

Abstract
Leptin, a hormone secreted from adipose tissue, was originally discovered to regulate body weight. The localization of the leptin receptor in limbic structures suggests a potential role for leptin in emotional processes. Here, we show that rats exposed to chronic unpredictable stress and chronic social defeat exhibit low leptin levels in plasma. Systemic leptin treatment reversed the hedonic-like deficit induced by chronic unpredictable stress and improved behavioral despair dose-dependently in the forced swim test (FST), a model widely used for screening potential antidepressant efficacy. The behavioral effects of leptin in the FST were accompanied by increased neuronal activation in limbic structures, particularly in the hippocampus. Intrahippocampal infusion of leptin produced a similar antidepressant-like effect in the FST as its systemic administration. By contrast, infusion of leptin into the hypothalamus decreased body weight but had no effect on FST behavior. These findings suggest that: (i) impaired leptin production and secretion may contribute to chronic stress-induced depression-like phenotypes, (ii) the hippocampus is a brain site mediating leptin's antidepressant-like activity, and (iii) elevating leptin signaling in brain may represent a novel approach for the treatment of depressive disorders.
Keywords: depression, hippocampus, stress, forced swim test, social defeat
Depression is a serious mental disorder that affects ≈17%-21% of the population in the United States (1). Current treatment is dominated by antidepressants that exert their therapeutic effects through an interaction with serotonin and/or norepinepherine systems (2). However, not all depressed patients respond to existing antidepressants and only ≈35-40% of drug-responsive patients achieve full remission of symptoms (3). Months of treatment are usually required for full therapeutic response (1). Even though newer types of antidepressants are tolerated better and are safer in overdose than the older tricyclic compounds, they still produce troublesome side effects (3). Thus, better antidepressants from the perspectives of efficacy and side effects are needed.
Leptin is an adipocyte hormone encoded by the obese (ob) gene. It circulates as a 16-kDa protein and is transported across the blood-brain barrier by a saturable system to exert its central effects (4). A large body of work has focused on the role of leptin in the control of energy homeostasis, in which it acts as a negative feedback adiposity signal by interacting with receptors in specific hypothalamic nuclei. Six isoforms of leptin receptors alternatively spliced from the primary leptin receptor transcript with different lengths of the intracellular domain have been identified (5). These receptor isoforms, particularly the long form having signaling ability, are widely distributed in brain areas related to emotional responses such as the hippocampus (6, 7). However, the role of leptin in the regulation of mood and emotion is largely unknown. To begin to study this possible function, we examined circulating leptin levels in rats exposed to chronic unpredictable stress (CUS) and chronic social defeat as these stress paradigms produce behavioral deficits thought to model aspects of depression (8-10). The antidepressant-like potential of leptin was then evaluated by using behavioral alterations caused by CUS and the forced swim test (FST). Furthermore, brain sites that are sensitive to leptin treatment and may mediate leptin's antidepressant-like activity were investigated.
Results
Plasma Leptin Levels After Chronic Stress. Previous studies have shown that CUS and chronic social stress induce a variety of behavioral deficits resembling some symptoms of human depression (9-11). Rats subjected to CUS had significantly lower leptin levels with concurrent elevation of corticosterone levels in plasma, at 24 h after the cessation of the final stressor, compared with nonstressed controls (Fig. 1A). Whereas acute restraint stress (30 min) produced no effects on plasma leptin in handled control rats, CUS rats showed a further decrease in plasma leptin levels in response to acute restraint (Fig. 1 A). In addition, there was a significant decrease in basal plasma leptin levels in chronically defeated rats as compared with handled controls (Fig. 1B). However, plasma corticosterone levels in the defeated rats were not significantly different from those in controls (Fig. 1B). Taken together, these results indicate that decreased leptin levels represent one abnormal endocrine feature shared by the CUS and chronic social defeat models.
Fig. 1.

Plasma leptin levels after CUS or chronic social defeat. (A) Effect of CUS for 14 days on plasma leptin levels and corticosterone at 24 h after the cessation of the final stressor or after 30 min of acute restraint stress (AS) on day 15. Control, n = 13; control with acute stress, n = 9; CUS, n = 12; CUS with acute stress, n = 11. ANOVAs indicated significant effects for CUS [F(1,40) = 13.37, P < 0.001 for leptin; F(1,40) = 5.55, P < 0.05 for corticosterone] and acute stress [F(1,40) = 4.923, P < 0.05 for leptin; F(1,40), P < 0.0001 for corticosterone]. *, P < 0.05 compared with controls; ++, P < 0.01 compared with control animals subjected to acute stress. (B) Effect of chronic social defeat (SD) for 10 days on plasma leptin levels at 24 h after the last SD. Control, n = 6; SD, n = 6. Two-tailed t test, P < 0.05.
