

Abstract
As part of highly active antiretroviral therapy, protease inhibitor treatment has significantly increased the lifespan of HIV infected individuals. Many patients, however, develop negative side effects including premature atherosclerosis. We have previously demonstrated that in male low-density lipoprotein receptor (LDL-R) null mice, HIV protease inhibitors induce atherosclerotic lesions and cholesterol accumulation in macrophages in the absence of changes in plasma lipid levels. We determined that these increases were due to an up-regulation of the scavenger receptor, CD36. In the present study, we examined the effects of HIV protease inhibitors in female LDL-R null mice. Female mice given ritonavir and amprenavir (23 and 10 μg/mouse/day, respectively) developed fewer atherosclerotic lesions than males. Furthermore, peritoneal macrophages isolated from ritonavir-treated females had reduced levels of cholesterol accumulation as compared to males, and CD36 protein levels were increased to a significantly lesser degree in females than in males. To investigate the molecular mechanisms of this gender difference, we examined the effect of genetically removing estrogen receptor-alpha (ERα). In female mice lacking both LDL-R and ERα, the protective effect of gender was lost. Additionally, the reduced levels of cholesterol accumulation in macrophages observed in females was reversed. Furthermore, the absence of ERα resulted in increased expression of CD36 protein in a macrophage-specific manner in mice treated with ritonavir. These data demonstrate that ERα is directly involved in the regulation of cholesterol metabolism in macrophages and plays an important role in the gender differences observed in HIV protease inhibitor-induced atherosclerosis.
INTRODUCTION
Protease inhibitors, as part of highly active anti-retroviral therapy, have greatly increased the lifespan of individuals infected with the human immunodeficiency virus (HIV). Protease inhibitors block the replication of HIV, thereby delaying or preventing the onset of AIDS. Unfortunately, one of the deleterious side effects of protease inhibitor therapy is the potential development of premature atherosclerosis (1). This is particularly true in younger patients (men under the age of 34 and women under the age of 44) (2). HIV protease inhibitor treatment also causes dyslipidemia which may contribute to the development of cardiovascular disease (3). We have recently demonstrated, however, that HIV protease inhibitors can affect macrophage lipoprotein metabolism and atherosclerotic lesion formation independent of changes in plasma triglycerides and cholesterol levels, suggesting that HIV protease inhibitors can have direct actions on macrophage function (4).
Macrophage recruitment into the vascular wall is one of the earliest events in atherogenesis. The scavenger receptor-dependent uptake of lipoprotein sterols by macrophages in the sub-endothelial space contributes to the formation of lipid-laden macrophages. The class B scavenger receptor, CD36, has been shown to mediate both the uptake and efflux of cholesterol in cell culture models (5,6) and is the primary mediator of cholesterol accumulation in atherosclerosis induced by HIV protease inhibitors (4).
Many physiological gender differences are the result of the female sex steroid hormones, estrogen and progesterone. Besides the well-studied effects of estrogen on the reproductive system, in recent years it has become apparent that estrogen has important actions in other tissues including non-reproductive areas of the brain, bone, and in the immune and cardiovascular systems (7,8). The mechanisms of action of estrogen in the cardiovascular system are not completely understood, although some protective actions have been attributed to decreasing plasma LDL levels (9). In addition, estrogen has been shown to have a potentially anti-atherogenic effect by causing vasodilation by increasing nitric oxide production in a non-genomic manner (10,11). The incidence of cardiovascular disease is greater in men than in women prior to menopause, however, following menopause the risk of cardiovascular disease in women increases dramatically (12). The mechanisms of this difference are not well understood. This lack of understanding is highlighted by the results of the Women's Health Initiative, which indicate that hormone replacement therapy (HRT) increases cardiovascular disease (13). It remains to be determined if the negative effects of hormone replacement would be see with endogenous estrogens (17β-estradiol) instead of the conjugated equine estrogens used in current HRT regimens. Additionally the age at which HRT is initiated is likely a critical factor in the effects of estrogen on cardiovascular disease (14).
Women infected with HIV have a higher rate of gonadal dysfunction resulting in a greater length between menstrual cycles and a higher rate of amenorrhea (15,16). These symptoms may all be related to a suppression of estrogen levels observed in a subpopulation of HIV infected women (17). A reduction in estrogen in some women could contribute to increasing atherosclerosis, explaining why younger HIV-infected women are no longer protected from cardiovascular disease as compared to males.
The cellular effects of estrogen are primarily mediated through one of two estrogen receptors; α or β (ERα or ERβ). Estrogen receptors are members of the nuclear receptor superfamily and act as ligand-activated transcription factors (18). ERα has been shown to be critical for estrogen atheroprotection in Apoe -/- mice that are prone to develop atherosclerotic lesions (19). In this model of atherosclerosis, estrogen lowers plasma lipid levels and reduces atherosclerotic lesion size. However, since estrogen modulates a variety of aspects of cardiovascular physiology, and the lipid-independent effects of estrogen on cardiovascular disease are largely unexplored. Such mechanisms were investigated in this study examining the development of atherosclerosis in a model where lesions are induced by HIV protease inhibitors.
