

Abstract
Isolates of human immunodeficiency virus type 1 (HIV-1) are classified according to the chemokine receptor (coreceptor) used in conjunction with CD4 to target and enter cells: viruses using CCR5 and CXCR4 are classified as R5 and X4, respectively. The major determinant of entry-related HIV-1 phenotypes is known to reside in the third variable region of gp120 (V3). It is clear, however, that positions outside of V3 play some role in influencing phenotype, although marked context dependence and extensive variability among HIV-1 isolates have made the identification of these positions difficult. We used the presence of previously described substitutions in V3 to classify a large set of HIV-1 subtype B gp120 sequences available in public databases as X4-like or R5-like. Using these classifications, we searched for positions outside of V3 where either amino acid composition or variability differed significantly among sequences of different inferred phenotypes. Our approach took the epidemiological relationships among sequences into account. A cluster of positions linked to changes in V3 was identified between amino acids 190 and 204 of gp120, immediately C-terminal of V2; changes at position 440 in C4 were also linked to inferred phenotype. Structural data place these positions at the coreceptor-binding face of gp120 in a surface-exposed location. We also noted a significant increase in net positive charge in a highly variable region of V2. This study both confirms previous observations and predicts specific positions that contribute to a functional relationship between V3, V2, and C4.
Isolates of human immunodeficiency virus type 1 (HIV-1) are classified according to their use of different cellular receptors, or coreceptors, in conjunction with CD4 for virus binding and entry (1, 14, 21, 23, 24, 28). Viruses using the seven-transmembrane chemokine receptor CCR5, CXCR4, or both are termed R5, X4, and R5X4, respectively (2). Other receptors have been implicated in virus entry in vitro, but their relevance in vivo has not been well documented, and all viruses at a minimum use CCR5 or CXCR4 (23, 33, 68, 81).
Strains of HIV-1 are also classified based on their ability to induce the formation of multinucleated giant cells (syncytia) in MT-2 cells: viruses that display this cytopathic effect are termed syncytium-inducing (SI); those that do not are called non-syncytium-inducing (NSI) (62, 71). X4 viruses are frequently SI, and MT-2 cell tropism has been shown to be an entry-related phenotype that is largely determined by coreceptor preference (5). Strong selection either during or shortly after transmission results in the predominance of R5 variants early in infection (17, 58, 82). Variants able to use CXCR4 ultimately appear in approximately one-half of HIV-infected individuals, and their detection has been associated with an accelerated loss of CD4+ T cells and an increased likelihood of progressing to AIDS (15, 39, 55).
The surface subunit of the HIV-1 Env glycoprotein, gp120, controls entry-related phenotype, though postentry factors may also play a role in determining cell tropism. The gp120 coding domain of env has been divided into alternating constant and variable regions, referred to as C1 through C5 and V1 through V5, respectively (70). The variable regions lie mostly within regions encoding disulfide-constrained, surface-exposed loops (45). It is clear, however, that considerable differences in position-by-position variability exist without respect to the boundaries of these regions (80).
The determinants of coreceptor usage (4, 33, 69, 73, 77) and MT-2 cell tropism (8, 12, 19, 35, 52, 66, 74, 76) lie largely within the 35 amino acids of V3. However, changes in this region alone are not always necessary or sufficient to confer a particular phenotype in viruses expressing engineered gp120 proteins, since changes in other regions of gp120, particularly in V1/V2 (6, 13, 37, 38, 59) and C4 (9), have been shown to influence phenotype either alone or in conjunction with V3. In addition, isolates with identical V3 sequences can have dissimilar patterns of coreceptor usage, cell tropism, or replication capacity (31, 37, 38, 53). Typically, specific changes in these other regions do not have consistent effects in a wide range of sequence contexts.
Although phenotype-associated variability in V3 has been studied extensively using statistical approaches (3, 40, 47, 48), other regions of gp120 have escaped similar scrutiny, due in part to the relative paucity of sequence information corresponding to isolates of known phenotype. Specific amino acid substitutions within V3 that are associated with different entry phenotypes can, however, be used to predict phenotype with reasonable success. In particular, previous studies have determined that the presence of at least one basic substitution at V3 position 11 or 25 (HXB gp160 306 and 322, respectively) is associated with the X4, R5X4, and SI phenotypes (18, 29). We have recently observed that this 11/25 rule is the most reliable of the available motif-based methods for predicting the X4/SI phenotype (54).
Here we have further refined this prediction method, resulting in an improved phenotypic classification scheme based on V3 sequence. Throughout this paper, inferred phenotypes are indicated in lowercase letters (x4 and r5), and experimentally determined phenotypes are indicated in uppercase letters (X4, R5, SI, and NSI).