Reversal of the CUS-Induced Reduction in Sucrose Preference by Leptin. One of the depression-like phenotypes induced by CUS is decreased sucrose preference, considered a sign of the hedonic deficit seen in depressed patients (8, 10). Rats subjected to the CUS paradigm shown in Table 1 showed decreased sucrose preference (Fig. 2). This effect induced by CUS was significantly reversed by acute systemic administration of leptin without affecting total fluid intake (Fig. 2). Sucrose preference in nonstressed rats was not affected by leptin treatment (data not shown), suggesting that leptin does not have hedonic-like effects in the absence of a hedonic deficit.
Table 1. The procedure of CUS.
| Day | Type of stressor | Time |
|---|---|---|
| 1 | 30-min restraint | a.m. |
| 2 | 1-h shaking/crowding, 240 Hz with 6 rats in a box (1 ft × 1 ft × 1 ft) | p.m. |
| 3 | 24-h separation | |
| 4 | 15-min warm swim (31°C) | a.m. |
| 5 | 24-h high density of housing (6 rats per cage) | |
| 6 | 10-min cold swim (18°C) | p.m. |
| 7 | 1.5-h shaking/crowding, 240 Hz with 6 rats in a box (1 ft × 1 ft × 1 ft) | a.m. |
| 8 | 10-min inescapable shock (1.5 mA, 15 s on, 150 s off) | p.m. |
| 9 | 24-h separation | |
| 10 | 15-min warm swim (31°C) | a.m. |
| 11 | 15-min inescapable shock (1.5 mA, 15 s on, 150 s off) | p.m. |
| 12 | 10-min tail pinch (a clothespin placed at 1 cm from the base of tail) | a.m. |
| 13 | 10-min cold swim (18°C) | p.m. |
| 14 | 15-min inescapable shock (1.5 mA, 15 s on, 150 s off) | a.m. |
Fig. 2.

Effect of leptin treatment on CUS-induced reduction in sucrose preference. (A) Sucrose preference expressed as a ratio of the volume of sucrose intake to the volume of water intake. CUS rats display a substantially decreased sucrose preference. This effect of CUS was reversed by leptin treatment (1 mg/kg, i.p.). ANOVA revealed a significant effect on sucrose preference for time after injection [F(1,26) = 37.38, P < 0.0001] and drug treatment [F(2,13) = 7.498, P < 0.01]. **, P < 0.01 compared with controls. (B) Total amount of fluid intake. The total fluid intake was not affected by leptin treatment. Rats subjected to CUS significantly drink less than handled controls [F(1,28) = 6.525, P < 0.05]. *, P < 0.05. The empty underbar indicates measurements in the light cycle; the filled underbar shows measurements in the dark cycle.
Antidepressant-Like Effects of Leptin in the FST. The most widely used behavioral assay for detecting potential antidepressant-like activity is the FST, which was originally developed by Porsolt and colleagues (12, 13) and modified by Lucki (14). This test has been shown to have high predictive validity for antidepressant activity. In this test, animals display “despair” behavior as indicated by immobility and escape-oriented behaviors, in particular, swimming and climbing (14). To evaluate further whether leptin possesses antidepressant-like activity, behavioral effects of leptin in the FST were measured. Leptin administered s.c. (three times within 24 h) produced a dose-dependent reduction in the duration of immobility in the FST (Fig. 3A). Desipramine (DMI), the positive control, significantly decreased immobility time as expected. The magnitude of the antiimmobility effect of leptin at 1.0 mg/kg was comparable to that of DMI at 15 mg/kg. The decreased immobility induced by leptin was accompanied by an increase in the duration of swimming time (Fig. 3A). By contrast, the DMI-induced decrease in immobility was associated with increased climbing time, as reported (15).
Fig. 3.

Behavioral and neuroendocrine effects of leptin treatment. (A) FST. Subacute leptin treatments (23.5, 2.5, and 0.5 h before the test session, s.c.) caused a reduction in immobility time [F(3,31) = 7.043, P = 0.001], accompanied by an increase in swimming time [F(3,31) = 10.11, P < 0.0001] with no significant change in climbing time [F(3,31) = 2.512, P > 0.05]. Saline, n = 14; leptin 0.1 mg/kg, n = 5; leptin 0.5 mg/kg, n = 5; leptin 1.0 mg/kg, n = 12; DMI 15 mg/kg, n = 5. Post hoc analyses indicate that leptin significantly decreases immobility at 1.0 mg/kg (**, P < 0.01) and increased swimming at 0.5 and 1.0 mg/kg (**, P < 0.01), as compared with saline treatment. DMI treatment significantly decreases immobility (**, P < 0.01) and increased climbing (**, P < 0.01). (B) Plasma corticosterone levels 30 min after the FST. The FST increased corticosterone levels markedly compared with nonstressed controls [n = 5-6 per group, F(1,19) = 120.5, P < 0.0001]. This effect was not attenuated by either leptin or DMI treatments [n = 5-6 per group, F(2,12) = 0.466, P = 0.639]. ***, P < 0.001. (C) Locomotor activity. At 24 h after the first exposure to forced swim, leptin treatment (23.5, 2.5, and 0.5 h before the locomotion test, 1.0 mg/kg, s.c.) did not significantly alter locomotor activity (10-min bin) [n = 5 per group; F(1,28) = 0.035, P = 0.852].
