

Abstract
Atherosclerosis, once believed to be a result of a slow, irreversible process resulting from lipid accumulation in arterial walls, is now recognized as a dynamic process with reversibility. Liver-directed gene therapy for dyslipidemia aims to treat patients who are not responsive to currently available primary and secondary prevention. Moreover, gene therapy strategies have also proved valuable in studying the dynamics of atherosclerotic lesion formation, progression, and remodeling in experimental animals. Recent results on the long-term effect of gene therapy suggest that hepatic expression of therapeutic genes suppresses inflammation and has profound effects on the nature of the atherogenic process.
Keywords: atherosclerosis, inhibition of lesion progression, lesion regression, inflammation, gene therapy, helper-dependent adenovirus
Abbreviations: AAV, adeno-associated virus; apo, apolipoprotein; CETP, cholesteryl ester transfer protein; EC, endothelial cell; FH, familial hypercholesterolemia; HDAd, helper-dependent adenovirus; HDL, high-density-lipoprotein; ICAM, intracellular adhesion molecule; INF-γ, interferon-γ; LDLR, low-density-lipoprotein receptor; PDGF, platelet-derived growth factor; SMC, smooth muscle cell; VCAM-1, vascular cell adhesion molecule-1; VLDLR, very-low-density lipoprotein receptor
The classical view of atherosclerosis was that it was a slow, irreversible process. However, mounting evidence now supports that it is a dynamic and reversible process. Animal studies in nonhuman primates and rabbits fed high cholesterol diets have revealed the possibility of lesion stabilization and even regression by an aggressive lipid lowering regimen (Armstrong & Megan, 1975; Aikawa et al., 1998a; 1999; Kockx et al., 1998). Furthermore, studies on thoracic aorta transplantation or transgenic mice carrying apolipoprotein E (apoE)-Mx1-Cre transgene have shown that long-term lipid normalization or stable expression of anti-atherogenic proteins induces regression of advanced lesions (Reis et al., 2001; Raffai et al., 2005). These studies also showed remodeling of arterial walls with decreased macrophage accumulation, reduced inflammation and increased collagen contents, providing a mechanistic explanation for the clinical benefit of lipid-lowering therapy (Libby & Aikawa, 2002).
Here we summarize current concepts of atherogenesis, recent progress made toward the development of liver-directed gene therapy for dyslipidemia, and how these approaches have contributed to our understanding of the dynamics of the atherosclerotic process. First, we will review the inflammatory response during progression of atherosclerosis, confer dynamics of atherosclerotic plaques, and discuss recent progress in vector biology. Finally, we will comment on our recent results on the inhibition, remodeling, and regression of atherosclerosis by liver-directed gene therapy in mouse models of dyslipidemia.
INFLAMMATION AND DEVELOPMENT OF ATHEROSCLEROSIS
Inflammation plays a central role in atherosclerosis (Glass & Witztum, 2001; Hansson, 2001; Libby, 2002; Nilsson et al., 2005). The initial step of atherosclerosis is monocyte invasion into the intima (Fig. 1). Under normal conditions, monocytes and T cells in blood have random contact with, but do not adhere to, endothelial cells (ECs). However, the situation is changed with the onset of inflammation (Blankenberg et al., 2003; Hoffman et al., 2004).
Figure 1. Development of atherosclerosis.
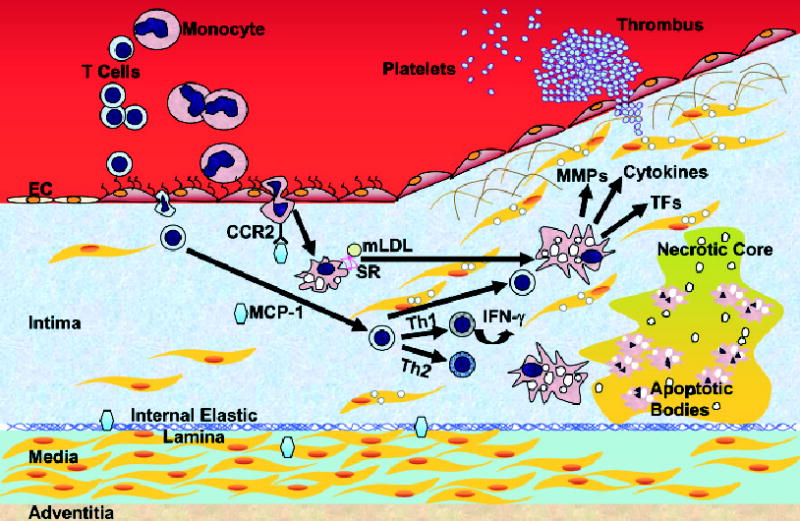
Monocytes and T-leukocytes do not adhere to ECs under normal conditions. 1. Adhesion. When ECs undergo inflammatory activation they express adhesion molecules, and leukocytes are trapped by ECs. 2. Transmigration. Once adherent, leukocytes migrate into the intima. 3. Foam cell formation. In the intima, monocytes differentiate into macrophages and take up lipids. 4. Progression. T cells are activated and further stimulate macrophages. SMCs of the media migrate to the top of the intima to form a fibrous cap over the lipid core. Further activation of macrophages produces MMPs that degrade the extracellular matrix and weaken the fibrous cap. 5. Plaque rupture. When the plaque ruptures, it allows the blood to contact to the procoagulant protein, tissue factor, and activate the coagulant cascade. Macrophages eventually die in a central core by apoptosis or necrosis, which forms a necrotic core in the lesion. IFN-γ, interferon-γ; MCP-1, monocyte chemoattractant protein-1; mLDL, modified LDL; MMP, matrix metalloproteinase; SR, scavenger receptors; TF, tissue factor. Note: Mast cells play an important role in atherogenesis. The interaction of the chemokine receptor CCR3 on the surface of mast cells and eotaxin, a chemoattractant, may facilitate the trans-migration of these cells. In the intima, they undergo degranulation and release factors that contribute to atherogenesis. Mast cells are not indicated.