We have searched for positions in gp120 that are linked to entry phenotype-associated changes in V3 in a large set of gp120 sequences obtained from the Los Alamos HIV Database (http://hiv-web.lanl.gov) spanning the V1 to C4 regions. After controlling for epidemiological and phylogenetic relationships among the sequences, we identified 15 positions in gp120 but outside of V3 that have significantly different variability or representation of specific amino acids among x4 and r5 sequences (i.e., sequences of different inferred phenotype). Six of these fall between positions 190 and 204 at the C-terminal end of V2. In addition, we have found that in x4 sequences, a significant accumulation of basic substitutions occurs nearby within V2, in a region of marked variability and length polymorphism (HXB positions 180 to 189).
The identified positions are especially attractive candidates for influencing gp120 function due to their hypothesized localization at the coreceptor binding face and oligomer interface in the gp120 trimer, their proximity to one another, and the nature of the specific changes at each position. Furthermore, several of these positions have also been described previously as playing a role in defining gp120 function.
MATERIALS AND METHODS
Sequence alignment and acquisition.
All available HIV-1 subtype B nucleotide sequences spanning either of two overlapping regions of env were downloaded from the Los Alamos HIV database (http://hiv-web.lanl.gov): V3 to C4 (V3C4), and V1 to V3 (V1V3) (Table 1). Sequences either lacking a continuous open reading frame or with more than two ambiguous codons were discarded. Multiple sequence alignments were constructed as follows. Amino acid translations of a randomly chosen subset of 50 sequences representing each region were aligned using ClustalW (72), followed by manual modification. Profiles created from each of these alignments were used to align the entire set of translated sequences corresponding to each region. Profile creation and sequence alignment were performed using Hmmbuild and Hmmalign (25). Positions in the resulting alignments containing gap characters in over 90% of sequences were discarded. The potential role of length variability in the variable regions was considered in a separate analysis.
TABLE 1.
Clade B env sequences available from the HIV database
| Parameter | V1V3 | V3C4 |
|---|---|---|
| Nucleotidesa | 6616-7217 | 7114-7589 |
| Amino acidsa | 131-331 | 296-456 |
| Length (amino acids) | 200 | 160 |
| No. of sequencesb | 1,198 | 3,519 |
| Total X4c | 232 | 273 |
| Total r5d | 934 | 3,134 |
HXB numbering.
Includes sequences with complete open reading frames for which epidemiological information was available.
R or K at V3 position 11 or K at position 25.
No basic amino acid at V3 position 11 or 25.
Data set construction.
Two sampling methods were used to choose subsets of unrelated sequences. First, one set each of V1V3 and V3C4 sequences was created by randomly choosing no more than one X4-like and one R5-like sequence representing each HIV-1-infected subject (set 1); multiple representatives of closely linked chains of transmission were treated as originating from the same individual. This was accomplished by using a database classifying sequences by patient of origin based mainly on annotations of GenBank references by Brian Foley of the Los Alamos HIV Sequence Database (available upon request).
For the second sampling method (set 2), V1V3 and V3C4 sequence sets were resampled separately 30 times to create subsets defined by both subject of origin and sequence similarity as follows. Nucleotide alignments were created by replacing each amino acid in the protein alignments described above with the corresponding codon from the nucleotide sequence. Aligned positions at which gap characters appeared in more than 97% of sequences were deleted. All pairwise distances between aligned nucleotide sequences were calculated with Dnadist (PHYLIP version 3.6α) according to the Jin-Nei method with the coefficient of variation = 1/a1/2 = 1, where a is the parameter of the gamma distribution (27). The resulting distance matrix was used to select groups of sequences in which no two sequences had a pairwise similarity score greater than 94 (where similarity = 100 − distance). The similarity value of 94 was chosen by determining the highest possible cutoff value that did not result in a significant number of sequence clusters (implying epidemiological linkage) supported by at least 50% of bootstrap replicates in neighbor-joining trees of representative subsets (see “Phylogenetic trees” below).
Approximately 57 and 40% of pairwise comparisons among V1V3 and V3C4 sequences, respectively, classified as originating in the same individual had scores of ≤94. Fewer than 1% of comparisons among unrelated sequences spanning either V1V3 or V3C4 had scores of >94. To prevent intrasubject sequences from being included in the same subset, these groups of sequences were also filtered using the patient database described above. As a result, each of the 30 subsets contained no two sequences which had either a pairwise similarity score of >94 or that were isolated from the same subject. The software used to perform this filtering process is available by request.
Phylogenetic trees.
