

Abstract
The discovery of the block-like structure of linkage disequilibrium (LD) in human populations holds the promise of delineating the etiology of common diseases. However, understanding the magnitude, mechanism, and utility of between-population LD sharing is critical for future genome-wide association studies. In this study, substantial LD sharing between six non-African populations was observed, although much less between African-American and non-African, based on 20,000 SNPs of chromosome 21. We also demonstrated the respective roles of recombination and demographic events in shaping LD sharing. Furthermore, we showed that the haplotype-tagged SNPs chosen from one population are portable to the others in East Asia. Therefore, we concluded that the magnitude of LD sharing between human populations justifies the use of representative populations for selecting haplotype-tagged SNPs in genome-wide association studies of complex diseases.
Keywords: bottleneck, genetic distance, association study, common disease, genetic variant
Comprehensive testing of the association between genetic variations in the human genome and common diseases holds the promise of delineating the genetic architecture of these diseases (1–5). Substantial sharing of the boundaries and specific haplotypes of linkage disequilibrium (LD) blocks between populations was observed (6). However, variations of haplotype and LD across populations were also reported, raising concerns on its practical hindrance for genomewide testing of association (7–9). Conflicting observations on the magnitude of LD sharing between human populations, therefore, call for a careful examination of the following three questions, which are fundamental in developing strategies for genomewide testing of association. First, measurement of LD sharing between populations should be made independent of the definition of LD blocks, which introduce inconsistent block boundaries (10). Second, the mechanisms that shape LD sharing between populations are yet to be fully explored although the roles of recombination hotspots and demographic events have been implicated (11, 12). Third, the portability of haplotype-tagged SNPs (tagSNPs, hereafter) selected in one population to the others requires a careful examination. This examination is of special importance considering that only three continental populations were included in the HapMap Project (13–15).
To address the aforementioned questions, we typed >20,000 SNPs on chromosome 21 in seven populations: three representative continental populations [African-American (AFR), European (EUR), and Han Chinese (HAN)] and four other major East Asian (EA) populations. This design allows a close examination of LD sharing between continental groups as well as those within East Asia. In this report, we measured the LD sharing between populations independent of the definition of LD block; and we showed that bottleneck events play a critical role in shaping the LD sharing between Africans and non-Africans, but much less so between non-Africans.
An important question for applying HapMap results to disease studies is how tagSNPs selected from a HapMap population will be ported to disease studies performed in other populations. In this study, we showed that tagSNPs selected from representative continental populations are indeed portable to the others in the same continent for association studies, at least in East Asia, with reasonable efficiency. In addition, we proposed a simple guideline that allows a quick evaluation of the portability of tagSNPs between populations by typing a small number of SNPs.
Results
Overall 26,112 SNPs were selected and typed in this study, and the data from 19,060 SNPs passed the quality control criteria and were used for further analyses. The SNPs and quality control criteria for SNP selection are described in Materials and Methods. Seven world populations, including EUR, AFR, and five EA populations, were studied. The five EA populations, i.e., HAN, Miao (HMJ), Zhuang (CCY), Wa (WBM), and Uighur (UIG), represent five major linguistic families spoken in East Asia.
Preservation of LD between populations, i.e., LD sharing (S, or SAB when the population A was given as reference), is measured by the proportion of SNP pairs in LD in one population (population A or the reference) that are also in LD in another (population B). In this study, LD sharing was estimated without invoking the inference of haplotype blocks; therefore, the measure is independent of the definition of haplotype blocks. LD between two loci was measured in r2 (16). Detail for the measure of LD sharing is described in Materials and Methods. LD sharing between EAs ranges from 63–74% for r2 ≥ 0.1 and 70–84% for r2 ≥ 0.5 (see Table 1). LD sharing between EUR and EAs is slightly smaller (≈56–60% for r2 ≥ 0.1 and ≈60–65% for r2 ≥ 0.5). S between EUR and UIG is higher due to the close connection of UIG and Central Asian populations. The LD sharing between EAs and EUR is approximately symmetric regardless of the selection of the reference, i.e., SAB ≈ SBA. However, the S values between AFR and other populations are asymmetric. When compared with S values between non-Africans, LD sharing between AFR and EAs is much smaller (45–47% for r2 ≥ 0.1 and 36–42% for r2 ≥ 0.5) in reference to any EA. Furthermore, such LD sharing is also much smaller than the LD sharing with AFR, which is the reference demonstrating the asymmetry (SAB > SBA). The asymmetry becomes much more pronounced when criteria for LD become more stringent (i.e., for r2 ≥ 0.5) than it is for r2 ≥ 0.1 (data not shown for r2 ≥ 0.8).
