

Abstract
HDM2 is a negative regulator of p53 that inhibits its transcriptional activity and subjects it to degradation by an E3 ligase activity. The primary binding site for HDM2 on p53 is located in its N-terminal domain. A second site on the p53 core domain (p53C) binds to an unidentified site in HDM2. We found that this site is in its acidic domain and part of the zinc finger domain by examining the interaction of full-length and domain constructs of p53 with the N-terminal region of HDM2 and peptide arrays derived from the full-length protein. NMR spectroscopy showed that peptides derived from this region of HDM2 bound to residues in the specific DNA-binding site of p53C. The peptides were displaced from the site by gadd45 sequence-specific DNA. Phosphorylation of single amino acids in the central domain of HDM2 did not abolish the interaction between the HDM2-derived peptides and p53C. We speculate that this second binding site helps in stabilizing the interaction between HDM2 and p53 during p53 degradation.
Keywords: isothermal titration calorimetry, Mdm2, NMR
The tumor suppressor protein p53 is important in maintaining genome stability and in preventing cancer development (1, 2). In response to various stress signals, p53 mediates cell-cycle arrest and apoptosis (3). p53 is a homotetramer consisting of an N-terminal transactivation domain, proline-rich regulatory domain, DNA-binding core domain (p53C), tetramerization domain, and C-terminal negative regulatory domain. Its major regulator, HDM2, is induced by p53 and acts as a feedback inhibitor (4). HDM2 regulates the activity of p53 in at least three ways. First, the N-terminal domain of HDM2 binds directly to p53's transactivation domain and inhibits its transcriptional function (5). Second, HDM2 acts as a ubiquitin ligase, targeting p53 and promoting its degradation (6, 7). Third, upon binding, HDM2 exports p53 from the nucleus to the cytoplasm (8).
The interaction between peptides derived from the p53 N terminus and the HDM2-N-terminal domain has been extensively studied (9–12), and several compounds have been proposed to abolish this interaction (13–16). Recently, a new HDM2-binding site has been reported in p53C that plays a regulatory role in modulating p53 ubiquitination (17), although the exact binding site on HDM2 involved in this interaction has not yet been identified. There are conflicting speculations on its location at either the HDM2 N terminus (18) or in the acidic domain (19).
Here, we examined the interaction of full-length and domain constructs of p53 with the N-terminal region of HDM2 and peptide arrays derived from the full-length protein. We found that the HDM2 N-terminal domain interacts only with p53's N-terminal domain, whereas from the peptide-array screen, we identified a second binding site for p53C located in the central domain (residues 221–302) of HDM2. We measured the binding affinity of the peptides to p53C by analytical ultracentrifugation. We identified the binding site on p53C by NMR and fluorescence anisotropy to be its DNA-binding site. The binding site in HDM2 is in its acidic domain. Phosphorylation of single amino acids in peptides derived from the central region of HDM2 did not abolish interaction between the two.
Results
Interaction of the HDM2 N Terminus with p53. The interaction of different constructs of p53, including its N terminus (p53N, residues 1–93), N terminus plus core (p53NC, residues 1–293), and full-length p53 (residues 1–393), with the HDM2 N-terminal domain (residues 2–125) was examined by isothermal titration calorimetry (ITC). All constructs bound tightly with a similar value of Kd (Fig. 1): 130 ± 30 nM for p53N, 180 ± 30 nM for p53NC, and 340 ± 10 nM for full-length p53. A control between the HDM2 N terminus and the p53 core domain detected no binding between them, indicating that the N terminus of HDM2 only binds to the N terminus of p53 at ≈100 nM.
Fig. 1.

Typical isothermal titration calorimetric (ITC) measurements of HDM2 N terminus with different constructs of p53 protein. (Upper) The original raw data. (Lower) The fit after integration. A 25 μM HDM2 N-terminal protein was loaded into the reaction cell and titrated with p53N (A), p53 NC (B), and full-length p53 (C).