To visualize the extent of relatedness among sequences in the sets constructed above, one of each of the 30 subsets of V1V3 and V3C4 nucleotide sequences were used to build phylogenetic trees. Sequences were codon-aligned as described above, and positions containing gap characters in >1% of entries were disregarded. Neighbor-joining trees were constructed, and bootstrap analysis was performed using PAUPSearch (GCG Wisconsin Package). Distances were calculated according to the general time-reversible model, with substitution rate variation across sites following the gamma distribution. The gamma shape parameter was set to alpha = 0.38, as estimated by Leitner et al. (44). Branches appearing in at least 50% of 100 bootstrap replicates were retained. Trees constructed using values of alpha ranging from 0.20 to 0.70 did not vary substantially in the number of supported clusters.
Sequence classification.
gp120 sequences were classified as either X4-like (x4) or R5-like (r5) based on amino acid composition at positions 11 and 25 of the V3 loop (HXB positions 306 and 322). Sequences with R or K at 11 and/or K at 25 were considered x4, and sequences with no basic amino acid at 11 and 25 were considered r5. Arginine at 25 does not discriminate between the two phenotypes, so sequences with 25R were not included in the analysis. Note that lowercase designations (x4 and r5) refer to inferred phenotypes, while uppercase letters (X4 and R5) denote experimentally determined coreceptor usage.
To determine the accuracy of this method for predicting MT-2 cell tropism and coreceptor usage, we classified sets of V3 sequences of known phenotype. In this test, the X4 reliability (positive predictive value) and R5 reliability (negative predictive value) were calculated using Bayes' theorem. Adjusted values for reliability were calculated using an estimated X4 prevalence in the database of 15% (54). The sequence sets used to perform this analysis as well as the methods used were described by Resch et al. (54).
Statistical analysis.
Three different measures were used to assess the difference in amino acid composition between x4 and r5 sequences. First, diversity of amino acids (D) was used as described by Yamaguchi-Kabata and Gojobori (80) to evaluate the extent of variability at each position in a population of HIV Env sequences and is computed as:
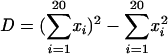 |
where xi is the frequency of amino acid i at a given position. Gaps are not included in this expression. Second, as another measure of variability, we calculated the Shannon entropy for each aligned position (64):
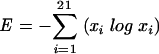 |
Entropy (E) measures how evenly represented the amino acids are; thus, a position with many amino acids equally represented will achieve a high entropy score (here gap characters and X are collapsed into a single, 21st, character in the alignment). Maximal hypothetical values for these two tests occur when all amino acids appear in equal proportions (Dmax = 0.95, Emax = 1.366); a minimum score of 0 results for both tests at a position with 100% conservation. In this study, we were interested in the difference in variability between x4 and r5 sequences at each position j in an alignment. This difference was calculated as either ΔDj= Dj(x4) − Dj(r5) or ΔEj = Ej(x4) − Ej(r5).
We used permutation tests to calculate probabilities that the above statistics described chance differences between x4 and r5 sequences. Let the diversity (ΔDj) and entropy (ΔEj) scores at each position be represented by a generic summary statistic, Sj. For each position j, a reference distribution of R = 1,000 scores for set Ak and Bk (k = 1, ... R) was created by randomly casting sequences into groups A and B, such that P(Ak) = P(x4) and P(Bk) = P(r5), and calculating a new summary statistic, S′jk. The probability that the summary statistic Sj describes a chance difference between x4 and r5 sequences can be estimated as P ≤ N/R, where N is the smaller of the two values NL (number of events in which Sj ≤ S′jk) and NG (number of events in which Sj ≥ S′jk), 1 ≤ k ≤ R. This results in a one-tailed test, where the P value is always calculated from the smaller tail. Put simply, the permutation test counts the number of (simulated) chance regroupings of sequences that results in a difference equal to or more extreme than the difference resulting from the classification of sequences as x4 and r5. If Sj fell outside the support of its reference distribution (i.e., N = 0), a P value of <0.001 (1/R) was assigned.
For the third measure of amino acid composition, a similar procedure was used to describe the representation of specific amino acids among x4 and r5 sequences. A binomial test of statistical significance was used to generate a Z score, Zij, describing the difference in the proportion of x4 and r5 sequences containing a given amino acid, i, at each aligned position, j (thus, there are potentially 21 such Z scores for each position: one for each amino acid, and one for the gap character). As described above, reference distributions of Z scores created by repartitioning the data sets (i.e., Z′ijk for 1 ≤ k ≤ R) were used to calculate a P value describing the probability that the observed difference in frequency of each amino acid at a position among x4 and r5 sequences was due to chance (thus, 21 reference distributions were created for each position). At each position, test results were retained only for amino acids representing at least 10% of either x4 or r5 sequences.
