by high-speed centrifugation of cellular debris and used as a crude preparation. The preparations were adjusted to 100 mM Hepes/1 mM CaCl2, divided into two aliquots, and incubated at 37°C for 7 h. One aliquot was incubated in the presence of 3.5 units of furin, and the other was incubated without furin. After this incubation, pseudovirions were added to HeLa cells and the infection was continued for 72 h in the absence or presence of 10 μM dec.-RVKR-cmk. GFP expression was quantified by flow cytometry.
Supplementary Material
Acknowledgments
We thank Dr. Christopher Buck for helpful insight and discussion, Dr. Stephen Leppla for providing the FD11 cell lines, and Dr. Walter Atwood (Brown University, Providence, RI) for providing BK and JC viruses. This research was supported by the Intramural Research Program of the National Cancer Institute Center for Cancer Research, National Institutes of Health.
Author contributions: R.M.R., D.R.L., J.T.S., and P.M.D. designed research; R.M.R. and P.M.D. performed research; R.M.R., D.R.L., J.T.S., and P.M.D. analyzed data; and D.R.L., J.T.S., and P.M.D. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PV, papillomavirus; BPV, bovine PV; HPV, human PV; PC, proprotein convertase; dec.-RVKR-cmk, decanoyl-RVKR-chloromethylketone; CHO, Chinese hamster ovary; BrdUrd, 5-bromodeoxyuridine; HA, hemagglutinin-derived; CHA, C-terminal HA epitope tag; NHA, N-terminal HA epitope tag.
References
- 1.Howley, P. M. & Lowy, D. R. (2001) in Fields Virology, eds. Knipe, P. M. & Howley, P. M. (Lippincott, Philadelphia), Vol. 2, pp. 2197-2229. [Google Scholar]
- 2.Day, P. M., Lowy, D. R. & Schiller, J. T. (2003) Virology 307, 1-11. [DOI] [PubMed] [Google Scholar]
- 3.Selinka, H. C., Giroglou, T. & Sapp, M. (2002) Virology 299, 279-287. [DOI] [PubMed] [Google Scholar]
- 4.Bousarghin, L., Touze, A., Sizaret, P. Y. & Coursaget, P. (2003) J. Virol. 77, 3846-3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Day, P. M., Baker, C. C., Lowy, D. R. & Schiller, J. T. (2004) Proc. Natl. Acad. Sci. USA 101, 14252-14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaemper, N., Day, P. M., Nowak, T., Selinka, H. C., Florin, L., Bolscher, J., Hilbig, L., Schiller, J. T. & Sapp, M. (2006) J. Virol. 80, 759-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bossis, I., Roden, R. B., Gambhira, R., Yang, R., Tagaya, M., Howley, P. M. & Meneses, P. I. (2005) J. Virol. 79, 6723-6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck, C. B., Pastrana, D. V., Lowy, D. R. & Schiller, J. T. (2004) J. Virol. 78, 751-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seidah, N. G. & Chretien, M. (1997) Curr. Opin. Biotechnol. 8, 602-607. [DOI] [PubMed] [Google Scholar]
- 10.Thomas, G. (2002) Nat. Rev. Mol. Cell Biol. 3, 753-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon, V. M. & Leppla, S. H. (1994) Infect. Immun. 62, 333-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon, V. M., Klimpel, K. R., Arora, N., Henderson, M. A. & Leppla, S. H. (1995) Infect. Immun. 63, 82-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinnix, I., Council, J. E., Roseberry, B., Onstead, L., Mallender, W., Sucic, J. & Sambamurti, K. (2001) FASEB J. 15, 1810-1812. [DOI] [PubMed] [Google Scholar]
- 14.Komada, M., Hatsuzawa, K., Shibamoto, S., Ito, F., Nakayama, K. & Kitamura, N. (1993) FEBS Lett. 328, 25-29. [DOI] [PubMed] [Google Scholar]
- 15.Seidah, N. G., Chretien, M. & Day, R. (1994) Biochimie 76, 197-209. [DOI] [PubMed] [Google Scholar]
- 16.Duckert, P., Brunak, S. & Blom, N. (2004) Protein Eng. Des. Sel. 17, 107-112. [DOI] [PubMed] [Google Scholar]