MATERIALS AND METHODS
Animals. All animals were housed in the AAALAC certified animal facilities at the University of Kentucky. Animals were maintained in constant temperature conditions on a 14:10 light/dark cycle (lights on at 0400h) and were provided food and water ad libitum. The LDL-R null and C57BL/6J mice were supplied by The Jackson Laboratory (Bar Harbor, Maine, USA). ERα -/+ mice were purchased from Taconic (Germantown, NY). Both LDL-R null and ERα -/+ mice are on a C57BL/6J background. To generate mice lacking both the low density lipoprotein receptor (LDL-R) gene and ERα, LDL-R null mice were bred with ERα-/+ to produce offspring heterozygous for both alleles (LDL-R-/+; ERα-/+). These mice were then crossed with LDL-R null mice to produce LDL-R/-; ERα-/+ offspring. Finally, these mice were then used as breeding pairs to produce offspring that were homozygous knockouts for both genes (LDL-R -/-; ERα-/-). At six weeks of age mice were placed on a normal chow diet and given vehicle control (0.01% ethanol) or protease inhibitors in their drinking water (amprenavir or ritonavir) for 8 weeks. Glaxo Wellcome Inc. (Toronto, Ontario, Canada) provided the amprenavir, and Abbott Laboratories (Abbott Park, Illinois, USA) provided the ritonavir. This regimen has previously been described to induce atherosclerotic lesions in male LDL-R null mice without altering plasma cholesterol levels (4).
Quantification of atherosclerotic lesions. To quantify atherosclerotic lesion area, the aorta from the arch to the ileal bifurcation was collected and fixed in 4% paraformaldehyde. The extraneous tissue was dissected away and the intimal surfaces were exposed by a longitudinal cut. Isolated aortas were secured with pins and placed under a dissecting microscope equipped with a CCD camera to capture the image of the aorta. Atherosclerotic lesions on the intimal aortic surface appear as opaque white areas compared with the thin, translucent aorta. Areas of intima covered by atherosclerotic lesions were quantified with ImagePro software (Media Cybernetics, Silver Spring, Maryland, USA) as previously described (20).
Cholesterol and cholesteryl ester mass quantification. Cholesterol measurements were determined as previously described (21). Mouse peritoneal macrophages were isolated by saline lavage, and cell lysates collected (22). The samples were extracted with isopropanol-hexane and cholesteryl heptadecanoate was added to each vessel preparation to serve as an internal standard. The extracted lipid was derivatized by suspending the dried lipid in N,O-bis(trimethylsilyl)trifluoroacetamide, trimethylchlorosilane, and acetonitrile (89:1:10). The material was heated at 80°C for 5 minutes, dried, and suspended in iso-octane. Pure cholesterol (Sigma-Aldrich) was dissolved in isooctane and used as a standard for the retention time of cholesterol. The samples were injected into a 6890 gas chromatograph G2579A system (Agilent Technologies, Palo Alto, California, USA). A mass-selective detector (model 5973; Agilent Technologies) was used in both scan and selected ion-monitoring modes to identify the samples.
SDS-PAGE and immunoblotting. Proteins were isolated from peritoneal macrophages by homogenization in lysis buffer (1X PBS, 1% Triton-X, 0.1% SDS, 50 mM sodium fluoride, 0.5% deoxycholate with Complete protease inhibitor cocktail (Roche)). Proteins were suspended in sample buffer containing 0.31 M Tris (pH6.8), 2.5% (wt/vol) SDS, 50% (vol/vol) glycerol, and 0.125% (wt/vol) bromophenol blue 1.2% (vol/vol) β-mercaptoethanol and heated at 95°C for 3 minutes. 20 μg of total cell protein was separated on a 12.5% SDS-polyacrylamide gel. The separated proteins were then transferred to PVDF membranes. Each membrane was blocked in Tris-buffered saline containing 5% dry milk for 1 hour at room temperature. Primary antibodies were diluted in TBS containing 1% dry milk and incubated with the PVDF membrane for 1 hour at room temperature. Anti-actin IgG was from Sigma-Aldrich, anti—mouse CD36 (IgM) was from BioDesign International Inc. (Kennebunkport, Maine, USA), and anti—scavenger receptor A (anti-SRA) was from Serotec Ltd. (Raleigh, North Carolina, USA). HRP-conjugated IgG's were purchased from Organon Teknika Corp. USA (West Chester, Pennsylvania, USA). Super Signal chemiluminescent substrate was purchased from Pierce Chemical Co. (Rockford, Illinois, USA). The PVDF membrane was washed four times, 10 minutes each, in TBS plus 1% dry milk. The secondary antibodies (conjugated to HRP) were diluted 1:20,000 in TBS plus 1% dry milk and incubated with the PVDF membrane for 1 hour at room temperature. The membrane was then washed and the labeled proteins were visualized by chemiluminescence autoradiography. Relative band densities were quantified using ImagePro Software (Medical Cybernetics, Silver Spring, Maryland).