Logistic regression analysis.
We assembled sets of unaligned protein sequence fragments corresponding to the hypervariable regions of V1, V2, and V4 (amino acids 131 to 157, 180 to 189, and 386 to 413, respectively). Subsets of sequences used in the analysis corresponded exactly to those used in the tests described above (e.g., V1V3-set 1 and the 30 subsets of unrelated V1V3 sequences). Values describing net charge (R + K − D − E) and length in amino acids were assigned to each sequence fragment. Logistic regression analysis was used to correlate inferred phenotype with each of these two sequence descriptors. A univariate regression was performed for each of these descriptors.
RESULTS
Sequence acquisition and alignment.
As a first test of the hypothesis that sequence changes outside the third variable region (V3) of HIV-1 gp120 are linked to entry phenotype, we searched for V3-linked changes at well-aligned amino acid positions in subtype B sequences spanning two overlapping regions of the env gene: V1 to V3 (V1V3) and V3 to the fourth constant region (V3C4) (Table 1). We chose these commonly sequenced regions because they include most positions associated with CCR5 binding and were well represented in the HIV database.
Alignment of certain regions was not possible due to extensive length polymorphism, including hypervariable regions of V1 (positions 134 to 152) and V2 (positions 185 to 189), loop E in C3 (positions 354 to 357), and V4 (positions 396 to 413) (HXB gp160 numbering is used throughout this paper except for V3, which is numbered 1 to 35, starting at the first cysteine). These regions were not included in the analysis of individual amino acid variability and were considered separately for length variability and charge using a different strategy (see below).
Genotypic classification of gp120 sequences.
The great majority of available gp120 sequences are not associated with an experimentally determined phenotype for either growth in MT-2 cells or coreceptor usage. One classification scheme that has been used as a surrogate for phenotype designates sequences with basic residues at V3 positions 11 and 25 (HXB positions 306 or 322) as X4-like or SI-like (x4) and those with no basic residues at 11 or 25 as R5-like or NSI-like (r5) (20, 29, 30, 63).
Using V3 sequences of known phenotype, we previously determined that this 11/25 rule used as a test has an estimated reliability of 0.85 and 0.48 for the SI and X4 phenotypes, respectively, and was the best published method available for predicting phenotype (54). However, the presence of 25R alone is nondiscriminating for both the SI/NSI and X4/R5 phenotypes (54). Therefore, to improve the accuracy of this method, we excluded all sequences with arginine at V3 position 25 and no basic residue at 11, resulting in the rejection of 4 and 5% of available V1V3 and V3C4 sequences, respectively. We have termed this classification method the 11KR/25K rule.
We evaluated the accuracy of these criteria as described by Resch et al. (54). Briefly, using sets of HIV-1 V3 sequences of known NSI/SI or X4/R5 phenotype and subject of origin, we selected 100 subsets in which each subject was represented by at most one sequence of each phenotype. A 2 × 2 contingency table describing the success of the sequence classification was determined for each subset; summaries of the resulting 100 contingency tables are shown in Table 2. Compared to the 11/25 rule, we observed a reduction in R5 and NSI sequences misclassified as x4 or SI, resulting in an improvement in the specificity of the test, or the fraction of correctly classified R5/NSI sequences. Because the reliability (positive and negative predictive values) of this test also depends on the prevalence of sequences in the database having the SI or X4 phenotype, we calculated these values using both the prevalence of X4 or SI sequences in the average 2 × 2 table and an estimated prevalence of X4/SI sequences in the database of 15% (54). The resulting estimate for the X4 reliability of the 11KR/25K rule is a substantially improved 0.96 to 0.98 and 0.65 to 0.78 for the SI and X4 phenotypes, respectively.
TABLE 2.
Success of the 11KR/25K rule for phenotypic classification
| Pheno- type | Contingency tablea
|
Sensitivity | Specificity | x4 reliabilityb | Adjusted x4 reliabilityc | r5 reliabilityb | Adjusted r5 reliabilityc | |
|---|---|---|---|---|---|---|---|---|
| X4/SI | R5/NSI | |||||||
| x4 | 14.2 (13-15) | 4.0 (4-4) | 0.49 (0.45-0.52) | 0.95 (0.95-0.95) | 0.78 (0.76-0.79) | 0.65 (0.63-0.67) | 0.85 (0.84-0.86) | 0.91 (0.91-0.92) |
| r5 | 14.8 (14-16) | 83.0 (83-83) | ||||||
| SI | 28.6 (28-29) | 0.6 (0-2) | 0.82 (0.80-0.83) | 0.99 (0.98-1.0) | 0.98 (0.93-1.0) | 0.96 (0.85-1.0) | 0.93 (0.92-0.93) | 0.97 (0.97-0.97) |
| NSI | 6.4 (6-7) | 82.4 (81-83) | ||||||
Average of 100 subsets of V3 sequences of known phenotype chosen so that no more than one sequence of either phenotype from a single individual was represented in each subset. Columns indicate experimentally determined phenotype; rows indicate inferred phenotype using the 11KR/25K rule. Sequences classified as either X4/R5 or SI/NSI comprise two different data sets, as described previously (54). Ranges show the minima and maxima of the 100 subsets.