Adhesion of leukocytes to endothelial cells (ECs)
When ECs undergo inflammatory activation in response to atherogenic stimuli such as low-density lipoprotein (LDL) accumulation in the arterial wall, they express various leukocyte adhesion molecules including P-selectin, E-selectin, intracellular adhesion molecules (ICAMs), vascular cell adhesion molecule-1 (VCAM-1), and integrins. Selectins are involved in the rolling and tethering of leukocytes. ICAMs, VCAM-1, and integrins induce firm adhesion of monocytes and T cells to ECs (Blankenberg et al., 2003).
Transmigration
Once adherent to ECs, monocytes and T cells migrate into the intima (innermost layer of the arterial wall) in response to chemokines. Monocyte chemoattractant protein-1 (MCP-1) is one of such chemokines and is expressed by the ECs and smooth muscle cells (SMCs) (Boring et al., 1998; Gu et al., 1998).
Monocyte maturation and T cell activation
In the intima, monocytes differentiate into macrophages upon induction by macrophage colony stimulating factor (M-CSF) (Ishibashi et al., 1990; Clinton et al., 1992). This causes the expression of scavenger receptors and the macrophages to engulf modified lipoproteins and fill with lipid droplets. Once in the intima, T cells encounter antigens such as modified LDL and heat-shock proteins. Upon activation by engagement of the receptor on T cells and antigens, the T cells produce cytokines such as interferon-γ (INF-γ) and further activate macrophages. The fat-laden macrophages so called foam cells and activated T cells are abundant in the fatty streak, which is the earliest stage of atherosclerosis.
Macrophages and smooth muscle cell (SMC) proliferation and plaque progression
Inflammatory molecules promote further growth of the atherosclerotic plaque. SMCs of the media migrate to the top of the intima to form a fibrous cap over the lipid core. The fibrous cap provides a shield between the thrombogenic material in the plaque’s lipid core and the coagulation factors present in the intima. Foam cells secrete pro-inflammatory cytokines that amplify the local inflammatory response in the lesion and reactive oxygen species. The production of these mediators provides a signal amplification loop between acquired immunity (T cells) and innate immunity (monocyte/macrophages). Engagement of activated T cells and macrophages induces the secretion of tissue factors (TFs), pro-inflammatory cytokines, and matrix metalloproteinases (MMPs) which degrade extracellular matrix necessary for strength of the plaque’s fibrous cap.
Plaque rupture
When the plaque ruptures, it allows the circulating blood to contact the pro-coagulant protein, TF. Eventually macrophages die in a central core in the atherosclerotic plaque by apoptosis or necrosis, resulting in a necrotic core (Glass & Witztum, 2001; Libby, 2002).
IS ATHEROSCLEROSIS A DYNAMIC AND REVERSIBLE PROCESS?
The normal coronary artery has a trilaminar structure (Fig. 2). The ECs are in direct contact with the blood in the lumen over a basement membrane. The intima contains few SMCs within its extracellular matrix. The elastic lamina forms the barrier between the intima and media. SMCs synthesize and secrete elastin polymer that inhibits proliferation and regulates migration of SMCs to the intima (Karnik et al., 2003). The media consists of multiple layers of SMCs tightly packed and embedded in an elastin-rich matrix and collagen. The adventitia is the outermost layer containing connective tissue.
Figure 2. Evolution of atheroma.
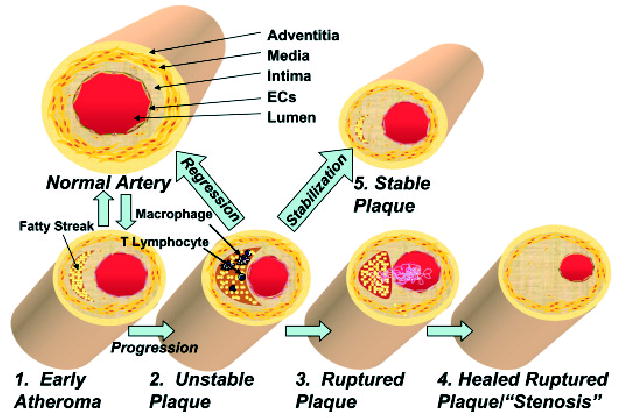
The normal human coronary artery has a trilaminar structure. 1. Early atherosclerotic lesions. In early atherogenesis, migration of immune cells and the accumulation of lipids in macrophages lead to the formation of fatty streaks. These early lesions frequently regress spontaneously. 2. Vulnerable plaque. If inflammatory conditions persist, the lipid core grows. Further activation of macrophages secretes matrix degrading enzymes and weakens the fibrous cap, which leads to vulnerable plaques prone to rupture. 3. Plaque rupture. Disruption of the fibrous cap causes the direct contact of blood components to tissue factor and initiates coagulation. This leads to the formation of the thrombus. 4. Stenosis. A wound healing response stimulates smooth muscle cell migration and collagen synthesis. This process results in a thick fibrous cap and expansion of the intima, which constricts the lumen. 5. Stabilization. Drug treatment or gene therapy for dyslipidemia remodels the nature of the vulnerable plaques, which reduces the incidence of acute coronary events. 6. Regression. Advanced atherosclerotic lesions could be regressed under aggressive lipid lowering or drug treatments.