Northern Blot Hybridization. Total RNA was extracted from cells using TRIzol (Molecular Research Center). RNA was quantified by spectrometry and 20 μg of RNA was loaded onto a 5% denaturing agarose gel. Following electrophoretic separation, the RNA was transferred to nylon membranes and probed for CD36, PPARγ and GAPDH mRNA as previously described (4).
Statistics. Least squares ANOVA was used to evaluate the data with respect to treatment and gender or genotype using Statistica v.6 (StatSoft, Tulsa, Oklahoma, USA). When appropriate, samples were compared within a given group using the Tukey HSD test. Means were considered statistically different at p < 0.05.
RESULTS
Protease inhibitors induce smaller lesions in intact female LDL-R null mice as compared to males. To determine the effect of gender on the development of atherosclerotic lesions by HIV protease inhibitors, six-week-old LDL-R null male and female mice were treated with vehicle (0.01% ethanol), amprenavir or ritonavir (23 or 10 μg/mouse/day, respectively). Following 8 weeks of treatment, aortas were removed and analyzed for the extent of atherosclerotic lesions as we previously described (4). Male mice given amprenavir had a 3-fold increase in lesion area while females had only a 1.5 fold increase (Figure 1). Male mice given ritonavir had a 5-fold increase in lesion area while females only had a 2-fold increase.
Figure 1.

HIV protease inhibitors promote atherosclerotic lesion formation in male mice to a greater degree than female mice. LDL-R null mice were treated with vehicle (0.01% ethanol), amprenavir (23 μg/day) or ritonavir (10 μg/day) for 8 weeks. Following 8 weeks of treatment, aortas were removed and atherosclerotic lesions quantified. Females exhibited a smaller surface area of atherosclerotic lesions in both amprenavir and ritonavir treated animals. Bars represent mean +/- SEM, n = 8. * = significantly different from vehicle, p<0.05. # = significantly different from males in the same treatment group, p<0.05. Open bars represent male values and closed bars represent female values.
Peritoneal macrophages from female LDL-R null mice treated with protease inhibitors accumulate less cholesterol than macrophages from males. To determine if the decrease in atherosclerotic lesions was associated with a decrease in macrophage cholesterol accumulation, we isolated peritoneal macrophages from animals given vehicle or ritonavir for 8 weeks (same animals as described in Figure 1). The mass of total cholesterol (free cholesterol plus cholesteryl esters) was determined by gas chromatography. Peritoneal macrophages isolated from animals receiving ritonavir contained significantly more total cholesterol than animals receiving vehicle only (Figure 2) (p<0.01, n=4). Macrophages from ritonavir treated female mice contained significantly less total cholesterol than their male counterparts (p<0.01, n=4).
Figure 2.

Peritoneal macrophages isolated from male LDL-R null mice accumulate more cholesterol mass than those isolated from female mice treated with ritonavir. LDL-R null mice were treated with vehicle (0.01% ethanol) or ritonavir (10μg/day) for 8 weeks. Peritoneal macrophages were isolated by saline lavage and processed to quantify total cholesterol mass by gas chromatography. Bars represent the mean +/- SEM, n = 4. * = significantly different from vehicle, p<0.01. # = significantly different from males in the same treatment group, p<0.01.
Peritoneal macrophages from female LDL-R null mice treated with ritonavir have reduced CD36 protein expression as compared to macrophages from males. We have previously determined that the scavenger receptor, CD36, is responsible for HIV protease-induced cholesteryl ester accumulation in macrophages (4). To determine if the effects of HIV protease inhibitors on the total amount of CD36 protein in peritoneal macrophages are different in male and female mice, we isolated peritoneal macrophages from animals given vehicle or ritonavir for 8 weeks. Cell lysates were generated from the isolated macrophages and equal amounts of protein resolved by SDS-PAGE and immunoblotted for the scavenger receptors CD36 and SR-A. Figure 3 demonstrates that peritoneal macrophages isolated from male LDL-R null mice receiving ritonavir contained more CD36 than mice receiving vehicle. SR-A levels were not altered by ritonavir treatment, suggesting that the effect is specific for CD36. Female mice also had an increase in CD36 levels, but it was significantly less compared to males. Blots were normalized for loading by blotting with actin. There was no difference in actin levels among the groups (data not shown). Relative levels were quantified and are represented in Figure 3B (p<0.05, n=4).
Figure 3.

Peritoneal macrophages isolated from male LDL-R null mice contain more CD36 than female mice treated with ritonavir. LDL-R null mice were treated with vehicle (0.01% ethanol) or ritonavir (10μg/day) for 8 weeks. Peritoneal macrophages were isolated and processed for immunoblots. Total cell lysates were generated and 20 μg of protein was resolved by SDS-PAGE and immunobloted with antibodies for CD36 and SR-A. Reactive material was visualized by chemiluminescence (Figure 3A). Relative densities were quantified and represented as fold increase over vehicle control (Figure 3B). Bars represent mean +/- SEM, n = 4, * = significantly different from vehicle, p<0.05.