Calculated using the X4 or SI prevalence in the contingency table.
Calculated according to Bayes' theorem using an estimated SI/X4 prevalence in the database of 15%.
Data set construction.
Although a large number of HIV-1 subtype B gp120 sequences are available in public databases, many of the sequences are phylogenetically related, having been isolated either from the same individual or from individuals involved in closely linked chains of transmission. Because studies of amino acid substitution linkage between sites are confounded by phylogenetic relationships, it was necessary to construct subsets of sequences that were separated from one another by at least a minimal evolutionary or epidemiological distance. We used two different sampling methods to generate these subsets. The first method resulted in a single subset from each of the larger V1V3 and V3C4 datasets in which no two x4 or r5 sequences originated from the same individual (these subsets are referred to as V1V3-set 1 and V3C4-set 1; Table 3). This approach prevented the overrepresentation of the large number of isolates collected from certain subjects but resulted in the exclusion of 77 and 90% of available V1V3 and V3C4 sequences, respectively.
TABLE 3.
Description of set 1 and 30 subsets of sequencesa
| Set | V1V3 | V3C4 | |
|---|---|---|---|
| Set 1 | Sequences | 275 | 365 |
| x4 | 66 | 75 | |
| r5 | 209 | 290 | |
| 30 subsets | Sequences | 242.5 (0.5) | 323.9 (1.3) |
| x4/subset | 48.6 (2.3) | 54.5 (2.1) | |
| r5/subset | 193.9 (2.3) | 269.4 (2.3) |
Each set contains no more than one x4 and one r5 sequence isolated from a single individual. Each subset contains no two sequences related by a similarity score of >94 and contains no more than one sequence per individual. Data for the 30 subsets are averages (standard deviation).
We also used a second sampling method based on a minimal genetic distance and implemented in a way that allowed us to sample a greater proportion of available sequences. This second approach employed distance matrices generated from nucleotide alignments to select subsets of V1V3 and V3C4 sequences in which no two sequences achieved a similarity score above 94 (Jin-Nei distance). To ensure further that these subsets of sequences did not contain closely related sequences, only one sequence originating from any given individual was retained in each subset. The process of removing similar sequences further reduced the number of sequences that could be included in a single subset.
To include as many sequences as possible in the analysis, we resampled all available sequences by constructing 30 subsets each of V1V3 and V3C4 sequences (Table 2). Among the 30 subsets thus constructed, 79% (948) of the available V1V3 sequences and 51% (1810) of V3C4 sequences were represented at least once. A greater proportion of V1V3 sequences were sampled because of the smaller total number of sequences and the presence of fewer large clusters of sequences isolated from single individuals. Of the sequences sampled from the V1V3 and V3C4 datasets, 24 and 43%, respectively, appeared in only one of the 30 subsets, and 68 and 79%, respectively, were present in five or fewer of the 30 subsets. As a result, a group of highly divergent sequences uniquely representing single individuals was somewhat overrepresented in this resampling method. These two sampling methods complemented each other, and positions displaying significant linkage to V3 in both groups appear to be strongly supported.
The fraction of sequences in common between all possible pairs of the 30 subsets was also calculated. Because the average overlap between any two subsets was 66 and 60% for the V1V3 and V3C4 sets, respectively, these subsets could not be considered independent samples of sequences. Accordingly, we made no attempt to aggregate results of statistical tests from different subsets; rather, we report the results of the analysis performed on each subset as a separate test.
Variability of certain positions outside of V3 is linked to V3 genotype.
Amino acid variability in gp120 is not uniformly distributed, suggesting that different regions of the protein are subject to dissimilar selective pressures and functional constraints (80). One such constraint could be imposed by the requirement for specific physical interactions with the cell-associated ligands of gp120. Since R5 and X4 strains use different coreceptors for entry, we hypothesized that some of the site-to-site variability might differ among gp120 sequences derived from viruses of different phenotype. We therefore used two measures of variability, entropy (E) and diversity (D), to compare the extent of heterogeneity at each aligned position in r5 and x4 protein sequences. Permutation tests were used to determine if the difference of entropy (ΔE) or diversity (ΔD) between the two phenotypes fell in the tails of the reference distributions for each position under the assumption of no difference.