Early atherosclerotic lesions
In early atherogenesis, migration of immune cells and the accumulation of lipids in macrophages lead to formation of fatty streaks (Libby, 2002; Libby & Aikawa, 2002). In humans, early atherosclerotic lesions are clinically silent, do not necessarily progress to advanced lesions (Stary et al., 1994; 1995; Virmani et al., 2000), and frequently regress spontaneously (Virmani et al., 2000).
Vulnerable plaques
If proinflammatory conditions such as dyslipidemia persist, the lipid core grows. MMPs are secreted by the activated macrophages and degrade the extracellular matrix, while pro-inflammatory cytokines such as INF-γ inhibit collagen synthesis. These changes lead to a thin fibrous cap, large lipid core, and few SMCs, a phenotype of vulnerable plaques prone to rupture.
Plaque rupture
Disruption of the thin fibrous cap causes the direct contact of blood components to TF, initiating coagulation, recruitment of platelets, and ultimately formation of a thrombus. If the thrombus occludes the vessel, an acute myocardial infarction occurs.
Stenosis
Endogenous thrombolysis may eventually dissolve the thrombus. The wound healing response triggered by thrombin produced during blood coagulation stimulates SMC proliferation. Platelet-derived growth factor (PDGF) released from activated platelets stimulates SMC migration and collagen synthesis. This process results in a thick fibrous cap and expansion of the intima, offen in an inward direction. Stenotic lesions may impede blood flow under increased cardiac work demand, leading to ischemia and angina. Symptoms of advanced stenotic plaques are less susceptible to rupture due to the presence of thick fibrous caps.
Stabilization
A clinically important therapeutic goal is to reduce the incidence of acute coronary events. This aim can be accomplished by remodeling the vulnerable plaques qualitatively or functionally. For example, HMG-CoA reductase inhibitors (statins) have been shown to lower LDL cholesterol and stabilize lesions (Libby & Aikawa, 2001; Shah, 2002).
Regression
“Vulnerable plaques” may potentially regress. This extraordinary possibility has been demonstrated in “thoracic aorta transplantation model” (Reis et al., 2001).
EXPERIMENTAL GENE THERAPY FOR DYSLIPIDEMIA VIA LIVER-DIRECTED GENE TRANSFER HAS PROVIDED A MEANS TO STUDY THE DYNAMICS OF ATHEROSCLROTIC LESION DEVELOPMENT
The development of cholesterol-lowering drugs has greatly contributed to current primary and secondary preventions of cardiovascular disease. However, a substantial proportion of patients does not respond adequately to or tolerate these drugs, leaving therapeutic goals unattained. The need for novel therapies for these patients prompted the investigation of somatic gene therapy as an alternative treatment. In gene therapy, a gene delivery vector with high efficiency, specificity, and safety profile is required (Thomas et al., 2003). The development of effective gene transfer vectors has further provided a useful tool to study the role of different circulating proteins and their receptors on the vascular biology of atherosclerosis in experimental animal models (Oka & Chan, 2002; 2004). The most popular vectors for liver-directed gene delivery are adeno-associated virus (AAV) and adenovirus (Ad).
AAV is a non pathogenic virus that encompasses a large number of serotypes. To date, studies on experimental gene therapy using AAV vectors have been based mainly on the use of eight different serotypes. The most commonly used serotype 2 AAV (AAV2) displays a relatively poor transduction efficiency, which severely limits its usefulness. Several AAV receptors and co-receptors have been identified, which include heparan sulphate proteoglycan, integrins, PDGF receptor, and sialic acid. The tissue distribution of these receptors or co-receptors determines cell type- or tissue-specific transduction (Grimm et al., 2003; Burger et al., 2004). A recently isolated AAV8 has been reported to be highly efficient in hepatic transduction (Gao et al., 2002); it efficiently transduces hepatocytes following intravenous administration (Sarkar et al., 2004; Nakai et al., 2005). A drawback of AAV as a gene transfer vector is its relatively small cloning capacity of approx. 4.7 kb. This size limitation can be overcome by an intermolecular joining method using two AAV vectors (Duan et al., 2000), but the value of such a procedure in practice remains to be determined. Nonetheless, AAV remains a popular viral gene delivery system. A major advantage of AAV is its ability to confer long-term transgene expression in vivo, by expressing transgene mainly as an extrachromosomal episome (McCarty et al., 2004).
Helper-dependent adenoviral vector (HDAd) is the latest Ad vector which has an improved safety profile with sustained transgene expression in vivo in comparison with early generation Ads (Kochanek, 1999). It also has a large cloning capacity of up to 37 kb. Despite substantial efforts directed at improving the safety profile and prolonging transgene expression of early generation Ad vectors, the toxicity and immunogenecity of these vectors severely limited their usefulness for liver-directed gene therapy. Like early generation Ads, HDAds are associated with acute toxicity, which appears to be caused by the hosts’ innate immune response (Schnell et al., 2001) (Brunetti-Pierri et al., 2004; Muruve et al., 2004), resulting from the direct interaction of viral particles with innate immune effector cells upon systemic vector administration. Transient depletion of Kupffer cells has been found effective to reduce toxicity as well as increase hepatic transduction efficiency (Schiedner et al., 2003). HDAd vector preferentially target the liver upon intravenous administration, and it is our preferred gene delivery vector in liver-directed experimental gene therapy in small animals.