ERα is required for reduced atherosclerotic lesion formation in female mice treated with ritonavir. To begin to investigate the molecular mechanisms responsible for the gender difference observed in HIV protease inhibitor-induced atherosclerosis, we investigated the role of ERα in mice lacking both the LDL-R and ERα. We used female LDL-R null mice with an intact ERα gene and female LDL-R null mice that we crossed with ERα knockout mice to produce LDL-R null mice that were also ERα -. Six-week old female mice were treated with vehicle (0.01%) or ritonavir (25μg/mouse/day) for 8 weeks and analyzed for atherosclerotic lesions. This dose of ritonavir is greater than that used in the previous experiment examining gender differences. This dose was chosen to maximize lesion formation and does not alter plasma lipid levels (Table 1). Ritonavir treatment induced a significant increase in lesion area in ERα + mice (Figure 4) (p<0.001, n=10) as we had observed previously. Mice lacking ERα, however, had an even greater increase in lesion area (p<0.001, n=10).
Table 1.
Physiological parameters of mice treated with ritonavir.
| LDL-R null |
LDL-R null/ ERα -/- |
|||
|---|---|---|---|---|
| Vehicle | Ritonavir | Vehicle | Ritonavir | |
| % body weight gain | 39.7 +/- 0.022 | 19.0 +/- 0.025* | 46.3 +/- 0.018 | 33.4 +/- 0.032*# |
| Glucose (mg/dL) | 81.2 +/- 0.814 | 81.4 +/- 1.477 | 82.0 +/- 0.803 | 81.9 +/- 1.130 |
| Insulin (ng/ml) | 0.32 +/- 0.007 | 0.32 +/- 0.004 | 0.33 +/- 0.009 | 0.33 +/- 0.007 |
| Total Cholesterol (mmol/l) | 8.9 +/- 0.35 | 9.2 +/- 0.41 | 8.7 +/- 0.21 | 9.4 +/- 0.56 |
| Triglycerides (mmol/L) | 2.83 +/- 0.014 | 2.82 +/- 0.013 | 2.80 +/- 0.009 | 2.81 +/- 0.014 |
= significantly different from Vehicle treated mice (p<0.01).
= significantly different from LDL-R null ritonavir- treated mice (p<0.01).
Figure 4.

Ritonavir promotes atherosclerotic lesion formation in female mice with an intact the ERα gene to a greater degree than in female mice with an intact ERα gene. ERα+ or ERα- mice were treated with vehicle (0.01% ethanol) or ritonavir (25μg/day) for 8 weeks. Following 8 weeks of treatment, aortas were removed and atherosclerotic lesions quantified. ERα- mice had a significantly larger area of atherosclerotic as compared to the ritonavir treated ERα + mice. Bars represent mean +/- SEM, n = 10, * = significantly different from vehicle, p<0.001. # = significantly different from ERα + mice treated with ritonavir, p<0.001.
HIV protease inhibitors do not cause dyslipidemia in LDL-R null/ ERα-/- mice. We have previously demonstrated that at low doses, ritonavir can induce atherosclerosis without causing changes in plasma levels of triglycerides or total cholesterol (4). To confirm that the same is true in the mice we generated which lack the ERα gene in addition to the LDL-R gene we measured plasma levels of glucose, insulin, triglycerides and total cholesterol from mice treated for 8 weeks with 25μg ritonavir/day (Table 1). Additionally, we monitored weight gain over the treatment period. There was no effect of ritonavir on any of the lipids or metabolic parameters we measured in the plasma. Furthermore, there was no difference in baseline levels between LDL-R null mice with or without the ERα gene. Mice treated with ritonavir gained significantly less weight (p<0.01, n=10) than vehicle and this was partially reversed in the ERα- mice (p<0.01).
ERα is responsible for reduced cholesterol accumulation in peritoneal macrophages from female mice given ritonavir. Peritoneal macrophages from animals given vehicle or ritonavir for 8 weeks (same animals as described in Figure 4) were isolated and lipids extracted. The mass of cholesterol was determined by gas chromatography. Peritoneal macrophages isolated from animals receiving ritonavir contained more total cholesterol than animals receiving vehicle (Figure 5). Peritoneal macrophages from ritonavir-treated ERα - mice contained nearly twice as much total cholesterol as macrophages from ERα + mice (p<0.01, n=4).
Figure 5.

Peritoneal macrophages isolated from female ERα - mice treated with ritonavir accumulate more total cholesterol mass than those isolated from female mice expressing ERα. Female mice were treated with vehicle (0.01% ethanol) or ritonavir (25μg/day) for 8 weeks. Peritoneal macrophages were isolated by saline lavage and processed to quantify total cholesterol mass by gas chromatography. Bars represent the mean +/- SEM, n = 4. * = significantly different from vehicle, p<0.001. # = significantly different from ERα + mice treated with ritonavir, p<0.001.