To determine whether effects of leptin treatment in the FST result from antagonizing acute stress responses to the FST procedure, plasma corticosterone was measured 30 min after initiation of the FST. As expected, the FST procedure produced a dramatic increase in plasma corticosterone levels. This response, however, was not altered by either leptin or DMI treatment (Fig. 3B), implying that the antidepressant-like activity of these compounds in the FST was not caused by acute blockade of the FST-induced stress response. Moreover, to rule out nonspecific motoric effects of leptin that could influence activity in the FST, locomotor activity of rats was evaluated for 30 min after administration of leptin (s.c., three times within 24 h). Leptin showed no significant effects on locomotor activity as compared with that seen in control rats (Fig. 3C). Such data imply that the immobility-antagonizing and swimming-enhancing effects of leptin in the FST are not related to its increasing locomotor activity.
Identification of Leptin-Sensitive Brain Areas in the FST by Mapping mRNA Expression of c-fos. To assess whether the antidepressant-like effect of leptin observed in the FST was associated with changes in activation of specific brain areas, we examined mRNA expression of c-fos, a neuronal activation marker, in rats exposed to the FST. In situ hybridization showed that mRNA expression of c-fos was significantly increased by leptin in the hippocampal formation, including CA1, CA3, and dentate gyrus and in the basolateral nucleus of the amygdala, as compared with saline-treated control rats (Fig. 4A). The most robust change in c-fos expression in response to DMI treatment was observed in the central nucleus of the amygdala (Fig. 4). By contrast, c-fos expression in this subregion was not altered by leptin treatment, although leptin elicited a significant increase in c-fos induction in the basolateral nucleus of the amygdala. These results link leptin's actions in the FST to activity of specific limbic structures. In particular, the robust increase in c-fos expression in the hippocampus in response to leptin treatment suggests that this brain area might be a target site of systemic leptin for its antidepressant-like actions. This notion is supported by high levels of expression of the leptin receptor in the hippocampus (6, 7) and its facilitatory effects on synaptic transmission in this brain region (16).
Fig. 4.

c-fos induction in limbic areas in response to treatments with leptin or DMI after the FST. (A) Changes in mRNA levels of c-fos in the hippocampal formation, prefrontal cortex, cingulate cortex, and amygdala after the FST in rats treated with saline, leptin (1.0 mg/kg), or DMI (15 mg/kg) (23.5, 2.5, and 0.5 h before testing, s.c.). IOD, integrated optical density; CA1, CA2, CA3, fields of the hippocampus; DG, dentate gyrus; PFC, prefrontal cortex; CeA, central nucleus of amygdala; BLA, basolateral nucleus of amygdala; MeA, medial nucleus of amygdala. n = 5 per group. *, P < 0.05; **, P < 0.01 compared with saline treatment. (B) Autoradiograms showing c-fos induction in the hippocampal formation and amygdala.
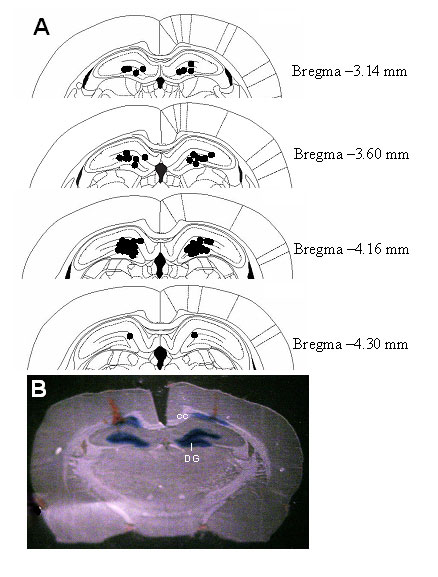
Intrahippocampal Infusion of Leptin Induces Antidepressant-Like Effects in the FST. To determine whether the hippocampus is a possible site mediating antidepressant-like effects of leptin, the behavioral effects of intrahippocampal infusion of leptin in the FST were studied. Behaviors of the animals in the FST were evaluated 3 days after stereotaxic microinjection of leptin or artificial cerebrospinal fluid into the hippocampus. During the first 5 min of the training session, intrahippocampal injection of leptin had no significant effect on immobility or active behaviors (Fig. 5A). A 5-min period was selected for study as it is the duration of the actual test session. By contrast, leptin dose-dependently decreased immobility during the test session with a significant increase in swimming behavior (Fig. 5B). This profile of behavioral effects is similar to that seen when leptin was administered systemically, suggesting that the leptin receptor in the hippocampus mediates the behavioral effects of leptin in the FST. The histology showed that intrahippocampal injections mainly targeted the dentate gyrus and the surrounding area (Fig. 7, which is published as supporting information on the PNAS web site).