We also analyzed the interaction between HDM2 and p53 by fluorescence anisotropy, using a fluorescein-labeled peptide, FL-PMD2, derived from p53 N-terminal domain from residues 17–26, labeled at its N terminus with fluorescein. FL-PMD2 binds tightly to the HDM2 N-terminal domain. The binding of different p53 domains was detected by the displacement of the FL-PMD2 from its complex with the HDM2 N terminus. The dissociation constants of the p53 constructs were measured from the decrease in amounts of the FL-PMD2/HDM2-N terminus complex present (Fig. 2). All p53 constructs were able to displace FL-PMD2 and bind tightly to the HDM2 N terminus. The values of Kd were 100 ± 8 nM, 100 ± 11 nM, and 230 ± 43 nM for p53N, p53NC, and full-length p53, respectively. These values were in good agreement with the ITC data. This finding indicates that there is tight binding of HDM2 N terminus only at the N terminus of p53.
Fig. 2.

Binding competition experiments. Different p53 constructs (20 μM) p53N, p53NC, and p53 full-length were titrated into a complex of HDM2 N-terminal domain (20 nM) with FL-PMD2 (10 nM).
p53C Binds to Peptide Sequences Derived from Central Domain of HDM2. Having failed to detect the binding of the HDM2 N-terminal domain to p53C, we investigated the binding of p53C to immobilized peptides derived from the full-length HDM2 (Fig. 3). We used an array consisting of 49 peptides spanning the complete HDM2 sequence of HDM2 (array A); and a second array consisting of 34 peptides derived from regions of HDM2 that had potential phosphorylation sites (array B).
Fig. 3.

Binding of p53C to immobilized peptide arrays, screening the HDM2 sequence. (A) Average of seven immunoblot experiments of p53C bound to full-length HDM2 array. (B) Average of six immunoblot experiments of p53C bound to peptide sequence motifs designed according to HDM2 phosphorylation sites. The phosphorylated peptide were marked with “p” and its corresponding nonphosphorylated version was positioned next to it. (C) Duplicated array of B. Average of six immunoblot experiments of full-length p53 bound to peptide sequence motifs. The 9-mer peptide CDB3 and 18-mer peptides HP13 and HP39 were used as positive controls.
Array A was of 20-mer peptides, corresponding to full-length HDM2 sequence with a 10-residue overlap. Array B was designed according to different phosphorylation sites occurring within HDM2 (20). Each phosphorylated peptide and its corresponding nonphosphorylated version were located alternately. The peptide arrays, immobilized on cellulose, were produced by Jerini (Berlin). The peptides were acetylated at their N termini and attached to a cellulose–PEG membrane via their C termini by an amide bond. Peptides CDB3, HP13, and HP19, which are known to bind to p53C with different affinities (21, 22), were used as controls and placed at the first and the last three positions in the arrays.
In array A, there were eight dominant sequence motifs spanning residues 221–310, which lie in the central domain of HDM2, and in array B, six major sequence motifs were also located in the central domain of HDM2, from amino acids 233 to 307 (Table 1). The results also showed that phosphorylation at a single site did not abolish binding to p53C and immunoblotting of the peptides containing the amino acids in question exhibited a similar intensity whether they were phosphorylated or not. When incubated with duplicated of array B, full-length p53 and p53C produced the same pattern of signals (Fig. 3).
Table 1. Peptide sequences of binding site on mapping array.