- 17.Krysan, D. J., Rockwell, N. C. & Fuller, R. S. (1999) J. Biol. Chem. 274, 23229-23234. [DOI] [PubMed] [Google Scholar]
- 18.de Villiers, E. M., Fauquet, C., Broker, T. R., Bernard, H. U. & zur Hausen, H. (2004) Virology 324, 17-27. [DOI] [PubMed] [Google Scholar]
- 19.Day, P. M., Roden, R. B., Lowy, D. R. & Schiller, J. T. (1998) J. Virol. 72, 142-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buck, C. B., Thompson, C. D., Pang, Y.-Y. S., Lowy, D. R. & Schiller, J. T. (2005) J. Virol. 79, 2839-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, W. J., Gissmann, L., Sun, X. Y., Kanjanahaluethai, A., Muller, M., Doorbar, J. & Zhou, J. (1997) Virology 227, 474-483. [DOI] [PubMed] [Google Scholar]
- 22.Nagai, Y. (1993) Trends Microbiol. 1, 81-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakayama, K. (1997) Biochem. J. 327, 625-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebert, D. H., Deussing, J., Peters, C. & Dermody, T. S. (2002) J. Biol. Chem. 277, 24609-24617. [DOI] [PubMed] [Google Scholar]
- 25.Chandran, K., Sullivan, N. J., Felbor, U., Whelan, S. P. & Cunningham, J. M. (2005) Science 308, 1643-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pillay, C. S., Elliott, E. & Dennison, C. (2002) Biochem. J. 363, 417-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayer, G., Boileau, G. & Bendayan, M. (2004) J. Histochem. Cytochem. 52, 567-579. [DOI] [PubMed] [Google Scholar]
- 28.Molloy, S. S., Anderson, E. D., Jean, F. & Thomas, G. (1999) Trends Cell Biol. 9, 28-35. [DOI] [PubMed] [Google Scholar]
- 29.Inocencio, N. M., Moehring, J. M. & Moehring, T. J. (1994) J. Biol. Chem. 269, 31831-31835. [PubMed] [Google Scholar]
- 30.Klimpel, K. R., Molloy, S. S., Thomas, G. & Leppla, S. H. (1992) Proc. Natl. Acad. Sci. USA 89, 10277-10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molloy, S. S., Bresnahan, P. A., Leppla, S. H., Klimpel, K. R. & Thomas, G. (1992) J. Biol. Chem. 267, 16396-16402. [PubMed] [Google Scholar]
- 32.Selinka, H. C., Giroglou, T., Nowak, T., Christensen, N. D. & Sapp, M. (2003) J. Virol. 77, 12961-12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pastrana, D. V., Gambhira, R., Buck, C. B., Pang, Y. Y., Thompson, C. D., Culp, T. D., Christensen, N. D., Lowy, D. R., Schiller, J. T. & Roden, R. B. (2005) Virology 337, 365-372. [DOI] [PubMed] [Google Scholar]
- 34.Roden, R. B., Day, P. M., Bronzo, B. K., Yutzy, W. H., IV, Yang, Y., Lowy, D. R. & Schiller, J. T. (2001) J. Virol. 75, 10493-10497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gensberg, K., Jan, S. & Matthews, G. M. (1998) Semin. Cell Dev. Biol. 9, 11-17. [DOI] [PubMed] [Google Scholar]
- 36.Jean, F., Stella, K., Thomas, L., Liu, G., Xiang, Y., Reason, A. J. & Thomas, G. (1998) Proc. Natl. Acad. Sci. USA 95, 7293-7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearton, D. J., Nirunsuksiri, W., Rehemtulla, A., Lewis, S. P., Presland, R. B. & Dale, B. A. (2001) Exp. Dermatol. 10, 193-203. [DOI] [PubMed] [Google Scholar]
- 38.Zhong, M., Munzer, J. S., Basak, A., Benjannet, S., Mowla, S. J., Decroly, E., Chretien, M. & Seidah, N. G. (1999) J. Biol. Chem. 274, 33913-33920. [DOI] [PubMed] [Google Scholar]
- 39.Kibler, K. V., Miyazato, A., Yedavalli, V. S., Dayton, A. I., Jacobs, B. L., Dapolito, G., Kim, S. J. & Jeang, K. T. (2004) J. Biol. Chem. 279, 49055-49063. [DOI] [PubMed] [Google Scholar]
- 40.Peinado, J. R., Kacprzak, M. M., Leppla, S. H. & Lindberg, I. (2004) Biochem. Biophys. Res. Commun. 321, 601-605. [DOI] [PubMed] [Google Scholar]
- 41.Sarac, M. S., Peinado, J. R., Leppla, S. H. & Lindberg, I. (2004) Infect. Immun. 72, 602-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}