Fig. 5.

Antidepressant-like effect in the FST induced by intrahippocampal injection of leptin. Artificial cerebrospinal fluid (vehicle) or rat leptin at different doses (0.25, 1.0, and 3.0 μg per side) was infused into the hippocampus. Three days later, the FST behavior recorded in the first 5 min of the training session and the 5-min test session was scored. Vehicle, n = 18; leptin 0.25 μg, n = 6; leptin 1.0 μg, n = 13; leptin 3.0 μg, n = 6. (A) Training session (first 5 min). Immobility, F(3,39) = 1.165, P = 0.3354; swimming, F(3,39) = 1.276, P = 0.2961; climbing, F(3,39) = 0.1427, P = 0.934. (B) Test session. Immobility, F(3,39) = 4.509, P < 0.01; swimming, F(3,39) = 3.472, P < 0.05; climbing, F(3,39) = 1.769, P = 0.1689. Post hoc tests, *, P < 0.05; **, P < 0.01 compared with vehicle controls.
Intrahypothalamic Infusion of Leptin Decreases Body Weight but Does Not Change FST Behaviors. In the FST, escape-oriented behaviors such as swimming and climbing are energy consuming, whereas immobility is energy conserving. It is possible, then, that the antidepressant-like efficacy of leptin in the FST reflects its ability to decrease energy conservation so as to facilitate active behaviors. To test this possibility, leptin, at an effective dose for the intrahippocampal injection, was stereotaxically injected into the hypothalamus, a brain site for energy balance. The behaviors in the FST during either the training session or the testing session were not affected by intrahypothalamic infusion of leptin (Fig. 6A). However, leptin in the hypothalamus positively regulated energy balance by decreasing body weight at 24 and 48 h postinjection (Fig. 6B). These findings suggest that the effect of leptin on the FST behavior dissociates from its effect on energy balance. The histology showed that intrahypothalamic injections mainly targeted at the lateral hypothalamus or the surrounding area (Fig. 8, which is published as supporting information on the PNAS web site).
Fig. 6.

Effect of intrahypothalamic injection of leptin on FST behaviors and body weight. Artificial cerebrospinal fluid (vehicle) or rat leptin (1 μg per side) was stereotaxically infused into the lateral hypothalamus. (A) FST behaviors recorded in the first 5 min of the training session (Left) and the 5-min test session (Right). Vehicle, n = 6; leptin 1 μg, n = 6. (B) Body weight. ANOVA with repeated measures revealed a significant effect of intrahypothalamic injection of leptin (1 μg per side) on body weight [F(1,30) = 13.98, P < 0.01]. **, P < 0.01 compared with vehicle controls.
Discussion
The present study demonstrates that plasma levels of endogenous leptin, a hormone produced by white adipose tissue, were decreased in rats subjected to CUS and chronic social defeat. The decreased sucrose preference in CUS rats was reversed by systemic administration of leptin. Further, leptin produced antidepressant-like activity in the FST. Its ability to reverse the CUS-induced hedonic-like deficit and attenuate behavioral despair in the FST supports the idea that leptin has antidepressant potential. Moreover, increased neuronal activation in the hippocampus and antidepressant-like effects produced by direct activation of leptin receptors in this region suggest that the hippocampus is a brain site mediating leptin's effect on the regulation of mood.
Chronic stress acts as a predisposing and participating factor in the onset of depression in humans (17, 18). Rats or mice exposed to CUS or chronic social defeat stress develop a number of behavioral and hormonal changes typical of depression and have been used as experimental models of depression (9-11). The present study shows decreased plasma leptin levels in both the CUS and chronic social defeat models. A baseline hypersecretion of corticosterone was observed in rats exposed to CUS but not to chronic social defeat, which is in agreement with previous reports using similar models (19, 20). The opposite changes in plasma leptin and corticosterone in CUS rats appear to be consistent with the reciprocal relationship between circulating leptin and glucocorticoids (21). Decreased circulating leptin in the CUS rats may contribute to increased corticosterone levels because leptin has been reported to blunt the response of adrenocorticotropic hormone and corticosterone to stress and inhibit glucocorticoid synthesis and secretion from adrenal cortical cells (22, 23). Further, this notion was supported by the inverse correlation of the sensitized decrease in the leptin response with the sensitized increase in the corticosterone response to acute stress in CUS rats.