DYSLIPIDEMIA AND THERAPEUTIC GENES
A number of potential therapeutic genes have been tested for their efficacy in experimental gene therapy for animal models of dyslipidemia. There are two broad mechanisms that affect atherosclerosis associated with dyslipidemia: 1) direct influence over lipoproteins, which includes lowering atherogenic lipoproteins, elevating anti-atherogenic lipoproteins, or facilitating reverse cholesterol transport; and 2) modulate the inflammatory reactions. In addition to the causative genes of dyslipidemia, genes that affect local inflammation in the arterial wall are now considered as potential therapeutic genes. Atherosclerosis is a slowly progressive disease that takes decades to develop in humans. Recent data from long-term experiments show that effects of long-term treatment almost always are different from those obtained from short-term experiments, underscoring the importance of such long-term observations (Oka & Chan, 2004).
INHIBITION OF ATHEROSCLEROSIS PROGRESSION
LDLR- and apoE-deficient mice are popular models of atherosclerosis. Familial hypercholesterolemia (FH) is an autosomal recessive genetic disorder caused by defective LDLR. Despite the development of improved primary and secondary interventions, treatment for homozygous FH is limited and often does not achieve the therapeutic goal (Hopkins, 2003; Naoumova et al., 2004). Therefore, FH is a good target for gene therapy. In contrast to FH, apoE deficiency in humans is a rare genetic disease with a mild phenotype compared with that in mice. This is considered to be due to differences in lipoprotein physiology between two species, notably the lack of cholesteryl ester transfer protein activity and the presence of hepatic apoB mRNA editing activity in mice. ApoE-deficient mice develop atherosclerosis spontaneously on a regular chow diet. Furthermore, it has been shown that in older apoE-deficient mice, atherosclerotic plaques in the brachiocephalic artery show considerable similarity to the vulnerable human plaques (Rosenfeld et al., 2000). Therefore, this model has been used in many studies of atherosclerosis (Meir & Leitersdorf, 2004).
Irrespective of the vector used, many gene therapy studies for dyslipidemia have found retardation of atherosclerotic lesion progression after delivery of appropriate therapeutic genes (Chen et al., 2000; Kawashiri et al., 2001; 2002; Oka et al., 2001; Belalcazar et al., 2003; Mertens et al., 2003; Nomura et al., 2004; Pastore et al., 2004). The most remarkable example showing the efficacy of a HDAd vector was reported in apoE deficient mice (Kim et al., 2001). A single intravenous administration of HDAd containing mouse apoE gene into apoE deficient mice resulted in lifetime correction of hypercholesterolemia. Plasma apoE levels were near normal 2.3 years after treatment. Aorta in control mouse was covered 100% by atherosclerotic lesions, whereas aortas in vector treated mice were essentially lesion-free. Furthermore, this study demonstrated the feasibility of re-administration of HDAd vector to re-stimulate transgene expression. Re-administration of the same HDAd vector is inhibited by generation of neutralizing antibodies against vector capsid proteins. However, these antibodies are serotype-specific, which allows re-administration of the same vector of a different serotype.
First generation Ad vector-mediated transfer of the LDLR gene to the liver of LDLR deficient mice (Ishibashi et al., 1993) or Watanabe heritable hyperlipidemic rabbits (Kozarsky et al., 1994; Li et al., 1995) resulted in significant but transient plasma cholesterol lowering. The transient nature of the lipid lowering after LDLR gene therapy is related in part to an immune response against the induced LDLR expression in mice lacking LDLR (Kozarsky et al., 1996; Chen et al., 2000). Lebherz and coworkers have used AAV vectors with serotype 2 (AAV2), 7 (AAV2/7) and 8 (AAV2/8) cap proteins. They have employed a liver-specific promoter to express the LDLR and have administered these viral constructs into LDLR deficient mice fed a high-fat western diet (Lebherz et al., 2003). They found that serum cholesterol levels were significantly lower in mice treated with AAV2/7 and AAV2/8 twenty weeks after treatment. Furthermore, mice treated with AAV2/7 and AAV 2/8 had 44% and 62% less lesions, respectively, than control mice. AAV2 LDLR treated mice had a 12% inhibition of lesion progression, which was not statistically significant. The success of the modified AAV2/8 is explained in part by the fact that nearly 85% of hepatocytes were transduced by AAV2/8 compared with less than 5% transduction by AAV2.
Our group has shown that HDAd directed anti-atherogenic gene transfer is a powerful way of inhibiting atherosclerosis development/progression both short- and long-term. A 24 week treatment with HDAd-mouse VLDLR inhibited the lesion progression by 87% in LDLR deficient mice fed a high cholesterol diet (Oka et al., 2001). A humoral anti-LDLR developed in a minority of mice treated with the vector. In mice that did not display such a response, LDLR gene therapy was even more effective in cholesterol lowering and protection against atherosclerosis (Nomura et al., 2004). Long-term efficacy of HDAd-LDLR gene therapy is also excellent. When LDLR deficient mice were treated at 12 weeks of age with a low dose of HDAd-LDLR, they responded with lowering of their plasma cholesterol that lasted at least 108 weeks. By that time, all of the control mice had died, and the aorta was heavily covered with thick lesions compared to the scattered lesions found in treated mice. In addition to inhibition of lesion progression, HDAd-LDLR treatment caused lesion remodeling from a vulnerable-looking to a more stable-appearing phenotype with a marked reduction in VCAM-1 expression, well-preserved collagen-rich extracellular matrix, and increased α-actin staining. These results are consistent with the studies that have shown that long-term lipid lowering contributes to plaque stabilization (Aikawa et al., 1998a; 1998b; Verhamme et al., 2002).