| Number | Peptide sequence | Residues |
|---|---|---|
| Array A | ||
| A26 | NPDLDAGVSEHSGDWLDQDS | 221-240 |
| A27 | HSGDWLDQDSVSDQFSVEFE | 231-250 |
| A28 | VSDQFSVEFEVESLDSEDYS | 241-260 |
| A29 | VESLDSEDYSLSEEGQELSD | 251-270 |
| A30 | LSEEGQELSDEDDEVYQVTV | 261-280 |
| A31 | EDDEVYQVTVYQAGESDTDS | 271-290 |
| A32 | YQAGESDTDSFEEDPEISLA | 281-300 |
| A33 | FEEDPEISLADYWKCTSCNE | 291-310 |
| Array B | ||
| B13p/13 | GDWLDQDSVSDQFSVEFEVE | 233-252 |
| B14p/14 | DQDSVSDQFSVEFEVESLDS | 237-256 |
| B15p/15 | VESLDSEDYSLSEEGQELSD | 251-270 |
| B16p/16 | ESLDSEDYSLSEEGQELSDE | 252-271 |
| B17p/17 | YSLSEEGQELSDEDDEVYQV | 259-278 |
| B18p/18 | TDSFEEDPEISLADYWKCTS | 288-297 |
Phosphorylated residues are shown in bold.
Among these, two peptide sequences, B17 and B18, gave the most intense signal and were thus determined to be the most dominant binding sites; subsequent experiments were based on peptides derived from these sequences.
Binding Affinity of HDM2-Derived Peptides to p53C. To improve solubility and afford an easier synthesis, four fluorescein-labeled peptide derivatives were made by deleting three amino acids from either the N or C termini of B17 and B18. We measured the affinity of these derivatives for p53C by analytical ultracentrifugation (AUC). All four peptides had 1:1 binding stoichiometry. Under the same buffer conditions used in the screening array, peptides derived from B17, residues 259–275 and 262–278, had stronger binding than did residues 288–304 and 291–307, derived from B18 (Table 2). There was no significant difference between residues 288–304 and 291–307, but sequence 259–275 had a higher affinity than 262–278. Furthermore, the absence of residues 276–278 strengthened p53C binding, whereas the absence of residues 259–261 weakened it (Table 2).
Table 2. Binding affinity of HDM2-derived peptides to p53C as measured by AUC.
| Peptide | Sequence | Kd, μM |
|---|---|---|
| 259-275 | FL-YSLSEEGQELSDEDDEV | 130 ± 6 |
| 262-278 | FL-SEEGQELSDEDDEVYQV | 180 ± 10 |
| 288-304 | FL-TDSFEEDPEISLADYWK | 300 ± 20 |
| 291-307 | FL-FEEDPEISLADYWKCTS | 270 ± 15 |
Peptide B17 corresponds to amino acids 259-278, peptide B18 corresponds to amino acids 288-307. All experiments were done in triplicate and the values correspond to the average of these results. Binding experiments were conducted in 50 mM HEPES (pH 7.2), 150 mM NaCl, and 5 mM DTT at 15°C. All peptides were fluorescein-labeled.
Defining the Sites of the Interaction Between p53C and HDM2-Derived Peptides. We used heteronuclear single quantum coherence NMR to locate the binding site on p53C of the HDM2-derived peptides, by examining the changes in the chemical shifts of the backbone amides in 15N-labeled p53C on addition of HDM2-derived peptides. These experiments were done at a higher ionic strength of I = 207 nM. The residues in p53C that changed chemical shift upon binding the peptides were mainly in the DNA-binding interface and were almost the same for all four peptides (see Fig. 5). The corrected changes in 1H–15N chemical shift, upon peptide binding, were calculated as described (23), and shifts of 0.04 ppm and above were considered significant. Changes were observed for Lys-101, Thr-102, and Tyr-103 (from the N terminus); Leu-114, His-115, Gly-117, and Thr-118 (from loop L1); Tyr-126 (from strand S2); Val-143 (from strand S3); Glu-198 and Gly-199 (from strand S5); Arg-248 (from loop L3); Asn-263 and Leu-265 (from strand S10); Gly-279, Arg-280, Arg-282, Arg-283, and Glu-286 (from helix H2); and Lys-291, Lys-292, Gly-293, and Gly-298 (from the C terminus). These residues can be divided into surface-exposed residues, which provide a binding site for HDM2 derived peptides and buried residues, which change their secondary conformation upon binding of peptides.
Fig. 5.