ERα plays a direct role in cholesterol metabolism in macrophages. To determine if the differences observed in cholesterol accumulation in peritoneal macrophages result from intrinsic differences at the cellular level caused by the absence of ERα, peritoneal macrophages were isolated from 6 week-old LDL-R null mice that were either ERα + or ERα -. Cells were plated and grown for 24 hours in the presence of ritonavir (30ng/ml) and or vehicle (0.01% ethanol) plus AgLDL (50μg/ml). Lipids were isolated and total cholesterol assayed by gas chromatography. Figure 6A shows that free cholesterol levels in the cells were not changed by the absence of ERα or the presence of ritonavir. However, when the cholesteryl esters were analyzed (i.e., those that are accumulated in fat droplets), there was a significant increase following ritonavir treatment (p<0.001, n=4). This increase was further exaggerated in the absence of ERα (Figure 6B) (p<0.001, n=4).
Figure 6.

Peritoneal macrophages isolated from ERα- mice accumulate more cholesteryl esters when exposed to ritonavir in vitro. Peritoneal macrophages were isolated by saline lavage and grown in culture for twenty-four hours in the presence of ritonavir (30 ng/ml) or vehicle (0.01% ethanol) plus AgLDL (50μg/ml). After twenty four hours, cell lysates were prepared and assayed for free and total cholesterol by gas chromatography. There were no significant differences in free cholesterol (Figure 6A). Total cholesterol (free cholesterol plus cholesteryl esters) dramatically increased following ritonavir treatment (Figure 6B). This increase was greater in peritoneal macrophages lacking the ERα. Bars represent the mean +/- SEM, n=4. * = significantly different from vehicle, p<0.001. # = significantly different from ERα + macrophages treated with ritonavir, p<0.001.
Ritonavir induces a greater increase in CD36 protein levels in peritoneal macrophages in the absence of ERα. Since we have previously determined that the CD36 mediates the accumulation of cholesterol esters in macrophages exposed to protease inhibitors, we investigated whether ERα is involved in the regulation of CD36 protein expression. Peritoneal macrophages were isolated from animals given vehicle or ritonavir for 8 weeks. Cell lysates were generated from isolated macrophages and adipocytes and equal amounts of proteins resolved by SDS-PAGE and immunobloted for the scavenger receptors CD36 and SR-A. Figure 7 demonstrates that peritoneal macrophages isolated from mice receiving ritonavir contained more CD36 protein than mice receiving vehicle. SR-A protein levels were not altered. In mice lacking both ERα and LDL-R, ritonavir had a greater increase in CD36 protein levels, although there was no difference in baseline CD36 expression. Additionally, SR-A levels were not changed. Blots were normalized for loading by blotting with actin. There was no difference in actin levels among the groups. The effect on CD36 was specific to macrophages as there was no effect in adipocytes with either ritonavir or the absence of ERα.
Figure 7.
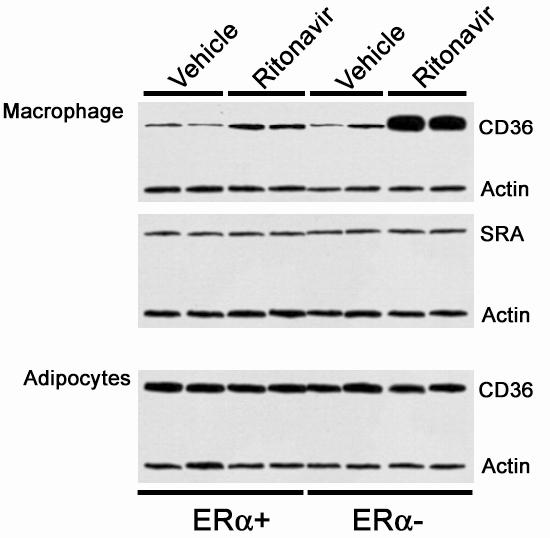
Peritoneal macrophages isolated from female ritonavir-treated ERα - mice contain more CD36 protein than ritonavir-treated ERα + mice. Female mice were treated with vehicle (0.01%) or ritonavir (25μg/day) for 8 weeks. Peritoneal macrophages and adipocytes were isolated and processed for immunoblots. Cell lysates were generated and 20 μg of protein was resolved by SDS-PAGE and immunobloted with antibodies for CD36, SR-A and actin. Actin was included as a control. Reactive material was visualized by chemiluminescence.
Ritonavir induces a greater increase in CD36 and peroxisome proliferator activated receptor gamma (PPARγ) mRNA expression in peritoneal macrophages from ERα - mice. We have previously demonstrated that PPARγ plays a critical role in regulating the expression of CD36 in macrophages (4). To determine if ERα mediates its expression, we examined PPARγ mRNA levels in peritoneal macrophages isolated from animals treated with ritonavir for 8 weeks. Total RNA was isolated and separated on a 5% denaturing agarose gel. The RNA was transferred to a nylon membrane and probed for CD36 and PPARυ using radiolabeled DNA. GAPDH mRNA served as a loading control. The absence of ERα had no effect on the baseline expression of CD36 mRNA, however, treatment with ritonavir greatly increased the levels of CD36 mRNA and these increases were further enhanced in ERα - mice (Figure 8). This increase paralleled that seen with the levels of CD36 protein. The levels of PPARγ mRNA are similarly elevated in the response to ritonavir in the absence of ERα.