REGRESSION OF ADVANCED ATHEROSCLEROTIC LESIONS
Promoting the regression of advanced atherosclerotic lesions or remodeling “vulnerable plaques” to a more stable phenotype undoubtedly is an important therapeutic goal. However, in contrast to inhibition of lesion progression, induction of lesion regression is more difficult to prove experimentally. Tangirara and coworkers have reported that the treatment of LDLR deficient mice with a second generation Ad vector expressing apoA-I resulted in a 70% reduction of pre-existing fatty streaks in en face lesion analysis after only 4 weeks treatment (Tangirala et al., 1999). This is in contrast to our long-term studies. After 24 weeks treatment with HDAd expressing human apoA-I (HDAd-hgAI), LDLR deficient mice having pre-existing advanced lesions showed 50% reduction of lesion progression. The plasma apoA-I levels were maintained at or above normal human levels for the duration of the study. Nevertheless, we found dramatic remodeling of the plaque to a more stable-looking phenotype (Belalcazar et al., 2003).
In addition to apoA-I, there is also considerable interest in apoE as an anti-atherogenic protein (Davignon, 2005). Two papers reported that second generation Ad vector expressing human apoE induced regression of early fatty streaks and advanced complex lesions only after 4–6 weeks treatment in apoE deficient mice (Tsukamoto et al., 1999; Harris et al., 2002). In another study, apoE deficient nude mice were treated with a first generation Ad expressing apoE at 17 weeks old. The authors found regression of fatty streaks 6 months after the treatment (Desurmont et al., 2000).
Given our success in using apoE3, we tested the efficacy of long-term hepatic apoE3 expression on advanced lesions in apoE deficient mice. Mice were fed a high cholesterol diet for 30 weeks to induce atherosclerosis and then treated with HDAd containing human apoE3 gene. After 36 weeks, lesion areas in treated mice had not significantly regressed compared to the baseline group that were sacrificed at vector administration, whereas those in control group had progressed over 340%. However, in spite of similarities in lesion area, the lesion thickness in treated mice was significantly smaller (a 28% reduction) than in baseline mice, suggesting that the induction of lesion regression occurs even in advanced lesions (Paul et al., manuscript in preparation). It should be noted that apoE3 is derived from the liver in this model. In contrast, apoE was derived from both the liver and macrophages in the thoracic aorta transplantation model (Reis et al., 2001).
ApoE could potentially be anti-atherogenic via mechanisms other than cholesterol lowering. This possibility has been tested in LDLR deficient mice. Hepatic apoE3 expression not only reduced lesions (Tsukamoto et al., 2000), but also stimulated a 61% regression after 6-week treatment without affecting plasma cholesterol levels. This lesion regression was accompanied by the reduction of urinary, LDL-associated and arterial wall isoprostane, an index of in vivo oxidant stress (Tangirala et al., 2001).
COMBINATION TREATMENT
Anti-atherogenic proteins and lipid lowering proteins work via different mechanisms. ApoA-I elevates high-density lipoprotein (HDL), which has anti-inflammatory and anti-oxidative effects, and also promotes reverse cholesterol transfer. ApoE may be anti-atherogenic by its diverse functions (e.g., anti-oxidant effect, inhibition of T cell proliferation, inhibition of SMC proliferation and migration, inhibition of platelet aggregation, promotion of reverse cholesterol transport through apoA-I or apoA-I-devoid HDL, etc.) (Davignon, 2005). LDLR works mainly by lowering atherogenic non-HDL lipoproteins. Therefore, there is considerable interest in the outcome of combined therapy. Although hepatic apoE3 expression alone inhibited atherosclerosis progression in LDLR deficient mice, the addition of LDLR with apoE3 did not show an additive or synergistic effect on either cholesterol lowering or atherosclerosis after 6-weeks treatment (Kawashiri et al., 2001). Similarly, the atherosclerotic lesion progression was reduced by LDLR or apoA-I single treatment, but the effects of combined treatment was not greater than those of a single gene treatment (Kawashiri et al., 2002). It should be noted that the expression of therapeutic genes was transient and the treatment lasted only for 6 weeks in both studies.
We hypothesized that long-term stable gene expression could allow more significant effects. LDLR deficient mice were fed a high cholesterol diet for 36 weeks and then treated with HDAd-hgAI or HDAd-LDLR vector alone or in combination. Atherosclerotic lesion area was measured by quantitative morphometry 28 weeks after the treatment. HDAd-hgAI treatment inhibited the lesion progression, but mice treated with HDAd-LDLR alone or combination induced lesion regression (Chao et al., manuscript in preparation). These results indicate that HDAd-mediated LDLR gene therapy is highly effective in inhibition of lesion progression and in inducing atherosclerotic lesion regression.
The progressive accumulation of macrophages and other immune cells in the atherosclerotic plaques is one of the hallmarks in atherosclerosis. Recruitment of monocytes into the intima and their retention there contributes to the progression of plaques (Llodra et al., 2004). Many studies including our results found that regression and remodeling of atherosclerotic lesions are associated with reduced macrophages in the lesions. The mechanism of macrophage retention and potential reverse transmigration into blood flow has not been well understood. Polansky has recently proposed a mathematical model which might explain cell motility in atherosclerotic lesion (Polansky, 2003).
CONCLUSIONS
Gene therapy was introduced with great expectations as the latest frontier in gene medicine. Despite the initial optimism, the field has experienced some setbacks. In the areas of dyslipidemia, the progress in liver-directed gene delivery has proved invaluable in studying vascular biology and the pathogenesis of atherosclerosis. Data on the long-term effects of gene delivery support the feasibility of achieving a clinical goal in stabilization or regression of vulnerable atherosclerotic plaques by a gene therapy approach. Although additional work is needed, the field is slowly but relentlessly moving toward fulfilling its promise made decades ago.