Binding of p53C to HDM2-derived peptides. (A) The change in corrected chemical shift of the heteronuclear single quantum coherence NMR spectrum of 15N-labeled p53C upon binding FL-peptides. Corrected chemical shift changes of 0.04 ppm and above were considered significant. (B) The binding site of FL-peptides in p53C. Residues with significant corrected chemical shifts are colored in red. (C) The space-filled model of the FL-peptides' binding site in p53C. The color code is the same as in B. Note that the protein orientation is rotated compared with B.
DNA Competition Binding Experiments. To confirm that the residues on p53C involved were in the DNA-binding site, we used the gadd45 DNA recognition element to compete with peptides bound to p53C. Competition binding experiments indicated that all four peptides had overlapping binding sites and that they bound to the DNA-binding site of p53C. The four peptides had a dissociation constant of ≈1 μM at low ionic strength (I = 20 mM), fitting to a simple 1:1 binding equation, which was consistent with data obtained from AUC. The gadd45 promoter DNA displaced the peptides from the complex with p53C. The replacement of the slowly tumbling complex by more rapidly tumbling free peptide was monitored as a gradual decrease in anisotropy (Fig. 4).
Fig. 4.

Fluorescence anisotropy studies of binding of HDM2-derived peptides to p53C. (A) Binding of HDM2-derived peptides to p53C, assayed by fluorescence anisotropy. (B) Fluorescence anisotropy competition assay between unlabeled p53 consensus DNA and a complex of fluorescein-labeled HDM2-derived peptides with p53C.
Discussion
We detected binding of the N terminus of HDM2 to only the N-terminal domain of p53. Binding was also identified between the core domain of p53 (p53C) and the central region of HDM2, which consists of the acidic domain and part of zinc finger domain. The interaction between the HDM2 N-terminal domain and different p53 constructs conformed to a 1:1 binding model, and the Kd fell within the range of that for the binding of other peptide fragments of p53 studied to date (≈100 nM) (9–12). The N-terminal domain of HDM2 did not appear to bind to any other domains of p53 because the dissociation constant was unaffected by the presence of other domains in the larger constructs of p53.
We detected additional binding sites using immobilized peptide arrays derived from full-length HDM2, and localized to a region within residues 221–307. Bioinformatic analysis and protease digestion of full-length HDM2 suggested that the acidic domain is unstructured (G.W.Y., unpublished data). The part of the zinc finger domain of HDM2 from residues 290–307 contains both unfolded and folded regions. The NMR solution structure of this domain revealed that residues 290–302 are not involved in maintaining the structure of the zinc finger domain of HDM2 (24). As seen from Table 2, deleting residues 305–307 from peptide B18 does not affect the dissociation constant, implying that residues 305–307 contribute structural stability without participating in binding to p53C, narrowing down the binding region to an area within residues 221–302. Because this region is highly unstructured, peptide fragments are likely to be particularly good models for mapping the binding sites.
Experiments in solution, such as AUC, confirmed that there were specific interactions between HDM2-derived peptides and p53C. DNA competition assays and NMR spectroscopy revealed that the binding site on p53C was located at its DNA-binding site, mainly involving residues from loop L1, helix H2, and residues responsible for direct DNA contact (Arg-248, Arg-280, and Arg-283), with secondary chemical shifts for Tyr-126, Glu-198, Gly-199, Arg-282, Val 143, and Gly-279 (Fig. 5). The changes in chemical shifts on binding HDM2-derived peptides were similar to those produced by other protein-derived peptides that bind p53C, e.g., Rad51 (25), HIF-1α (22), and CDB3 (21). As suggested by Friedler et al. (25), p53C provides a multipurpose promiscuous binding site that allows many protein–protein interactions.