As described in previous studies, notably in that by Yamaguchi-Kabata and Gojobori (80), we found marked differences in variability from site to site in gp120. To emphasize global patterns of variability, we plotted entropy scores for sequences in V1V3-set 1 and V3C4-set 1 averaged over a 5-amino-acid sliding window (Fig. 1A). Note that this analysis excludes positions in the alignment with no corresponding amino acid in the HXB2 numbering standard. Differences in diversity and entropy among x4 and r5 sequences were calculated at each well-aligned position.
FIG. 1.

Entropy at each aligned amino acid corresponding to its position in HXB-2 from V1 to C4 among subtype B gp120 sequences. Entropy values were calculated from alignments of sequences in V1V3-set 1 (V1 to C3) and V3C4-set 1 (V3 to C4). (A) Entropy values averaged over a 5-amino-acid sliding window (i.e., the value plotted at position 3 is the average entropy for positions 1 through 5). Regions of poor alignment are included in the plot. (B) Differences in entropy among x4 and r5 sequences at each position. Positive values correspond to positions at which variability is higher among x4 sequences. Black bars indicate positions at which entropy differed significantly between x4 and r5 sequences. Positions within regions of poor sequence alignment (134 to 152, 185 to 189, 354 to 357, and 396 to 413) were excluded from the statistical analysis and are set at zero in this plot.
Figure 1B shows the difference in entropy (regions of V1, V2, and V4 which display length variability and align poorly are set to zero in this figure). Clearly, the greatest concentration of differences between the two inferred phenotypes lies in V3. Phenotype-associated sequence variability within V3 has been described elsewhere (reference 54 and references therein); therefore, we have provided results here only for positions outside of the third variable loop. For tests measuring both variability and amino acid frequency using V1V3-set 1 and V3C4-set 1, we identified positions that exhibited significant linkage to a pattern of substitutions in V3 as those with P values of <0.01. Positions considered significant in tests using the 30 subsets of V1V3 or V3C4 had P values of <0.05 for all 30 subsets.
We identified six positions for which either ΔD or ΔE achieved significance according to the above criteria using both sequence sampling methods; an additional six positions achieved significance using either one sampling method or the other (Fig. 1B and Table 4). Both measures of variability gave comparable results for all positions in achieving significance except for 424, where 11 versus 30 subsets reached P < 0.05 for ΔD and ΔE, respectively. Overall, variability increased at each of these positions among x4 sequences except at positions 345 and 365, both of which were more homogeneous for the consensus amino acid among x4 compared to r5 sequences (Fig. 1).
TABLE 4.
Summary of all test results for V3-linked variability
| Domain | Position | Testa | P, set 1b |
P, 30 subsetsc
|
||||
|---|---|---|---|---|---|---|---|---|
| 0.05 | 0.01 | 0.005 | 0.001 | *d | ||||
| V2 | 166 | Sig | 0.003 | 18 | 1 | — | — | — |
| Ent | 0.436 | — | — | — | — | — | ||
| Div | 0.295 | — | — | — | — | — | ||
| 177 | Sig | 0.007 | 30 | 21 | 12 | 4 | 1 | |
| Ent | 0.036 | 26 | 8 | 2 | — | — | ||
| Div | 0.015 | 30 | 19 | 12 | 2 | — | ||
| V1/V2 stem | 190 | Sig | <0.001 | 30 | 30 | 30 | 30 | 30 |
| Ent | <0.001 | 30 | 30 | 30 | 30 | 28 | ||
| Div | <0.001 | 30 | 30 | 30 | 30 | 29 | ||
| 191 | Sig | 0.004 | 30 | 28 | 24 | 11 | 9 | |
| Ent | 0.002 | 30 | 29 | 26 | 17 | 10 | ||
| Div | 0.002 | 30 | 29 | 26 | 15 | 10 | ||
| 195 | Sig | <0.001 | 30 | 30 | 30 | 19 | 11 | |
| Ent | 0.087 | 20 | 5 | 1 | — | — | ||
| Div | 0.100 | 22 | 8 | 3 | 1 | — | ||
| 198 | Sig | <0.001 | 30 | 28 | 25 | 18 | 16 | |
| Ent | 0.002 | 30 | 25 | 21 | 12 | 8 | ||
| Div | <0.001 | 30 | 28 | 25 | 17 | 11 | ||
| 200 | Sig | <0.001 | 30 | 30 | 29 | 17 | 11 | |
| Ent | 0.244 | — | — | — | — | — | ||
| Div | 0.331 | — | — | — | — | — | ||
| C2 | 204 | Sig | 0.006 | 22 | 3 | 1 | — | — |
| Ent | 0.004 | 27 | 6 | 2 | 1 | — | ||
| Div | 0.004 | 27 | 6 | 2 | 1 | — | ||
| 211 | Sig | 0.019 | 4 | 1 | — | — | — | |
| Ent | 0.016 | 5 | 1 | — | — | — | ||