A number of studies report that rats or mice exposed to the CUS or chronic mild stress (CMS) procedures exhibit impaired rewarding behavior such as decreased sucrose intake and preference, resembling a state of anhedonia, which is a core symptom of depression in humans (10). The decreased sucrose intake and preference in CUS/CMS rats can be reversed by administration of clinically effective antidepressants including tricyclics, mono-amine uptake inhibitors, atypical antidepressants, and mono-amine oxidase inhibitors (8, 10). The present study used a CUS procedure with modifications including elimination of those stressors affecting metabolism such as food deprivation and the use of some more severe stressors (e.g., electric shock, tail pinch, and cold swim). Rats subjected to this CUS paradigm exhibit a consistent decrease in sucrose preference with other behavioral changes such as increased immobility in the swim test and decreased exploration in an open field. However, inconsistent findings with different CUS/CMS procedures have been reported by various research groups, summarized in a recent review by Willner (10). Differences in CUS/CMS schedules, stress administration (type, intensity, and duration), strains of animals, housing conditions, and nutritive status may contribute to the discrepancies among these studies.
The antidepressant-like properties of leptin were evaluated by measuring the effects of leptin on sucrose preference in the CUS model and despair-like behaviors in the FST. Systemic administration of leptin reversed the reduced sucrose preference in chronically stressed rats. This effect is comparable to clinically active antidepressants tested in similar animal models (8, 24, 25). However, the rapid onset of leptin's actions is in contrast to the longer treatment (2-5 weeks) needed with traditional antidepressants (8, 24, 25). Further, the antidepressant-like activity of leptin was confirmed in the FST, a test widely used for screening potential antidepressants (12, 13). Leptin produced a dose-dependent behavioral effect in the FST, indicated by decreasing immobility and increasing swimming time. Leptin and DMI, a norepinepherine uptake inhibitor, had similar inhibitory effects on immobility but produced differential active behaviors in the FST with the former increasing swimming and the latter increasing climbing, implying that different mechanisms may underlie their behavioral effects. The behavioral profile of leptin in the FST suggests functional similarity with those antidepressants targeting the serotonin system (14).
The analyses and comparison of c-fos mRNA expression in the brain in rats exposed to the FST provide neural correlates of leptin-induced antidepressant-like behavioral effects in the FST. It is not surprising that neuronal activation was observed in several limbic structures implicated in the pathophysiology of major depressive disorders, particularly the hippocampus, a target site of existing antidepressants (26, 27). Direct activation of the leptin receptor in the hippocampus but not in the hypothalamus produced antidepressant-like behavioral effects in the FST, suggesting that the hippocampus is a neuroanatomical substrate for leptin's antidepressant-like actions. The behavioral profiles produced by single intrahippocampal infusion of leptin are similar to those reported previously for neurotrophic factors, such as brain-derived neurotrophic factor and neurotrophin-3 (28, 29). Whether leptin functionally interacts with these neurotrophic factors or leptin itself elicits a neurotrophic action in the hippocampus to mediate its antidepressant-like activity needs to be further investigated.
Clinical data on the possible association of leptin levels with depression are very limited and divergent (30-33). Two studies reported patients with major depression to have low leptin levels in plasma or cerebrospinal fluid (30, 33). However, another clinical study reported that plasma leptin levels did not differ between depressed patients and healthy controls (31). It may be that leptin deficiency occurs in a subgroup of depressed patients. A very recent study reported that improvement from depression with antidepressant treatment is associated with an elevation of plasma leptin levels (34). The antidepressant-like activity of leptin identified by the present study in animal models suggests that leptin might be effective in the treatment of depression, particularly for those patients with low leptin levels. Of course, this suggestion has to be tempered by the realization that there are concerns about the validity of animal models for symptoms of depression (35). On the other hand, considerable thought has been expended on criteria needed to be met for an animal model to be considered valid (36, 37). Importantly, the CUS and chronic social stress models have been suggested to meet these criteria (9-11). Given the prevalence of depressive disorders in the population and the proportion of depressed patients that are not responsive to existing antidepressants, further clinical studies are needed to evaluate the antidepressant properties of leptin.
In summary, our study demonstrates that leptin, a circulating hormone secreted from adipocytes, is dysregulated in both the CUS and chronic social defeat models. Leptin treatment overcomes two types of behavioral deficits, i.e., decreased sucrose preference in the CUS model and immobility in the FST. In addition, evidence is provided suggesting that the hippocampus is one brain site that may mediate leptin's actions in the regulation of mood. Thus, leptin may function as a novel antidepressant.
Methods
Animals. Adult male Sprague-Dawley rats (Charles River Laboratories), weighing 200-300 g, were housed in groups of three on a 12-h light schedule (7 a.m. on), with access to water and food ad libitum, and were allowed to acclimate to the housing conditions for 7 days before experiments began. Long-Evans rats (Harlan Labs, Indianapolis), weighing 400-450 g, that were used as resident attackers were housed individually in a large cage (80 × 55 × 40 cm) with one ovariectomized female rat on the same light-dark cycle. All procedures were carried out in accordance with the National Institutes of Health Guide.