The central region of HDM2 binds much more weakly to p53C than does the N terminus to that of p53. This may be of relevance as the central part of HDM2 is involved in the degradation and ubiquitination of p53 (19, 26, 27). The central region of HDM2 interacts also with several other proteins that are responsible for p53 regulation, such as L5 (28), L23 (29), L11 (30), p14ARF (31), TBP (32), and Retinoblastoma (Rb) protein (33). All of these inhibit HDM2 polyubiquitination and degradation of p53, and hence stabilize p53 in cells. The existence of weak binding between the central region of HDM2 and p53C increases the possibility for those proteins to disrupt their interaction.
Protein–protein interactions can also be regulated by phosphorylation. There are several phosphorylation sites on HDM2 and the region within its central domain is particularly highly phosphorylated (reviewed in ref. 20); this may affect the interaction between HDM2 and p53. Our peptide array revealed that, with a single phosphorylated amino acid residue on HDM2-derived peptides, p53C was still able to bind to (Fig. 3). However, phosphorylation may alter the binding affinity of HDM2 to other proteins that enhance the ubiquitination and degradation of p53, e.g., phosphorylated HDM2 interacts more efficiently with p300 (34) and thereby stimulates p53 ubiquitination and degradation (35).
Materials and Methods
Purification of Human HDM2 N-Terminal (Residues 2–125) Domain. The p53-binding domain of HDM2 (residues 2–125) was purified as described (12). 15N-labeled ammonium chloride was used to produce isotopically labeled protein for NMR studies.
Purification of p53N (Amino Acids 1–93). p53N (amino acids 1–93) cDNA was cloned into a modified pRSETa vector encoding an N-terminal 6× His tag, lipoyl domain, and thrombin cleavage site to make the plasmid pHLT1–93. The expression and purification method were performed as described (36).
Purification of p53NC (Residues 1–296). The cDNA clone of p53 (residues 1–296) was kindly provided by Zippi Shakked (Weizmann Institute of Science, Rehovot, Israel). The protein was expressed in Escherichia coli C41(DE3) cells at 22°C and purified on a nickel nitrilotriacetic Superflow column as described above. The eluent was subjected to thrombin digestion and purified by gel filtration chromatography.
DNA Annealing for Competition Binding Assays. The oligonucleotides used for competition binding assays were: 5′-GAG CCC AGC ATG CTT AGA CAT GTT CTG CTC-3′ and 5′-GAG CAG AAC ATG TCT AAG CAT GCT GGG CTC-3′. The oligonucleotides were annealed in 10 mM Tris (pH 7.4), 100 mM NaCl, and 1 mM EDTA. DNA was placed in a heating block at 95°C for 5 min and then allowed to cool to 22°C.
Synthesis and Purification of Peptides. Peptide were synthesized and purified as described (21).
Isothermal Titration Calorimetry. All ITC measurements were performed as described (12, 37, 38). Full-length p53 used here is a superstable mutant of wild-type full-length p53 in its core domain (39), and was a kind gift from Caroline Blair (Centre for Protein Engineering, Cambridge, U.K.).
Fluorescence Anisotropy. All anisotropy measurements were performed with fluorescein-labeled PMD2 peptide (FL-PMD2, a derived peptide from p53 N terminus from amino acids 17–26, sequence FL-ETFSDLWKLL-NH2) at 15°C, using a PerkinElmer LS55 luminescence spectrometer equipped with a Hamilton Microlab titrator controlled by software. The excitation and emission were at 480 and 530 nm, respectively, with slit widths of 15 and 20 nm. The photomultiplier voltage used was 950 V with an integration time of 5 s for each measurement.
To determine the dissociation constant for FL-PMD2 complexed with HDM2 N-terminal domain, FL-PMD2 (900 μl, 10 nM) was placed in a cuvette and the HDM2 N-terminal domain (200 μl, 600 nM) was placed in the dispenser. Serial additions of 5 μl of protein were titrated into the peptide solution at interval of 1 min, the solution was stirred for 30 s, and the anisotropy was measured. Dissociation constants for the FL-PMD2-HDM2 N terminus complex were calculated by fitting the data (corrected for dilution) to a simple 1:1 equilibrium model.