Table 1. LD sharing between populations (SAB).
| r2 | HAN | HMJ | CCY | WBM | UIG | EUR | AFR | |
|---|---|---|---|---|---|---|---|---|
| ≥0.1 | HAN | 0.739 (1.0) | 0.720 (1.0) | 0.718 (1.1) | 0.708 (1.1) | 0.597 (1.1) | 0.468 (0.9) | |
| HMJ | 0.715 | 0.698 (1.0) | 0.692 (1.0) | 0.676 (1.0) | 0.589 (1.0) | 0.452 (0.8) | ||
| CCY | 0.728 | 0.735 | 0.703 (1.1) | 0.699 (1.1) | 0.602 (1.1) | 0.470 (0.9) | ||
| WBM | 0.677 | 0.682 | 0.661 | 0.660 (1.0) | 0.574 (1.0) | 0.448 (0.8) | ||
| UIG | 0.653 | 0.645 | 0.634 | 0.640 | 0.663 (1.0) | 0.467 (0.8) | ||
| EUR | 0.567 | 0.574 | 0.560 | 0.572 | 0.681 | 0.462 (0.8) | ||
| AFR | 0.538 | 0.536 | 0.530 | 0.540 | 0.585 | 0.562 | ||
| ≥0.5 | HAN | 0.827 (1.1) | 0.837 (1.0) | 0.800 (1.1) | 0.746 (1.0) | 0.634 (1.0) | 0.387 (0.5) | |
| HMJ | 0.786 | 0.798 (1.0) | 0.747 (1.0) | 0.700 (0.9) | 0.603 (0.9) | 0.365 (0.5) | ||
| CCY | 0.819 | 0.825 | 0.776 (1.0) | 0.731 (1.0) | 0.642 (1.0) | 0.381 (0.5) | ||
| WBM | 0.751 | 0.752 | 0.751 | 0.677 (0.9) | 0.609 (0.9) | 0.364 (0.5) | ||
| UIG | 0.770 | 0.758 | 0.765 | 0.734 | 0.785 (1.0) | 0.424 (0.5) | ||
| EUR | 0.647 | 0.642 | 0.654 | 0.647 | 0.772 | 0.407 (0.5) | ||
| AFR | 0.732 | 0.723 | 0.731 | 0.726 | 0.780 | 0.764 |
The references (population A) are listed in the first column. The symmetric index (T) is presented in parentheses.
LD sharing between populations is largely due to shared ancestry and demographic events (17). The magnitude of asymmetry can be measured by a symmetric index (T = SAB/SBA). The T values between non-African populations are approximately close to 1, but they are much smaller between AFR and other populations studied, especially when r2 ≥ 0.5 (see Table 1). We showed that T is a measure of the effect of a bottleneck event that occurred in one of the two populations (see Fig. 1). Therefore, we argued that the observed asymmetry associated with AFR is attributable to the demographic history of human populations, in particular, the bottleneck event that occurred during the separation of African and non-Africans (18). In contrast, the T values between non-African populations (T ≈ 1) suggest that the LD sharing between these populations is much less affected by their respective bottleneck events, although gene flow between these populations may have attenuated the signature of such events.
Fig. 1.

Relationship of two populations. O, shared ancestral population; P, population after bottleneck event; A and B, extant populations derived from O and P, respectively.