CUS Procedure. The stress procedure was modified from procedures used by Katz (38) and Willner et al. (24). This paradigm was designed to maximize unpredictability. It consisted of a variety of stressors applied randomly and at varying times of day for 14 days (Table 1). Stressors that directly influence energy and water balance such as food deprivation and water deprivation were excluded. All stressors were applied to animals in a separate procedure room. After being stressed, animals were kept in the procedure room for 1-2 h until stress odor disappeared. All control and stressed animals were housed in groups of three throughout the experiment unless when they were subjected to isolation, high-density housing, or sucrose preference test. Control rats were handled daily in the housing room for 14 days. On day 15, animals were killed in the early light cycle, between 8 and 10 a.m., by decapitation with or without 30 min of acute restraint stress. Trunk blood was collected and plasma was separated by centrifugation for measuring leptin and corticosterone levels.
Chronic Social Defeat Procedure. Social defeat was obtained by using a resident-intruder paradigm. On the test day, the ovariectomized female was removed, and an adult male Sprague-Dawley rat (the intruder) was introduced into the home cage of an unfamiliar, aggressive Long-Evans rat (the resident). The intruder was rapidly attacked and defeated as indicated by fleeing, freezing, and submissive behavior. When full submissive posture occurred, usually within 1-3 min, as indicated by the intruder lying motionless on its back for 4 s, it was protected from further physical attacks for the rest of a 30-min period by placing it within a small wire mesh cage (30 × 15 × 15 cm) within the home cage of the resident. This approach allowed auditory, olfactory, and visual contact between the two rats, but minimized physical injury, while emphasizing the psychosocial component of the stress. After this procedure, the intruder rats were returned to their home cages and housed individually. The intruder rats were subjected to social defeat daily for 10 days. Each day the intruder rats were introduced to a different resident in a Latin square design. The defeated and control rats were housed in a separate room from the Long-Evans rats. Twenty four hours after the last social defeat, the defeated and control rats were killed by decapitation. Trunk blood was collected for hormone analyses.
Sucrose Intake. Animals subjected to CUS were randomly assigned to leptin or saline treatment. After animals habituated to two water bottles for 3 days in their home cages, water bottles were removed 2.5 h before treatment. A free choice between plain water and 1% sucrose solution was provided to each animal 30 min after i.p. injection of recombinant rat leptin (1.0 mg/kg) or saline. The positions of bottles were counterbalanced across the left or right side of the testing cages. Water intake and sucrose intake were measured for 1 h before the dark cycle. Then, the positions of the weighed bottles were reversed. Water intake and sucrose intake were measured during the next 12-h dark cycle. The preference for sucrose over water was used as a hedonic measure.
FST. This test consists of a 15-min pretest swim and a 5-min test swim 24 h later (12, 13). The stressful pretest swim facilitates the development of immobility during the test session and increases the sensitivity to antidepressants (39). Rats were handled and sham-injected before the experiment began. On the first day of the experiment, the rats were placed in a cylindrical tank (60 cm tall × 30 cm in diameter) for the 15-min pretest. The tank contained water at a 35-cm depth at a temperature of 25°C. Animals received three s.c. injections of saline, recombinant rat leptin (0.1-1.0 mg/kg), or DMI (15 mg/kg), a tricyclic antidepressant used as a positive control, 23.5, 2.5, and 0.5 h before the testing session. Water was changed between subjects. Both pretest and test sessions were recorded by two video cameras. One was positioned on the top of the swimming tank, the other positioned on the side of the swimming tank. The videotaped behaviors of rats in the FST were scored by using a time sampling technique to rate the predominant behavior over a 5-s interval as described by Lucki (14). Immobility was defined as floating or no active movements made other than those necessary to keep the nose above the water. Swimming consisted of active motions throughout the swim tank and crossing into another quadrant. Climbing was defined as upward-directed movements of the forepaws against the wall.
One subset of rats that were treated with saline or leptin (1 mg/kg) and those that received DMI (15 mg/kg) were used to determine corticosterone levels and c-fos expression patterns. After the 5-min swim test, animals were returned to the home cage and remained undisturbed until they were decapitated 30 min after initiation of the swim test. Trunk blood was collected and plasma was separated by centrifugation for measuring corticosterone. Brains were quickly removed and frozen in an isopentane dry-ice bath (-40°C). Brains were sectioned at 14 μm on a Leica cryostat and used for in situ hybridization.
Locomotor Activity. Rats underwent the first day of the 15-min swim training session and received three injections of leptin (1 mg/kg) or saline (s.c. 23.5, 2.5, and 0.5 h before the locomotion test). At 24 h after the first exposure to forced swim, the rats were placed in a clear cage on a locomotor activity meter for 30 min instead of being tested in the FST. Cumulative activity counts were recorded every 2 min for 30 min. The activity counts per 2 min were calculated and pooled over 10-min bins.