Anisotropy was measured in competition experiments to study how the different p53 constructs competed with the FL-PMD2. A stock solution of the appropriate p53 construct (20 μM, 200 μl) was titrated into a cuvette containing 900 μl of 20 nM HDM2 N-terminal domain and 10 nM FL-PMD2. The dissociation constants were derived from the equilibrium equations.
The DNA competition assays for HDM2-derived peptides used the same instrument and methods with modifications. Briefly, 20–25 μM p53C was titrated against 0.5 μM HDM2-derived peptides in a cuvette. The competition was then monitored by titrating 5 μM gadd45 DNA into the cuvette containing of a mixture of 0.5 μM peptide and 1 μM p53C.
Screening of p53C Binding to Peptide Arrays. Human p53C (residues 94–312) was kindly provided by Caroline Blair. The analysis of binding to cellulose-bound peptides (40) was modified slightly. The peptide arrays were prewashed in binding buffer (50 mM Hepes, pH 7.2/150 mM NaCl/5 mM DTT/5% wt/vol sucrose/0.05% vol/vol Tween 20). The p53 constructs were incubated with the arrays in binding buffer for at least 30 min at 15°C with gentle shaking. The cellulose sheets were washed rapidly in ice-cold binding buffer four times and transferred to poly(vinylidene difluoride) (PVDF) membrane (Bio-Rad) with the peptide side against the membrane. Bound p53C was transferred to the membrane (anode side) by semidry blotting at a current of 0.8 mA/cm2 for 10–20 min. The solution used for transblot and the immunodetection methods were the same as described in ref. 22.
AUC. Equilibrium sedimentation experiments were performed in a Beckman Optima XL-I ultracentrifuge with the Ti-60 rotor and six-sector cells at speeds of 30,000 and 40,000 rpm. All experiments were performed at 15°C. The sample volume was 100 μl. Samples were measured at equilibrium, as judged by comparing several scans at each speed. Buffer conditions were 50 mM Hepes (pH 7.2), 5 mM DTT, and 150 mM NaCl. To measure the dissociation constant (Kd) for peptide–protein binding, fluorescein-labeled peptides at a concentration of 5 μM were loaded in a cell with 100 μM p53C, and the absorbance at 495 nm and interference data were recorded on reaching equilibrium (typically after 6–12 h). The calculation method was as described (41).
NMR Spectroscopy of Peptides with p53C. All 1H, 15N heteronuclear single quantum coherence spectra were acquired at 25°C on a Bruker DRX 500-MHz spectrometer. Samples for NMR experiments contained 15N-labeled wild-type p53 core domain at a concentration of 200 μM and the corresponding peptides at concentrations ranging from 800 μM to 1 mM. The buffer was 150 mM KCl, 5 mM DTT, and 2% D2O in 25 mM sodium phosphate, pH 7.2 (I = 207 mM). The combined chemical shift differences were calculated according to the following equation (23):
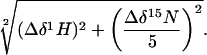 |
Acknowledgments
We thank Dr. Assaf Friedler and Karoly Von Gloss for peptide synthesis, Dr. Christopher Johnson for ITC technical advice, and Dr. Trevor Rutherford for NMR technical help. G.W.Y. is supported by a Croucher Foundation Scholarship (Hong Kong).
Author contributions: G.W.Y. and A.R.F. designed research; G.W.Y. and S.F. performed research; S.R. and D.V. contributed new reagents/analytic tools; G.W.Y., S.F., M.R.F.-F., and A.R.F. analyzed data; and G.W.Y. and M.R.F.-F. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: ITC, isothermal titration calorimetry; AUC, analytical ultracentrifugation.