Figure 8.
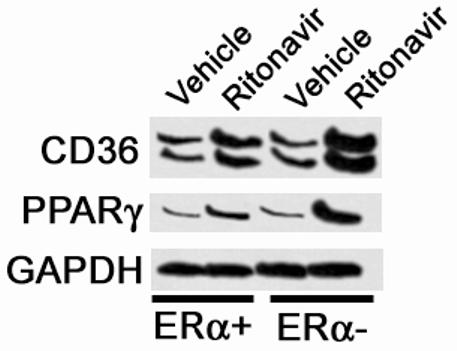
Peritoneal macrophages isolated from ritonavir-treated ERα - mice contain more CD36 and PPARγ mRNA than ritonavir-treated ERα + mice. Peritoneal macrophages were removed by saline lavage from animals treated as described in Figure 4. Total RNA was isolated and separated on 5% denaturing agarose gel. RNA was transferred to nylon membranes and probed for CD36, PPARγ and GAPDH mRNA as a control with radioactively labeled oligonucleotides. A representative Northern blot is shown.
DISCUSSION
Clinical evidence suggests that HIV infection and HIV protease inhibitors can induce lipodystrophy, hyperlipidemia and accelerated coronary heart disease in both male and female patients, especially in young patients (1-3). Healthy young women are generally protected against atherosclerosis as compared to men. However, following menopause this protection is lost, suggesting a protective role of the female hormonal milieu (23). The mechanisms of this gender difference are not completely understood, but estrogen appears to have a beneficial role in reducing plasma LDL levels as well as numerous direct effects on the vascular wall (11) (24). In light of recent data from the Women's Health Initiative concerning the negative effects of the current HRT regimens on cardiovascular disease (13), the mechanism of action of endogenous estrogens needs to be elucidated. While, the Women's Health Initiative investigated the actions of conjugated equine estrogens used in HRT, a better understanding of the mechanism of action of endogenous estrogens will eventually lead to improved hormonal replacement therapies.
We have previously shown that HIV protease inhibitors can induce significant atherosclerosis in male mice prone to developing lesions (LDL-R null and Apoe -/- mice) at doses that do not increase plasma lipid concentrations (4). In the present study, females develop lesions following treatment with HIV protease inhibitors, but to a greatly reduced extent as compared to males. Furthermore, the source of this gender difference involves ERα since genetic removal of the receptor completely reverses the protective effect in females and even worsens atherosclerosis induced by ritonavir without altering plasma lipid levels or metabolic markers associated with insulin resistance commonly seen in patients on HIV protease inhibitor therapy. Reduced cholesteryl ester accumulation, CD36 protein and mRNA and PPARγ mRNA expression in macrophages from female mice requires ERα and suggests a novel role of estrogen and ERα in the development of atherosclerosis.
Atherosclerosis is a multi-faceted, complex process, but macrophage cholesterol accumulation plays a critical role in the development of foam cells and fatty streaks. Since we have used a model where atherosclerosis develops in the absence of changes in plasma lipid levels, we are able to remove that complicating influence from our studies. Our data suggest that the gender differences observed occur at least, in part, at the level of cholesterol accumulation in macrophages. Peritoneal macrophages from female mice contain less cholesteryl ester mass and have reduced alterations in the scavenger receptor system responsible for cholesterol uptake. The effect on CD36 expression is specific to macrophages as demonstrated by a lack of an effect in adipocytes. Furthermore, macrophages from ER- mice have increased cholesteryl ester accumulation in response to ritonavir treatment in vitro. These data suggest that in females, estrogen can influence cholesterol accumulation directly at the level of the lipoprotein metabolism machinery in macrophages.
In several animal models of diet-induced atherosclerosis, females tend to have larger lesions (25). The different effect in females in these models and the current model of atherosclerosis induced by HIV protease inhibitors is likely due to the pharmacological initiation of atherosclerosis in the absence of increase plasma lipid concentrations. The female gonadal steroid hormone, 17β-estradiol, has been shown to protect against atherosclerosis in Apoe -/- mice (19). In that study, ERα was responsible for atheroprotection and this may be, in part, due to the positive effects on plasma lipid levels. In the study we present here, the absence of ERα exacerbated lesion size following the HIV protease inhibitor insult, reversing the normally protective effect of gender. These data confirm and extend the findings that ERα is involved in the gender differences observed in different animal models of atherosclerosis, and that estrogen action in the cardiovascular system occurs multiple mechanisms.
The levels of circulating 17β-estradiol in ERα- mice are elevated due to a lack of negative feedback (26). Elevated estrogen levels may influence atherosclerosis progression by the antioxidant properties of estrogens (27). Similar levels of 17β-estradiol, however, have been shown previously to prevent atherosclerosis in the Apoe-/- model of atherosclerosis in a receptor dependent fashion (19). Furthermore, in the mice lacking ERα, protection is lost and any antioxidative effects of elevated estrogen are swamped in this model of atherosclerosis induced by HIV protease inhibitors.