Acknowledgments
The work performed in the authors’ laboratories described in this review was supported by US National Institutes of Health Grant HL-59314 and HL-73144.
We thank A. Paul, K. Ko, H. Chao, S. Cormier, and E. Nour for their assistance in the study; S. Samson for helpful comments on the manuscript.
Footnotes
Presented as invited lecture at the XXXII Winter School, 3–7 March 2005, Zakopane, Poland.
References
- Aikawa M, Rabkin E, Okada Y, Voglic SJ, Clinton SK, Brinckerhoff CE, Sukhova GK, Libby P. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation. 1998a;97:2433–2444. doi: 10.1161/01.cir.97.24.2433. [DOI] [PubMed] [Google Scholar]
- Aikawa M, Rabkin E, Voglic SJ, Shing H, Nagai R, Schoen FJ, Libby P. Lipid lowering promotes accumulation of mature smooth muscle cells expressing smooth muscle myosin heavy chain isoforms in rabbit atheroma. Circ Res. 1998b;83:1015–1026. doi: 10.1161/01.res.83.10.1015. [DOI] [PubMed] [Google Scholar]
- Aikawa M, Voglic SJ, Sugiyama S, Rabkin E, Taubman MB, Fallon JT, Libby P. Dietary lipid lowering reduces tissue factor expression in rabbit atheroma. Circulation. 1999;100:1215–1222. doi: 10.1161/01.cir.100.11.1215. [DOI] [PubMed] [Google Scholar]
- Armstrong ML, Megan MB. Arterial fibrous proteins in cynomolgus monkeys after atherogenic and regression diets. Circ Res. 1975;36:256–261. doi: 10.1161/01.res.36.2.256. [DOI] [PubMed] [Google Scholar]
- Belalcazar LM, Merched A, Carr B, Oka K, Chen KH, Pastore L, Beaudet A, Chan L. Long-term stable expression of human apolipoprotein A-I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation. 2003;107:2726–2732. doi: 10.1161/01.CIR.0000066913.69844.B2. [DOI] [PubMed] [Google Scholar]
- Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170:191–203. doi: 10.1016/s0021-9150(03)00097-2. [DOI] [PubMed] [Google Scholar]
- Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N, Palmer DJ, Beaudet AL, Carey KD, Fine-gold M, Ng P. Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates. Hum Gene Ther. 2004;15:35–46. doi: 10.1089/10430340460732445. [DOI] [PubMed] [Google Scholar]
- Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10:302–317. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- Chen SJ, Rader DJ, Tazelaar J, Kawashiri M, Gao G, Wilson JM. Prolonged correction of hyperlipidemia in mice with familial hypercholesterolemia using an adeno-associated viral vector expressing very-low-density lipoprotein receptor. Mol Ther. 2000;2:256–261. doi: 10.1006/mthe.2000.0122. [DOI] [PubMed] [Google Scholar]
- Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. 1992;140:301–316. [PMC free article] [PubMed] [Google Scholar]
- Davignon J. Apolipoprotein E and atherosclerosis: beyond lipid effect. Arterioscler Thromb Vasc Biol. 2005;25:267–269. doi: 10.1161/01.ATV.0000154570.50696.2c. [DOI] [PubMed] [Google Scholar]
- Desurmont C, Caillaud JM, Emmanuel F, Benoit P, Fruchart JC, Castro G, Branellec D, Heard JM, Duverger N. Complete atherosclerosis regression after human ApoE gene transfer in ApoE-deficient/nude mice. Arterioscler Thromb Vasc Biol. 2000;20:435–442. doi: 10.1161/01.atv.20.2.435. [DOI] [PubMed] [Google Scholar]
- Duan D, Yue Y, Yan Z, Engelhardt JF. A new dual-vector approach to enhance recombinant adeno-associated virus-mediated gene expression through intermolecular cis activation. Nat Med. 2000;6:595–598. doi: 10.1038/75080. [DOI] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Grimm D, Kay MA, Kleinschmidt JA. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol Ther. 2003;7:839–850. doi: 10.1016/s1525-0016(03)00095-9. [DOI] [PubMed] [Google Scholar]
- Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- Hansson GK. Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol. 2001;21:1876–1890. doi: 10.1161/hq1201.100220. [DOI] [PubMed] [Google Scholar]
- Harris JD, Graham IR, Schepelmann S, Stannard AK, Roberts ML, Hodges BL, Hill V, Amalfitano A, Hassall DG, Owen JS, Dickson G. Acute regression of advanced and retardation of early aortic atheroma in immunocompetent apolipoprotein-E (apoE) deficient mice by administration of a second generation [E1(-), E3(-), polymerase(-)] adenovirus vector expressing human apoE. Hum Mol Genet. 2002;11:43–58. doi: 10.1093/hmg/11.1.43. [DOI] [PubMed] [Google Scholar]
- Hoffman M, Blum A, Baruch R, Kaplan E, Benjamin M. Leukocytes and coronary heart disease. Atherosclerosis. 2004;172:1–6. doi: 10.1016/s0021-9150(03)00164-3. [DOI] [PubMed] [Google Scholar]
- Hopkins PN. Familial hypercholesterolemia — improving treatment and meeting guidelines. Int J Cardiol. 2003;89 :13–23. doi: 10.1016/s0167-5273(02)00420-5. [DOI] [PubMed] [Google Scholar]
- Ishibashi S, Inaba T, Shimano H, Harada K, Inoue I, Mokuno H, Mori N, Gotoda T, Takaku F, Yamada N. Monocyte colony-stimulating factor enhances uptake and degradation of acetylated low density lipoproteins and cholesterol esterification in human monocyte-derived macrophages. J Biol Chem. 1990;265:14109–14117. [PubMed] [Google Scholar]
- Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883–893. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130:411–423. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- Kawashiri M, Zhang Y, Usher D, Reilly M, Pure E, Rader DJ. Effects of coexpression of the LDL receptor and apoE on cholesterol metabolism and atherosclerosis in LDL receptor-deficient mice. J Lipid Res. 2001;42:943–950. [PubMed] [Google Scholar]
- Kawashiri MA, Zhang Y, Pure E, Rader DJ. Combined effects of cholesterol reduction and apolipoprotein A-I expression on atherosclerosis in LDL receptor deficient mice. Atherosclerosis. 2002;165:15–22. doi: 10.1016/s0021-9150(02)00103-x. [DOI] [PubMed] [Google Scholar]