References
- 1.Lane, D. P. (1992) Nature 358, 15–16. [DOI] [PubMed] [Google Scholar]
- 2.Vousden, K. H. & Lu, X. (2002) Nat. Rev. Cancer 2, 594–604. [DOI] [PubMed] [Google Scholar]
- 3.Levine, A. J. (1997) Cell 88, 323–331. [DOI] [PubMed] [Google Scholar]
- 4.Woods, D. B. & Vousden, K. H. (2001) Exp. Cell Res. 264, 56–66. [DOI] [PubMed] [Google Scholar]
- 5.Momand, J., Zambetti, G. P., Olson, D. C., George, D. & Levine, A. J. (1992) Cell 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- 6.Honda, R. & Yasuda, H. (2000) Oncogene 19, 1473–1476. [DOI] [PubMed] [Google Scholar]
- 7.Fang, S., Jensen, J. P., Ludwig, R. L., Vousden, K. H. & Weissman, A. M. (2000) J. Biol. Chem. 275, 8945–8951. [DOI] [PubMed] [Google Scholar]
- 8.Roth, J., Dobbelstein, M., Freedman, D. A., Shenk, T. & Levine, A. J. (1998) EMBO J. 17, 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kussie, P. H., Gorina, S., Marechal, V., Elenbaas, B., Moreau, J., Levine, A. J. & Pavletich, N. P. (1996) Science 274, 948–953. [DOI] [PubMed] [Google Scholar]
- 10.Lai, Z., Auger, K. R., Manubay, C. M. & Copeland, R. A. (2000) Arch. Biochem. Biophys. 381, 278–284. [DOI] [PubMed] [Google Scholar]
- 11.Sakaguchi, K., Saito, S., Higashimoto, Y., Roy, S., Anderson, C. W. & Appella, E. (2000) J. Biol. Chem. 275, 9278–9283. [DOI] [PubMed] [Google Scholar]
- 12.Schon, O., Friedler, A., Bycroft, M., Freund, S. M. & Fersht, A. R. (2002) J. Mol. Biol. 323, 491–501. [DOI] [PubMed] [Google Scholar]
- 13.Stoll, R., Renner, C., Hansen, S., Palme, S., Klein, C., Belling, A., Zeslawski, W., Kamionka, M., Rehm, T., Muhlhahn, P., et al. (2001) Biochemistry 40, 336–344. [DOI] [PubMed] [Google Scholar]
- 14.Zhao, J., Wang, M., Chen, J., Luo, A., Wang, X., Wu, M., Yin, D. & Liu, Z. (2002) Cancer Lett. 183, 69–77. [DOI] [PubMed] [Google Scholar]
- 15.Duncan, S. J., Gruschow, S., Williams, D. H., McNicholas, C., Purewal, R., Hajek, M., Gerlitz, M., Martin, S., Wrigley, S. K. & Moore, M. (2001) J. Am. Chem. Soc. 123, 554–560. [DOI] [PubMed] [Google Scholar]
- 16.Vassilev, L. T., Vu, B. T., Graves, B., Carvajal, D., Podlaski, F., Filipovic, Z., Kong, N., Kammlott, U., Lukacs, C., Klein, C., et al. (2004) Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu, H., Burch, L. R., Smith, A. J., Dornan, D., Wallace, M., Ball, K. L. & Hupp, T. R. (2002) J. Biol. Chem. 277, 28446–28458. [DOI] [PubMed] [Google Scholar]
- 18.Dawson, R., Muller, L., Dehner, A., Klein, C., Kessler, H. & Buchner, J. (2003) J. Mol. Biol. 332, 1131–1141. [DOI] [PubMed] [Google Scholar]
- 19.Kawai, H., Wiederschain, D. & Yuan, Z. M. (2003) Mol. Cell. Biol. 23, 4939–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meek, D. W. & Knippschild, U. (2003) Mol. Cancer Res. 1, 1017–1026. [PubMed] [Google Scholar]