To study other factors that may shape LD sharing, such as recombination, drift, and mutation, we investigated the relationship of S and the time of divergence between the populations (measured by FST) (19). Strong negative correlation of S and FST between populations (ρ = -0.94 for r2 ≥ 0.1 and ρ = -0.95 for r2 ≥ 0.5, excluding AFR) was observed. Therefore, LD sharing (S) between populations is a decreasing function of the time after divergence. When the populations are sufficiently large where the new LD introduced by drift can be ignored (20), the factors that might be involved in shaping LD sharing are recombination and mutation, both of which accumulate with time. However, because the SNPs used for estimating LD sharing are shared between the populations, the effect of mutation can be excluded. Therefore, recombination is the primary factor that leads to the decreasing of LD sharing with time. Furthermore, strong correlation of the block size and FST between non-African populations and AFR (data not shown) further implicates the role of recombination in shaping LD sharing.
It was proposed that tagSNPs, selected from a set of SNPs in a DNA segment, can recapitulate LD information of the segment (5) with a preset requirement (e.g., r2 ≥ 0.5). To evaluate the portability of tagSNPs selected from one population to another, we introduced the recovery rate of tagSNPs (R), which is measured by the proportion of SNPs that can be represented by the tagSNPs. When two populations are considered, the recovery rate (RAB) provides the measurement of the portability of tagSNPs selected from population A to population B. In this study, an efficient algorithm (21) was used to infer tagSNPs without losing generality. The number of tagSNPs for non-Africans ranges from 554–664 for r2 ≥ 0.1 and from 2,366–3,120 for r2 ≥ 0.5. However, the number of tagSNPs for AFR is much greater (945 for r2 ≥ 0.1, and 5,473 for r2 ≥ 0.5), indicating a much stronger LD in non-Africans than in AFR. In this study, only those loci with minor allele frequency (MAF) ≥ 0.1 were used in estimating RAB (Table 2), with RAA = 1.0 indicating a full recovery. For r2 ≥ 0.1, RAB of tagSNPs selected from any non-African population is reasonably high in the others (83–93%), except for AFR (65–75%). For r2 ≥ 0.5, the RAB are lower (72–89% for non-African and 41–52% for AFR). Most importantly, any EA could be used for tagSNPs selection for other EAs. For example, tagSNPs selected from HAN render the highest efficiency considering both their numbers (628 for r2 ≥ 0.1 and 2,540 for r2 ≥ 0.5) and RABs (91% for r2 ≥ 0.1 and 84% for r2 ≥ 0.5). The tagSNPs selected from EUR also perform reasonably well in EAs (88% for r2 ≥ 0.1 and 81% for r2 ≥ 0.5). The excessive number of tagSNPs from AFR leads to an improved RAB in all populations (93–94% for r2 ≥ 0.1, and 93–95% for r2 ≥ 0.5) at a cost of drastically increased the number of tagSNPs for genotyping (945 for r2 ≥ 0.1 and 5,473 for r2 ≥ 0.5). Therefore, this strategy is not practically advisable.
Table 2. Recovery rate of tagSNPs.
| r2 | HAN | HMJ | CCY | WBM | UIG | EUR | AFR | N | |
|---|---|---|---|---|---|---|---|---|---|
| ≥0.1 | HAN | 1 | 0.928 | 0.91 | 0.917 | 0.89 | 0.863 | 0.71 | 628 |
| HMJ | 0.885 | 1 | 0.881 | 0.898 | 0.856 | 0.836 | 0.65 | 554 | |
| CCY | 0.899 | 0.93 | 1 | 0.907 | 0.89 | 0.86 | 0.704 | 618 | |
| WBM | 0.874 | 0.901 | 0.875 | 1 | 0.867 | 0.834 | 0.668 | 571 | |
| UIG | 0.898 | 0.921 | 0.894 | 0.919 | 1 | 0.903 | 0.745 | 664 | |
| EUR | 0.865 | 0.889 | 0.874 | 0.89 | 0.902 | 1 | 0.739 | 654 | |
| AFR | 0.931 | 0.941 | 0.931 | 0.931 | 0.94 | 0.934 | 1 | 945 | |
| ≥0.5 | HAN | 1 | 0.881 | 0.863 | 0.843 | 0.769 | 0.733 | 0.439 | 2540 |
| HMJ | 0.823 | 1 | 0.827 | 0.815 | 0.73 | 0.696 | 0.408 | 2366 | |
| CCY | 0.859 | 0.877 | 1 | 0.84 | 0.751 | 0.731 | 0.442 | 2530 | |
| WBM | 0.834 | 0.852 | 0.833 | 1 | 0.743 | 0.719 | 0.423 | 2452 | |
| UIG | 0.894 | 0.888 | 0.873 | 0.882 | 1 | 0.853 | 0.518 | 3120 | |
| EUR | 0.8 | 0.824 | 0.8 | 0.826 | 0.821 | 1 | 0.48 | 2936 | |
| AFR | 0.943 | 0.945 | 0.938 | 0.945 | 0.928 | 0.931 | 1 | 5473 |
The tagSNPs were selected from reference populations that were listed in the first column. The last column shows number of tagSNPs of the reference populations.