Peritoneal macrophages from female LDL-R null mice treated with ritonavir had less of an increase in CD36 mRNA as compared to their male littermates. We have previously demonstrated that in this model, cholesterol accumulation is dependent on CD36 activity. CD36 is regulated by several transcription factors including PPARγ. The mechanism by which ritonavir increases CD36 levels is not completely understood, but we have shown that blocking the increase in PPARγ induced by ritonavir prevents the upregulation of CD36, and that this upregulation involves protein kinase C (PKC) activation (4). While the absence of ERα alone has no effect on CD36 or PPARγ mRNA levels, the absence of ERα prevents the female hormones from suppressing CD36 levels in response to ritonavir treatment. Total cholesteryl ester accumulation within macrophages can be influenced by both uptake and efflux of cholesterol by the ATP-binding cassette transporter A1 (ABCA1) (28). PPARγ has also been shown to regulate ABCA1 expression (29). ABCA1, however, is likely not the target of estrogen action via PPARγ since estrogen has been shown to regulate ABCA1 in an ERα-independent fashion (30). Our data demonstrate a clear role for ERα.
One possible mechanism by which estrogen can modulate the cellular response to ritonavir is by preventing the activation of the PKC pathway and ultimately CD36 expression. We have observed a similar effect of estrogen on the phosphatidylinositide 3'-OH kinase/ Akt and mitogen activated kinase pathways (MAPK) in neurons (31,32). For example, following stroke-like conditions, estrogen enhances the response of the Akt pathway without any observable differences in total Akt levels (31). These data suggest that estrogen can act to modify early signaling events within target cells resulting in an altered response to a negative stimulus.
Additionally, ERα may modulate the expression of PPARγ mRNA directly. As we noted, baseline levels of PPARα mRNA were not altered by a loss of ERα expression, however the change in expression in response to ritonavir treatment was altered. The PPARγ promoter has several putative estrogen receptor response elements (33). Furthermore, estrogen has been shown to suppress PPARγ mRNA expression in osteoblasts and adipocytes (34). Additionally, ritonavir may directly alter estrogen receptor activity. Ritonavir has been shown to inhibit the activity of the proteaseome (35). Interestingly, proteasomal degradation of ERα is required for efficient receptor activity (36). In the case of PPARγ, if ERα is not efficiently degraded, the negative effects on PPARγ mRNA expression could be sustained in the presence of ritonavir. This is a positive event in terms of cholesterol accumulation in macrophages. The signaling events stimulated by ritonavir treatment that result in an increase in CD36 mRNA expression could be counteracted in the presence of ERα.
The data presented here demonstrate a novel role of ERα in regulating proteins involved in lipoprotein metabolism in macrophages. This role of estrogen and ERα is critical for the prevention of atherosclerosis induced by HIV protease inhibitors. This may be of particular importance in patients as HIV and HIV protease inhibitor therapy can alter gonadal hormone levels as well as the metabolism of estrogen (17,37). Alterations in endogenous estrogen levels may contribute to the loss of the protection against cardiovascular disease in females on HIV protease inhibitor therapy.
ACKNOWLEDGEMENTS
This work was supported in part by a COBRE grant (P20 RR15592) from the National Center for Research Resources at NIH in addition to NIH R01 HL73693 (MEW) and R01 HL68509 (EJS).
REFERENCES
- 1.Currier JS, Taylor A, Boyd F, Dezii CM, Kawabata H, Burtcel B, Maa JF, Hodder S. J Acquir Immune Defic Syndr. 2003;33:506–512. doi: 10.1097/00126334-200308010-00012. [DOI] [PubMed] [Google Scholar]
- 2.Mooser V. Aids. 2003;17(Suppl 1):S65–69. doi: 10.1097/00002030-200304001-00009. [DOI] [PubMed] [Google Scholar]
- 3.Carr A, Samaras K, Chisholm DJ, Cooper DA. Lancet. 1998;351:1881–1883. doi: 10.1016/S0140-6736(98)03391-1. [DOI] [PubMed] [Google Scholar]
- 4.Dressman J, Kincer J, Matveev SV, Guo L, Greenberg RN, Guerin T, Meade D, Li XA, Zhu W, Uittenbogaard A, Wilson ME, Smart EJ. J Clin Invest. 2003;111:389–397. doi: 10.1172/JCI16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC, Rothblat GH, Swaney JB, Tall AR. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 6.Graf GA, Connell PM, van der Westhuyzen DR, Smart EJ. J Biol Chem. 1999;274:12043–12048. doi: 10.1074/jbc.274.17.12043. [DOI] [PubMed] [Google Scholar]