Stereotaxic Injection. Stereotaxic surgery was performed under anesthesia with a mixture of 34 mg/kg ketamine, 1 mg/kg acepromazine, and 7 mg/kg xylazine in saline, injected intramuscularly. With a 28-gauge stainless steel microinjection cannula (Small Parts, Miami), rats were injected bilaterally with artificial cerebrospinal fluid or different doses of recombinant rat leptin into the hippocampus (-3.8 mm anteroposterior, ±2.0 mm mediolateral, -3.1 mm from dura), 0.25-3.0 μg per side, or hypothalamus (-2.5 mm anteroposterior, ±1.8 mm mediolateral, -8.7 mm from dura), 1.0 μg per side. A total volume of 1 μl per side was infused over 10 min. After completion of the infusion, the injector was left in place for 5 additional min before withdrawal. After stereotaxic injection, rats were returned to their home cages and housed in groups of three (intrahippocampal injection) or two (intrahypothalamic injection) without shifting cage partners. Body weight was measured after stereotaxic injection. The training session (pretest, 15 min) was performed 3 days later followed by the testing session (5 min) on day 4. Recorded behaviors within the first 5 min of the training session and the 5-min testing session were evaluated as described above. At the end of experiment, the microinjection sites were examined by using histological staining (see Supporting Text, which is published as supporting information on the PNAS web site).
Plasma Hormone Analysis. See Supporting Text.
In Situ Hybridization. The in situ hybridization method was performed as described (40). See Supporting Text.
Statistical Analysis. Results are expressed as mean ± SEM. Statistical analyses were performed as follows: two-factor ANOVA (CUS × acute stress) on plasma leptin and corticosterone; two-tailed t test on plasma leptin and corticosterone (chronic social defeat) and c-fos mRNA expression; two-factor ANOVA (treatment × time) with repeated measures on sucrose intake; one-factor ANOVA on behaviors in the FST; two-factor ANOVA (drug × swim) on plasma corticosterone after the FST; one-factor ANOVA with repeated measures on locomotor activity and body weight; and two-tailed t test on c-fos mRNA expression. Student Newman Keuls or Tukey/Kramer (for unequal n) post hoc comparisons followed ANOVAs. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Honghong Yao for her excellent work on the in situ hybridization and assistance with microinjection and Dr. Georgianna Gould for assistance with social defeat. This work was supported by American Heart Association Grant AHA0530345N (to X.-Y.L.) and the Executive Research Committee Research Fund (to X.-Y.L.).
Author contributions: X.-Y.L., C.S.K., A.F., and W.Z. designed research; X.-Y.L., C.S.K., and W.Z. performed research; X.-Y.L., C.S.K., and W.Z. analyzed data; and X.-Y.L. and A.F. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CUS, chronic unpredictable stress; FST, forced swim test; DMI, desipramine.
References
- 1.Wong, M. L. & Licinio, J. (2001) Nat. Rev. Neurosci. 2, 343-351. [DOI] [PubMed] [Google Scholar]
- 2.Lenox, R. H. & Frazer, A. (2002) Mechanism of Action of Antidepressants and Mood Stabilizers (Williams & Wilkins, Philadelphia).
- 3.Cassano, P. & Fava, M. (2004) Ann. Clin. Psychiatry 16, 15-25. [DOI] [PubMed] [Google Scholar]
- 4.Banks, W. A., Kastin, A. J., Huang, W., Jaspan, J. B. & Maness, L. M. (1996) Peptides 17, 305-311. [DOI] [PubMed] [Google Scholar]
- 5.Ahima, R. S. & Osei, S. Y. (2004) Physiol. Behav. 81, 223-241. [DOI] [PubMed] [Google Scholar]
- 6.Elmquist, J. K., Bjorbaek, C., Ahima, R. S., Flier, J. S. & Saper, C. B. (1998) J. Comp. Neurol. 395, 535-547. [PubMed] [Google Scholar]
- 7.Mercer, J. G., Hoggard, N., Williams, L. M., Lawrence, C. B., Hannah, L. T. & Trayhurn, P. (1996) FEBS Lett. 387, 113-116. [DOI] [PubMed] [Google Scholar]
- 8.Katz, R. J. (1982) Pharmacol. Biochem. Behav. 16, 965-968. [DOI] [PubMed] [Google Scholar]
- 9.Rygula, R., Abumaria, N., Flugge, G., Fuchs, E., Ruther, E. & Havemann-Reinecke, U. (2005) Behav. Brain Res. 162, 127-134. [DOI] [PubMed] [Google Scholar]