For any pair of non-African populations, we observed a strong correlation between R and S (ρ = 0.968 for r2 ≥ 0.1 and ρ = 0.983 for r2 ≥ 0.5), indicating that R between populations is largely dictated by the magnitude of their LD sharing in non-African populations. R was estimated by taking an arithmetic average of RAB and RBA. S is an arithmetic average of SAB and SBA. The R also correlates well with FST, as expected. Therefore, we suggested that both S and FST can be used as indices to evaluate the portability of preselected tagSNPs in other populations. Empirically, for FST = 0.10, a 75% and 85% recovery rate can be achieved for r2 ≥ 0.1 and r2 ≥ 0.5, respectively; for FST = 0.05, an 80% and 90% recovery rate can be achieved for r2 ≥ 0.1 and r2 ≥ 0.5, respectively. For practical purposes, when a new population is being considered for an association study, the portability of tagSNPs selected from one of the continental populations to this population can be evaluated by estimating their FST based on a small number of SNPs that are not in linkage disequilibrium. This guideline is important when using the data from the HapMap Project in future genome-wide association studies.
Discussion
Our study showed that the LD sharing between human populations is substantial when using a measure that is independent of the definition of haplotype block, validating the observation made by Gabriel et al. (6). This finding was achieved by estimating LD sharing surrounding each SNP individually without invoking the process of inferring the block structure of LD, which can be subjective and equivocal. Although the practical utility of such an approach is yet to be carefully explored, it serves the objective of this study well.
The sharing of common ancestry is the primary source of LD sharing between populations, but the maintenance of LD sharing between populations is affected by the interplay of both recombination and demographic events (22). The analytical framework we proposed allowed us to investigate the primary mechanisms underlying the magnitude of LD sharing. The strong bottleneck of the ancestors of non-Africans out of Africa played an important role in shaping the LD sharing between Africans and non-Africans. However, our observations are consistent with a mechanism that LD sharing between non-African populations has been primarily affected by historical recombination events.
We also showed that the tagSNPs selected from a representative population can be used in the genomewide association study of other populations in which the LD levels are yet to be fully characterized, at least in EA populations. This problem cannot be directly addressed by the data of the HapMap Project (15), but this study provides a unique opportunity to evaluate the utility of the Project for tagSNP selection. We also proposed an empirical approach to evaluate the recovery rate or portability of tagSNPs quickly and inexpensively.
Materials and Methods
SNP Selection and Genotyping. Overall, 26,112 SNPs, selected from all SNPs on chromosome 21 listed in dbSNP (build 117), passed the criteria for Illumina assay. Most of them are double-hits. These SNPs were mapped to human genome build 34 (Golden Path), and the average distance between two adjacent SNPs is ≈1,300 bp. Genotyping was performed on the Illumina SNP genotyping BeadLab platform. This platform combines a high-density oligonucleotide array and a multiplex thermocycled primer extension. The 26,112 SNPs were partitioned into 17 oligonucleotide primer sets, and 17 independent reactions were performed to type all 26,112 SNPs. Three main criteria were used in quality control procedures. First, all data from one sample that showed low signal-noise ratio in most loci were dropped. Second, if the typing result from one SNP was inconsistent with the known relationship of the trios or blind duplicates, data from this locus were dropped. Third, data that showed significant deviation from Hardy-Weinberg expectation were dropped. Overall 19,060 SNPs passed the quality control criteria and were subjected to further analysis. The data from the children of the trios and duplicated samples were also excluded from further analysis.