- 7.Davidson NE. N Engl J Med. 1995;332:1638–1639. doi: 10.1056/NEJM199506153322409. [DOI] [PubMed] [Google Scholar]
- 8.Mendelsohn ME, Karas RH. Science. 2005;308:1583–1587. doi: 10.1126/science.1112062. [DOI] [PubMed] [Google Scholar]
- 9.Barrett-Connor E, Bush TL. Jama. 1991;265:1861–1867. [PubMed] [Google Scholar]
- 10.Mendelsohn ME, Karas RH. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 11.Hodgin JB, Maeda N. Endocrinology. 2002;143:4495–4501. doi: 10.1210/en.2002-220844. [DOI] [PubMed] [Google Scholar]
- 12.Wingard DL, Suarez L, Barrett-Connor E. Am J Epidemiol. 1983;117:165–172. doi: 10.1093/oxfordjournals.aje.a113527. [DOI] [PubMed] [Google Scholar]
- 13.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Jama. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 14.Barrett-Connor E. J Clin Endocrinol Metab. 2003;88:4031–4042. doi: 10.1210/jc.2003-030876. [DOI] [PubMed] [Google Scholar]
- 15.Chirgwin KD, Feldman J, Muneyyirci-Delale O, Landesman S, Minkoff H. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;12:489–494. doi: 10.1097/00042560-199608150-00008. [DOI] [PubMed] [Google Scholar]
- 16.Minkoff HL, Eisenberger-Matityahu D, Feldman J, Burk R, Clarke L. Am J Obstet Gynecol. 1999;180:824–836. doi: 10.1016/s0002-9378(99)70653-8. [DOI] [PubMed] [Google Scholar]
- 17.Grinspoon S, Corcoran C, Miller K, Biller BM, Askari H, Wang E, Hubbard J, Anderson EJ, Basgoz N, Heller HM, Klibanski A. J Clin Endocrinol Metab. 1997;82:1332–1337. doi: 10.1210/jcem.82.5.3907. [DOI] [PubMed] [Google Scholar]
- 18.Bjornstrom L, Sjoberg M. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 19.Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N. J Clin Invest. 2001;107:333–340. doi: 10.1172/JCI11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daugherty A, Pure E, Delfel-Butteiger D, Chen S, Leferovich J, Roselaar SE, Rader DJ. J Clin Invest. 1997;100:1575–1580. doi: 10.1172/JCI119681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uittenbogaard A, Everson WV, Matveev SV, Smart EJ. J Biol Chem. 2002;277:4925–4931. doi: 10.1074/jbc.M109278200. [DOI] [PubMed] [Google Scholar]
- 22.Whitman SC, Daugherty A, Post SR. J Biol Chem. 2000;275:35807–35813. doi: 10.1074/jbc.M001797200. [DOI] [PubMed] [Google Scholar]
- 23.Chae CU, Ridker PM, Manson JE. Thromb Haemost. 1997;78:770–780. [PubMed] [Google Scholar]
- 24.Simoncini T, Mannella P, Fornari L, Caruso A, Varone G, Genazzani AR. Steroids. 2004;69:537–542. doi: 10.1016/j.steroids.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 25.VanderLaan PA, Reardon CA, Getz GS. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 26.Couse JF, Korach KS. Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 27.Park D, Huang T, Frishman WH. Cardiol Rev. 2005;13:13–17. doi: 10.1097/01.crd.0000126084.68791.32. [DOI] [PubMed] [Google Scholar]
- 28.Rothblat GH, de la Llera-Moya M, Atger V, Kellner-Weibel G, Williams DL, Phillips MC. J Lipid Res. 1999;40:781–796. [PubMed] [Google Scholar]
- 29.Duval C, Chinetti G, Trottein F, Fruchart JC, Staels B. Trends Mol Med. 2002;8:422–430. doi: 10.1016/s1471-4914(02)02385-7. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava RA. Mol Cell Biochem. 2002;240:67–73. doi: 10.1023/a:1020604610873. [DOI] [PubMed] [Google Scholar]
- 31.Wilson ME, Liu Y, Wise PM. Molecular Brain Research. 2002;102:48–54. doi: 10.1016/s0169-328x(02)00181-x. [DOI] [PubMed] [Google Scholar]
- 32.Wilson ME, Dimayuga FO, Reed JL, Curry TE, Anderson CF, Nath A, Bruce-Keller AJ. Endocrine. 2005. [DOI] [PubMed] [Google Scholar]
- 33.Bajic VB, Tan SL, Chong A, Tang S, Strom A, Gustafsson JA, Lin CY, Liu ET. Nucleic Acids Res. 2003;31:3605–3607. doi: 10.1093/nar/gkg517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dang ZC, van Bezooijen RL, Karperien M, Papapoulos SE, Lowik CW. J Bone Miner Res. 2002;17:394–405. doi: 10.1359/jbmr.2002.17.3.394. [DOI] [PubMed] [Google Scholar]
- 35.Schmidtke G, Holzhutter HG, Bogyo M, Kairies N, Groll M, de Giuli R, Emch S, Groettrup M. J Biol Chem. 1999;274:35734–35740. doi: 10.1074/jbc.274.50.35734. [DOI] [PubMed] [Google Scholar]
- 36.Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Mol Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 37.Ouellet D, Hsu A, Qian J, Locke CS, Eason CJ, Cavanaugh JH, Leonard JM, Granneman GR. Br J Clin Pharmacol. 1998;46:111–116. doi: 10.1046/j.1365-2125.1998.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]