- 21.Friedler, A., Hansson, L. O., Veprintsev, D. B., Freund, S. M., Rippin, T. M., Nikolova, P. V., Proctor, M. R., Rudiger, S. & Fersht, A. R. (2002) Proc. Natl. Acad. Sci. USA 99, 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansson, L. O., Friedler, A., Freund, S., Rudiger, S. & Fersht, A. R. (2002) Proc. Natl. Acad. Sci. USA 99, 10305–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hajduk, P. J., Dinges, J., Miknis, G. F., Merlock, M., Middleton, T., Kempf, D. J., Egan, D. A., Walter, K. A., Robins, T. S., Shuker, S. B., et al. (1997) J. Med. Chem. 40, 3144–3150. [DOI] [PubMed] [Google Scholar]
- 24.Yu, G. W., Allen, M. D., Andreeva, A., Fersht, A. R. & Bycroft, M. (2006) Protein Sci., in press. [DOI] [PMC free article] [PubMed]
- 25.Friedler, A., Veprintsev, D. B., Rutherford, T., von Glos, K. I. & Fersht, A. R. (2005) J. Biol. Chem. 280, 8051–8059. [DOI] [PubMed] [Google Scholar]
- 26.Argentini, M., Barboule, N. & Wasylyk, B. (2001) Oncogene 20, 1267–1275. [DOI] [PubMed] [Google Scholar]
- 27.Meulmeester, E., Frenk, R., Stad, R., de Graaf, P., Marine, J. C., Vousden, K. H. & Jochemsen, A. G. (2003) Mol. Cell. Biol. 23, 4929–4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai, M. S. & Lu, H. (2004) J. Biol. Chem. 279, 44475–44482. [DOI] [PubMed] [Google Scholar]
- 29.Jin, A., Itahana, K., O'Keefe, K. & Zhang, Y. (2004) Mol. Cell. Biol. 24, 7669–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang, Y., Wolf, G. W., Bhat, K., Jin, A., Allio, T., Burkhart, W. A. & Xiong, Y. (2003) Mol. Cell. Biol. 23, 8902–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bothner, B., Lewis, W. S., DiGiammarino, E. L., Weber, J. D., Bothner, S. J. & Kriwacki, R. W. (2001) J. Mol. Biol. 314, 263–277. [DOI] [PubMed] [Google Scholar]
- 32.Leveillard, T. & Wasylyk, B. (1997) J. Biol. Chem. 272, 30651–30661. [DOI] [PubMed] [Google Scholar]
- 33.Sdek, P., Ying, H., Zheng, H., Margulis, A., Tang, X., Tian, K. & Xiao, Z. X. (2004) J. Biol. Chem. 279, 53317–53322. [DOI] [PubMed] [Google Scholar]
- 34.Zhou, B. P., Liao, Y., Xia, W., Zou, Y., Spohn, B. & Hung, M. C. (2001) Nat. Cell Biol. 3, 973–982. [DOI] [PubMed] [Google Scholar]
- 35.Ogawara, Y., Kishishita, S., Obata, T., Isazawa, Y., Suzuki, T., Tanaka, K., Masuyama, N. & Gotoh, Y. (2002) J. Biol. Chem. 277, 21843–21850. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Puig, N., Veprintsev, D. B. & Fersht, A. R. (2005) Mol. Cell 17, 11–21. [DOI] [PubMed] [Google Scholar]
- 37.Wiseman, T., Williston, S., Brandts, J. F. & Lin, L. N. (1989) Anal. Biochem. 179, 131–137. [DOI] [PubMed] [Google Scholar]
- 38.Cooper, A. & Johnson, C. M. (1994) Methods Mol. Biol. 22, 137–150. [DOI] [PubMed] [Google Scholar]
- 39.Joerger, A. C., Allen, M. D. & Fersht, A. R. (2004) J. Biol. Chem. 279, 1291–1296. [DOI] [PubMed] [Google Scholar]
- 40.Rudiger, S., Germeroth, L., Schneider-Mergener, J. & Bukau, B. (1997) EMBO J. 16, 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandez-Fernandez, M. R., Veprintsev, D. B. & Fersht, A. R. (2005) Proc. Natl. Acad. Sci. USA 102, 4735–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]