DNA Samples and Populations. Overall, 318 samples were included in this study. They are 48 AFR, 40 EUR, 50 Han, 46 Miao [HMJ, following Ethnologue: Languages of the World (23); www.ethnologue.com/ethno_docs/contents.asp], 44 CCY, 45 UIG, and 45 WBM. Purified genomic DNA of EUR and AFR was purchased from the Coriell Institute (Camden, NJ), whereas EA samples were collected with informed consent. Trios (two parents and an adult child) and duplicated samples were also included in typing in each population for quality control.
Statistical Analyses. In each population, two SNPs were considered in LD if r2 exceeded the preset criterion (0.1 or 0.5 in this study). r2 was estimated following Devlin et al. (16). The frequencies of two-locus haplotypes were estimated for all pairs of SNPs (24). This measurement does not require inference of haplotypes of >2 loci. The preservation of LD between two populations (A and B) can be measured by LD sharing (S), which is defined by the proportion of SNP pairs, in reference to those in LD in population A, that are in LD in both (SAB). For each SNP (target), the SNPs in a segment of 200 kb are included in the estimation of SAB with the target in the center of the segment. The number of SNPs that are in LD with the target are counted in both population A and population B. SAB is the ratio of the number of LDs shared in both populations (A and B) and the number of LDs in population A. For SBA, the number of LDs in population B was used as denominator. FST was estimated by an unbiased statistic (25) by using 19,060 loci.
Model. To facilitate the presentation, only two populations, i.e., A and B, are considered in this model. Fig. 1 presents a schematic illustration of the relationship of two populations. O is the ancestral population shared by both A and B. P is the population derived from O and ancestral to B. To simplify the model, we assume that the bottleneck event that led to an origin of a new population (P) occurred in a short period, the duration of which is negligible. Therefore, the relationship of the LD sharing between the populations A and B is as follows:
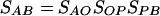 |
and
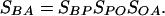 |
When the effective population sizes for both A and B have been large since divergence, no new LD will be generated, which leads to SAO = SBP = 1. The symmetric index T is defined as SBA/SAB. Again, under the assumption of large effective population size for both populations A and B, the decrease of LD is only a function of time; therefore, SOA = SPB. This equation leads to SBA/SAB = SPO/SOP. This result shows that the asymmetry between A and B is due to that between the ancestral populations O and P under the aforementioned assumption. Therefore, the asymmetry of LD sharing observed between African and non-African populations is dictated by the bottleneck event involved in the origin of non-Africans. In the absence of the bottleneck event, i.e., SOP = SPO = 1, we have T = 1 or SAB = SBA.
Acknowledgments
We thank the associates from Shanghai South Gene Technology Co., Ltd. and Shanghai Biochip Co., Ltd. for their technical support. This work was supported by Chinese High-Tech Program Grant (863) (2002BA711A10), National Key Project for Basic Research (973) (2004CB518605), Shanghai Science and Technology Committee Grants 03DJ14008 and 04DJ14003, the Chinese Ministry of Education and Health Science Center Innovation Fund, the Shanghai Institutes of Biological Sciences, the Chinese Academy of Sciences, and School of Medicine, Shanghai Jiaotong University.
Author contributions: W.H., L.J., and Z.C. designed and coordinated research; W.H., Y.H., H.W., Ying Wang (CNHGC), Y.L., Yi Wang (Fudan), X.C., Ying Wang (SJTU), L.X., Y.S., X.X., H.L., B.W., J.Q., W.Y., C.Z., Yi Wang (CNHGC), and H.J. performed research; L.J., Y.H. and Yi Wang (Fudan) contributed new reagents/analytic tools; L.J., Y.H., and Yi Wang (Fudan) analyzed data; L.J., Y.H., and W.H. wrote the paper; and G.Z. and Z.C. revised the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: LD, linkage disequilibrium; tagSNP, haplotype-tagged SNP; EA, East Asian; HAN, Han Chinese; HMJ, Miao; CCY, Zhuang; WBM, Wa; UIG, Uighur; EUR, European; AFR, African-American.