- Kim IH, Jozkowicz A, Piedra PA, Oka K, Chan L. Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc Natl Acad Sci USA. 2001;98:13282–13287. doi: 10.1073/pnas.241506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek S. High-capacity adenoviral vectors for gene transfer and somatic gene therapy. Hum Gene Ther. 1999;10:2451–2459. doi: 10.1089/10430349950016807. [DOI] [PubMed] [Google Scholar]
- Kockx MM, De Meyer GR, Buyssens N, Knaapen MW, Bult H, Herman AG. Cell composition, replication, and apoptosis in atherosclerotic plaques after 6 months of cholesterol withdrawal. Circ Res. 1998;83:378–387. doi: 10.1161/01.res.83.4.378. [DOI] [PubMed] [Google Scholar]
- Kozarsky KF, Mckinley DR, Austin LL, Raper SE, Stratford-Perricaudet LD, Wilson JM. In vivo correction of low density lipoprotein receptor deficiency in the Watanabe heritable hyperlipidemic rabbit with recombinant adenoviruses. J Biol Chem. 1994;269:13695–13702. [PubMed] [Google Scholar]
- Kozarsky KF, Jooss K, Donahee M, Strauss JF, 3rd, Wilson JM. Effective treatment of familial hypercholesterolaemia in the mouse model using adenovirus-mediated transfer of the VLDL receptor gene. Nat Genet. 1996;13 :54–62. doi: 10.1038/ng0596-54. [DOI] [PubMed] [Google Scholar]
- Lebherz C, Gao G, Louboutin J, Millar J, Rader DJ, Wilson J. AAV2/7 and AAV2/8 mediated liver directed gene therapy enables long-term expression of the human LDL receptor and substantially diminishes atherosclerosis in LDL receptor deficient mice. Circulation. 2003;108:IV–143. [Google Scholar]
- Li J, Fang B, Eisensmith RC, Li XH, Nasonkin I, Lin-Lee YC, Mims MP, Hughes A, Montgomery CD, Roberts JD, et al. In vivo gene therapy for hyperlipidemia: phenotypic correction in Watanabe rabbits by hepatic delivery of the rabbit LDL receptor gene. J Clin Invest. 1995;95:768–773. doi: 10.1172/JCI117725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Libby P, Aikawa M. Evolution and stabilization of vulnerable atherosclerotic plaques. Jpn Circ J. 2001;65:473–479. doi: 10.1253/jcj.65.473. [DOI] [PubMed] [Google Scholar]
- Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med. 2002;8:1257–1262. doi: 10.1038/nm1102-1257. [DOI] [PubMed] [Google Scholar]
- Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci USA. 2004;101:11779–11784. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty DM, Young SM, Jr, Samulski RJ. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet. 2004;38:819–845. doi: 10.1146/annurev.genet.37.110801.143717. [DOI] [PubMed] [Google Scholar]
- Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein-E-deficient mouse: a decade of progress. Arterioscler Thromb Vasc Biol. 2004;24:1006–1014. doi: 10.1161/01.ATV.0000128849.12617.f4. [DOI] [PubMed] [Google Scholar]
- Mertens A, Verhamme P, Bielicki JK, Phillips MC, Quarck R, Verreth W, Stengel D, Ninio E, Navab M, Mackness B, Mackness M, Holvoet P. Increased low-density lipoprotein oxidation and impaired high-density lipoprotein antioxidant defense are associated with increased macrophage homing and atherosclerosis in dyslipidemic obese mice: LCAT gene transfer decreases atherosclerosis. Circulation. 2003;107:1640–1646. doi: 10.1161/01.CIR.0000056523.08033.9F. [DOI] [PubMed] [Google Scholar]
- Muruve DA, Cotter MJ, Zaiss AK, White LR, Liu Q, Chan T, Clark SA, Ross PJ, Meulenbroek RA, Maelandsmo GM, Parks RJ. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J Virol. 2004;78:5966–5972. doi: 10.1128/JVI.78.11.5966-5972.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay MA. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol. 2005;79:214–224. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoumova RP, Thompson GR, Soutar AK. Current management of severe homozygous hypercholesterolaemias. Curr Opin Lipidol. 2004;15:413–422. doi: 10.1097/01.mol.0000137222.23784.2a. [DOI] [PubMed] [Google Scholar]
- Nilsson J, Hansson GK, Shah PK. Immunomodulation of atherosclerosis: implications for vaccine development. Arterioscler Thromb Vasc Biol. 2005;25:18–28. doi: 10.1161/01.ATV.0000149142.42590.a2. [DOI] [PubMed] [Google Scholar]
- Nomura S, Merched A, Nour E, Dieker C, Oka K, Chan L. Low-density lipoprotein receptor gene therapy using helper-dependent adenovirus produces long-term protection against atherosclerosis in a mouse model of familial hypercholesterolemia. Gene Ther. 2004;11:1540–1548. doi: 10.1038/sj.gt.3302310. [DOI] [PubMed] [Google Scholar]
- Oka K, Chan L. Recent advances in liver-directed gene therapy for dyslipidemia. Curr Atheroscler Rep. 2002;4:199–207. doi: 10.1007/s11883-002-0020-8. [DOI] [PubMed] [Google Scholar]
- Oka K, Chan L. Liver-directed gene therapy for dyslipidemia and diabetes. Curr Atheroscler Rep. 2004;6:203–209. doi: 10.1007/s11883-004-0033-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka K, Pastore L, Kim IH, Merched A, Nomura S, Lee HJ, Merched-Sauvage M, Arden-Riley C, Lee B, Finegold M, Beaudet A, Chan L. Long-term stable correction of low-density lipoprotein receptor-deficient mice with a helper-dependent adenoviral vector expressing the very low-density lipoprotein receptor. Circulation. 2001;103:1274–1281. doi: 10.1161/01.cir.103.9.1274. [DOI] [PubMed] [Google Scholar]
- Pastore L, Belalcazar LM, Oka K, Cela R, Lee B, Chan L, Beaudet AL. Helper-dependent adenoviral vector-mediated long-term expression of human apolipoprotein A-I reduces atherosclerosis in apo E-deficient mice. Gene. 2004;327:153–160. doi: 10.1016/j.gene.2003.11.024. [DOI] [PubMed] [Google Scholar]
- Polansky H (2003). Microcompetition with Foreign DNA and the Origin of Chronic Disease. The Center for the Biology and Chronic Disease. Rochester.