| Div | 0.009 | 5 | 2 | 1 | — | — | ||
| C3 | 345 | Sig | 0.016 | 25 | 3 | — | — | — |
| Ent | 0.006 | 28 | 4 | 2 | — | — | ||
| Div | 0.010 | 27 | 4 | 2 | — | — | ||
| 347 | Sig | 0.037 | 21 | 1 | — | — | — | |
| Ent | 0.019 | 29 | 17 | 11 | — | — | ||
| Div | 0.005 | 30 | 20 | 13 | 1 | — | ||
| 365 | Sig | 0.007 | 13 | — | — | — | — | |
| Ent | 0.007 | 17 | — | — | — | — | ||
| Div | 0.007 | 15 | — | — | — | — | ||
| V4 | 382 | Sig | 0.001 | 3 | — | — | — | — |
| Ent | 0.001 | 9 | 1 | — | — | — | ||
| Div | 0.001 | 4 | — | — | — | — | ||
| C4 | 424 | Sig | 0.001 | 30 | 19 | 13 | 5 | 2 |
| Ent | 0.059 | 11 | — | — | — | — | ||
| Div | 0.004 | 30 | 16 | 9 | 3 | 1 | ||
| 440 | Sig | <0.001 | 30 | 30 | 30 | 30 | 30 | |
| Ent | <0.001 | 30 | 30 | 30 | 30 | 30 | ||
| Div | <0.001 | 30 | 30 | 30 | 30 | 30 | ||
Sig, significance test (value is the lowest of the scores for all amino acids represented at a position); Ent, entropy; Div, diversity.
P values for indicated tests using a single set each of V1V3 and V3C4 sequences containing no more than one x4 and one r5 sequence from an individual. P values of ≤0.01 are bold.
Number of 30 total subsets achieving P values less than or equal to the indicated value for each test. Each subset is a sample of all available sequences, so that no two sequences were isolated from the same individual or have a pairwise similarity score of >94. Zeros have been replaced by dashes.
*, test result fell outside the range of the reference distribution generated by the permutation test (i.e., P < 0.001).
Amino acid composition of certain positions outside of V3 is linked to genotype.
We next determined the significance of observed differences in the distributions of specific amino acids between x4 and r5 sequences at each aligned position in gp120. In this test, the magnitude of the difference in frequency of each amino acid was compared to a reference distribution of amino acid frequencies among randomly partitioned sequences to infer a P value. The set of positions identified using the test (Sig) largely overlapped those that demonstrated V3-linked changes in variability. A total of eight positions contained at least one amino acid whose frequency was significantly different for r5 and x4 sequences with both sampling methods, and an additional four achieved significance using one of the two sampling methods (Table 4). Amino acid composition for x4 and r5 sequences in set 1 at all positions achieving significance for any of the three tests (entropy, diversity, or amino acid composition) is presented in Fig. 2.
FIG. 2.

Amino acid composition at positions varying significantly between x4 and r5 sequences with respect to entropy, diversity, or representation of specific amino acids. The percentage of sequences containing the indicated amino acid is indicated on the y axis. Black bars, amino acid composition of x4 sequences; lightly shaded bars, amino acid composition of r5 sequences. Values shown describe V1V3-set 1 and V3C4-set 1. P values of specific amino acids achieving significantly different representation in x4 and r5 sequences are indicated with asterisks or pluses as specified in the legend. A summary of the P values obtained for each of the sampling methods is shown in Table 4. Note that amino acids with a P value between 0.05 and 0.01 in set 1 were not considered significant but were marked with a single + for comparison.
Position 440, located within C4, showed the most dramatic linkage with inferred entry phenotype of any position outside of V3. The r5 genotype was strongly associated with arginine at position 440; glutamic acid was significantly overrepresented in x4 sequences, with a trend toward glycine, serine, threonine, and glutamine in x4 sequences. The change in charge at this position is in the opposite direction from that typically seen in V3, where basic amino acids accumulate in the evolution of X4 variants. This linkage with 440 was not unexpected, as this position has had consistent support as a determinant for viral phenotype in multiple env backgrounds (9, 48). Another position in C4, 424, also showed linkage to V3 genotype, with valine twice as abundant (28 versus 16%) among x4 sequences compared to r5 sequences (amino acid composition of set 1 and P values are described unless noted).