References
- 1.Risch, N. & Merikangas, K. (1996) Science 273, 1516-1517. [DOI] [PubMed] [Google Scholar]
- 2.Collins, F. S., Guyer, M. S. & Charkravarti, A. (1997) Science 278, 1580-1581. [DOI] [PubMed] [Google Scholar]
- 3.Daly, M. J., Rioux, J. D., Schaffner, S. F., Hudson, T. J. & Lander, E. S. (2001) Nat. Genet. 29, 229-232. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein, D. B. (2001) Nat. Genet. 29, 109-111. [DOI] [PubMed] [Google Scholar]
- 5.Rioux, J. D., Daly, M. J., Silverberg, M. S., Lindblad, K., Steinhart, H., Cohen, Z., Delmonte, T., Kocher, K., Miller, K., Guschwan, S., et al. (2001) Nat. Genet. 29, 223-228. [DOI] [PubMed] [Google Scholar]
- 6.Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B., Higgins, J., DeFelice, M., Lochner, A., Faggart, M., et al. (2002) Science 296, 2225-2229. [DOI] [PubMed] [Google Scholar]
- 7.Pritchard, J. K. & Przeworski, M. (2001) Am. J. Hum. Genet. 69, 1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boehnke, M. (2000) Nat. Genet. 25, 246-247. [DOI] [PubMed] [Google Scholar]
- 9.Crawford, D. C., Carlson, C. S., Rieder, M. J., Carrington, D. P., Yi, Q., Smith, J. D., Eberle, M. A., Kruglyak, L. & Nickerson, D. A. (2004) Am. J. Hum. Genet. 74, 610-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding, K., Zhou, K., Zhang, J., Knight, J., Zhang, X. & Shen, Y. (2005) Mol. Biol. Evol. 22, 148-159. [DOI] [PubMed] [Google Scholar]
- 11.Jeffreys, A. J., Kauppi, L. & Neumann, R. (2001) Nat. Genet. 29, 217-222. [DOI] [PubMed] [Google Scholar]
- 12.Wang, N., Akey, J. M., Zhang, K., Chakraborty, R. & Jin, L. (2002) Am. J. Hum. Genet 71, 1227-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.International HapMap Consortium (2003) Nature 426, 789-796. [DOI] [PubMed] [Google Scholar]
- 14.Mueller, J. C., Lohmussaar, E., Magi, R., Remm, M., Bettecken, T., Lichtner, P., Biskup, S., Illig, T., Pfeufer, A., Luedemann, J., et al. (2005) Am. J. Hum. Genet. 76, 387-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.International HapMap Consortium (2005) Nature 437, 1299-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devlin, B. & Risch, N. (1995) Genomics 29, 311-322. [DOI] [PubMed] [Google Scholar]
- 17.Cavalli-Sforza, L. L. & Feldman, M. W. (2003) Nat. Genet. 33, Suppl., 266-275. [DOI] [PubMed] [Google Scholar]
- 18.Excoffier, L. (2002) Curr. Opin. Genet. Dev. 12, 675-682. [DOI] [PubMed] [Google Scholar]
- 19.Nei, M. (1987) Molecular Evoluationary Genetics (Columbia Univ. Press, New York), 1st Ed., pp. 216-218.
- 20.Kruglyak, L. (1999) Nat. Genet. 22, 139-144. [DOI] [PubMed] [Google Scholar]
- 21.Carlson, C. S., Eberle, M. A., Rieder, M. J., Yi, Q., Kruglyak, L. & Nickerson, D. A. (2004) Am. J. Hum. Genet. 74, 106-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wall J. D. & Pritchard J. K. (2004) Nat. Rev. Genet. 8, 587-597. [DOI] [PubMed] [Google Scholar]
- 23.Grimes, B. F., ed. (2000) Ethnologue: Languages of the World (Int. Acad. Bookstore, Dallas), 14th Ed.
- 24.Hill W. G. (1974) Heredity 33, 229-239. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds J., Weir B. S. & Cockerham C. C. (1983) Genetics 105, 767-779. [DOI] [PMC free article] [PubMed] [Google Scholar]