- Raffai RL, Loeb SM, Weisgraber KH. Apolipoprotein E promotes the regression of atherosclerosis independently of lowering plasma cholesterol levels. Arterioscler Thromb Vasc Biol. 2005;25:436–441. doi: 10.1161/01.ATV.0000152613.83243.12. [DOI] [PubMed] [Google Scholar]
- Reis ED, Li J, Fayad ZA, Rong JX, Hansoty D, Aguinaldo JG, Fallon JT, Fisher EA. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein E-deficient mouse in a novel transplantation model. J Vasc Surg. 2001;34:541–547. doi: 10.1067/mva.2001.115963. [DOI] [PubMed] [Google Scholar]
- Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587–2592. doi: 10.1161/01.atv.20.12.2587. [DOI] [PubMed] [Google Scholar]
- Sarkar R, Tetreault R, Gao G, Wang L, Bell P, Chandler R, Wilson JM, Kazazian HH., Jr Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood. 2004;103:1253–1260. doi: 10.1182/blood-2003-08-2954. [DOI] [PubMed] [Google Scholar]
- Schiedner G, Hertel S, Johnston M, Dries V, Van Rooijen N, Kochanek S. Selective depletion or blockade of Kupffer cells leads to enhanced and prolonged hepatic transgene expression using high-capacity adeno-viral vectors. Mol Ther. 2003;7:35–43. doi: 10.1016/s1525-0016(02)00017-5. [DOI] [PubMed] [Google Scholar]
- Schnell MA, Zhang Y, Tazelaar J, Gao GP, Yu QC, Qian R, Chen SJ, Varnavski AN, Leclair C, Raper SE, Wilson JM. Activation of innate immunity in nonhuman primates following intraportal administration of adeno-viral vectors. Mol Ther. 2001;3:708–722. doi: 10.1006/mthe.2001.0330. [DOI] [PubMed] [Google Scholar]
- Shah PK. Low-density lipoprotein lowering and atherosclerosis progression: does more mean less? Circulation. 2002;106:2039–2040. doi: 10.1161/01.cir.0000037742.25731.3d. [DOI] [PubMed] [Google Scholar]
- Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. 1994;14:840–856. doi: 10.1161/01.atv.14.5.840. [DOI] [PubMed] [Google Scholar]
- Tangirala RK, Pratico D, Fitzgerald GA, Chun S, Tsukamoto K, Maugeais C, Usher DC, Pure E, Rader DJ. Reduction of isoprostanes and regression of advanced atherosclerosis by apolipoprotein E. J Biol Chem. 2001;276:261–266. doi: 10.1074/jbc.M003324200. [DOI] [PubMed] [Google Scholar]
- Tangirala RK, Tsukamoto K, Chun SH, Usher D, Pure E, Rader DJ. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation. 1999;100:1816–1822. doi: 10.1161/01.cir.100.17.1816. [DOI] [PubMed] [Google Scholar]
- Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- Tsukamoto K, Tangirala R, Chun SH, Pure E, Rader DJ. Rapid regression of atherosclerosis induced by liver-directed gene transfer of ApoE in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:2162–2170. doi: 10.1161/01.atv.19.9.2162. [DOI] [PubMed] [Google Scholar]
- Tsukamoto K, Tangirala RK, Chun S, Usher D, Pure E, Rader DJ. Hepatic expression of apolipoprotein E inhibits progression of atherosclerosis without reducing cholesterol levels in LDL receptor-deficient mice. Mol Ther. 2000;1:189–194. doi: 10.1006/mthe.2000.0028. [DOI] [PubMed] [Google Scholar]
- Verhamme P, Quarck R, Hao H, Knaapen M, Dymarkowski S, Bernar H, Van Cleemput J, Janssens S, Vermylen J, Gabbiani G, Kockx M, Holvoet P. Dietary cholesterol withdrawal reduces vascular inflammation and induces coronary plaque stabilization in miniature pigs. Cardiovasc Res. 2002;56:135–144. doi: 10.1016/s0008-6363(02)00515-1. [DOI] [PubMed] [Google Scholar]
- Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]