- 10.Willner, P. (2005) Neuropsychobiology 52, 90-110. [DOI] [PubMed] [Google Scholar]
- 11.Blanchard, D. C., Spencer, R. L., Weiss, S. M., Blanchard, R. J., McEwen, B. & Sakai, R. R. (1995) Psychoneuroendocrinology 20, 117-134. [DOI] [PubMed] [Google Scholar]
- 12.Porsolt, R. D., Bertin, A. & Jalfre, M. (1977) Arch. Int. Pharmacodyn. Ther. 229, 327-336. [PubMed] [Google Scholar]
- 13.Porsolt, R. D., Anton, G., Blavet, N. & Jalfre, M. (1978) Eur. J. Pharmacol. 47, 379-391. [DOI] [PubMed] [Google Scholar]
- 14.Lucki, I. (1997) Behav. Pharmacol. 8, 523-532. [DOI] [PubMed] [Google Scholar]
- 15.Detke, M. J. & Lucki, I. (1996) Behav. Brain Res. 73, 43-46. [DOI] [PubMed] [Google Scholar]
- 16.Harvey, J. (2003) Ann. Med. 35, 197-206. [DOI] [PubMed] [Google Scholar]
- 17.Akil, H. (2005) Nat. Med. 11, 116-118. [DOI] [PubMed] [Google Scholar]
- 18.Kessler, R. C. (1997) Annu. Rev. Psychol. 48, 191-214. [DOI] [PubMed] [Google Scholar]
- 19.Herman, J. P., Adams, D. & Prewitt, C. (1995) Neuroendocrinology 61, 180-190. [DOI] [PubMed] [Google Scholar]
- 20.Bhatnagar, S. & Vining, C. (2003) Horm. Behav. 43, 158-165. [DOI] [PubMed] [Google Scholar]
- 21.Licinio, J., Mantzoros, C., Negrao, A. B., Cizza, G., Wong, M. L., Bongiorno, P. B., Chrousos, G. P., Karp, B., Allen, C., Flier, J. S. & Gold, P. W. (1997) Nat. Med. 3, 575-579. [DOI] [PubMed] [Google Scholar]
- 22.Bornstein, S. R., Uhlmann, K., Haidan, A., Ehrhart-Bornstein, M. & Scherbaum, W. A. (1997) Diabetes 46, 1235-1238. [DOI] [PubMed] [Google Scholar]
- 23.Heiman, M. L., Ahima, R. S., Craft, L. S., Schoner, B., Stephens, T. W. & Flier, J. S. (1997) Endocrinology 138, 3859-3863.9275075 [Google Scholar]
- 24.Willner, P., Towell, A., Sampson, D., Sophokleous, S. & Muscat, R. (1987) Psychopharmacology 93, 358-364. [DOI] [PubMed] [Google Scholar]
- 25.Muscat, R., Papp, M. & Willner, P. (1992) Psychopharmacology 109, 433-438. [DOI] [PubMed] [Google Scholar]
- 26.Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., Weisstaub, N., Lee, J., Duman, R., Arancio, O., et al. (2003) Science 301, 805-809. [DOI] [PubMed] [Google Scholar]
- 27.Duman, R. S. (2004) Biol. Psychiatry 56, 140-145. [DOI] [PubMed] [Google Scholar]
- 28.Hoshaw, B. A., Malberg, J. E. & Lucki, I. (2005) Brain Res. 1037, 204-208. [DOI] [PubMed] [Google Scholar]
- 29.Shirayama, Y., Chen, A. C., Nakagawa, S., Russell, D. S. & Duman, R. S. (2002) J. Neurosci. 22, 3251-3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westling, S., Ahren, B., Traskman-Bendz, L. & Westrin, A. (2004) J. Affect. Disord. 81, 41-48. [DOI] [PubMed] [Google Scholar]
- 31.Deuschle, M., Blum, W. F., Englaro, P., Schweiger, U., Weber, B., Pflaum, C. D. & Heuser, I. (1996) Horm. Metab. Res. 28, 714-717. [DOI] [PubMed] [Google Scholar]
- 32.Antonijevic, I. A., Murck, H., Frieboes, R. M., Horn, R., Brabant, G. & Steiger, A. (1998) J. Psychiatry Res. 32, 403-410. [DOI] [PubMed] [Google Scholar]
- 33.Kraus, T., Haack, M., Schuld, A., Hinze-Selch, D., Koethe, D. & Pollmacher, T. (2002) Pharmacopsychiatry 35, 220-225. [DOI] [PubMed] [Google Scholar]
- 34.Esel, E., Ozsoy, S., Tutus, A., Sofuoglu, S., Kartalci, S., Bayram, F., Kokbudak, Z. & Kula, M. (2005) Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 565-570. [DOI] [PubMed] [Google Scholar]
- 35.Matthews, K., Christmas, D., Swan, J. & Sorrell, E. (2005) Neurosci. Biobehav. Rev. 29, 503-513. [DOI] [PubMed] [Google Scholar]
- 36.Geyer, M. A. (1995) Animal Models of Psychiatric Disorders (Raven, New York).
- 37.Willner, P. (1997) Psychopharmacology 134, 319-329. [DOI] [PubMed] [Google Scholar]
- 38.Katz, R. J. (1981) Neurosci. Biobehav. Rev. 5, 231-246. [DOI] [PubMed] [Google Scholar]
- 39.Borsini, F., Lecci, A., Sessarego, A., Frassine, R. & Meli, A. (1989) Psychopharmacology 97, 183-188. [DOI] [PubMed] [Google Scholar]
- 40.Lu, X. Y., Barsh, G. S., Akil, H. & Watson, S. J. (2003) J. Neurosci. 23, 7863-7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}