A cluster of positions at which amino acid composition was linked to inferred entry phenotype, including positions 190, 191, 195, 198, 200, and 204, appeared in the relatively well- conserved region at the C-terminal end of V2 (Table 4 and Fig. 2). Position 190 is the first well-conserved amino acid after the region of extensive length polymorphism in V2 (V2hv) and falls within a motif (defined as NXS/T) (41) that is predicted to direct the N-linked glycosylation of position 188N. This motif appears in 61% of sequences in V1V3-set 1 (although positions 188 and 189 were not aligned, their composition could be determined by examining the two amino acids in each sequence preceding position 190). Substitution of residues other than S or T at position 190 occurred more often among x4 sequences (39%) than r5 sequences (13%). When both positions 188 and 190 were considered, the difference in the predicted frequency of glycosylation of 188 among x4 (38%) and r5 (68%) sequences remained highly significant (P < 2 × 10−5, Fisher's exact test). In addition, x4-associated residues at position 190 were most frequently basic, with 23% of x4 sequences in V1V3-set 1 containing R190 or K190, compared to 4% of r5 sequences. The shift from a large, negatively charged carbohydrate complex to a positively charged amino acid represents a dramatic chemical change in the x4 sequences, one that is in the same direction as in V3.
Position 191 showed a significant change away from the consensus amino acid, tyrosine, among x4 sequences. At position 195, x4 sequences were more likely to contain a histidine than were R5 sequences (15% versus 3%). A change away from the consensus amino acid (threonine) also occurred at position 198, where nonconsensus substitutions were found in 3% of r5 sequences and 18% of x4 sequences. At position 200, we observed a significantly increased frequency of T among x4 sequences. Just outside of the V1/V2 stem, an additional position, 204, was less likely to contain A among x4 sequences; this result was significant in set 1 but not in the 30 subsets of V1V3 sequences.
Four other positions, 166, 177, 365, and 382, were significantly linked to V3 genotype in set 1 but not the 30 subsets of sequences. Positions 166 and 177 are N-terminal of the region of extensive length polymorphism in V2. Position 166 was more likely to contain K among r5 sequences (12%) than x4 sequences (2%). A shift from tyrosine to asparagine at position 177 was associated with the x4 genotype. Position 365 was almost always serine among x4 sequences, while a low level of substitution of A, L, or T was found in r5 sequences; this pattern of higher conservation among x4 sequences was notable, as most genotype-linked positions were better conserved among r5 sequences. Finally, 382F was present in 100% of r5 sequences, while low levels of substitution of Y, L, or G were observed among sequences with the x4 genotype.
Apparent linkage of positions to basic substitutions in V3 cannot be explained by phylogenetic relationships among sequences.
A theoretical source of bias in this analysis is phylogenetic relatedness among sequences. For example, substitutions shared by clusters of related sequences of the same phenotype could be misconstrued as being linked to phenotype rather than due to common ancestry. To estimate the relatedness of sequences in V1V3-set 1 and V3C4-set 1, neighbor-joining trees were constructed from nucleotide sequence alignments. Few clusters of more than two sequences were supported by 50% of bootstrap replicates (7.6 and 6.0% of V1V3 and V3C4 sequences, respectively), and no cluster of more than five sequences was observed. Among pairs and clusters of sequences of the same V3 genotype, there was no pattern of substitution at any of the V3-linked positions that suggested that phylogenetic relationships among sequences biased the results of the statistical analyses (data not shown).
Increased net positive charge in the V2 hypervariable region among x4 sequences.
Although poor alignment prevented the examination of position-by-position differences in highly variable regions of gp120, we were able to test the correlation between the V3 11KR/25K genotype and global descriptors of these regions. Previous studies have suggested that length, charge, and number of potential glycosylation sites in the V1 or V2 loops vary with coreceptor usage, MT-2 phenotype, or disease progression (32, 67). Some or all of these associations were not confirmed in other studies (34, 67, 75). We used logistic regression to test the correlation between the V3 genotype and net charge or length in sequences spanning the hypervariable portions of V1 (V1hv; positions 131 to 157), V2 (V2hv; positions 180 to 189), and V4 (V4hv; positions 386 to 413). We assembled protein sequence fragments representing V1hv and V2hv from both V1V3-set 1 and the 30 subsets of V1V3 sequences described above; likewise, V4hv sequence fragments corresponded to V3C4-set 1 and the 30 subsets of V3C4 sequences.
An inferred x4 phenotype was found to be correlated with an increased net positive charge in V2hv with a significance of P < 0.003 for sequences in V1V3-set 1 in a univariate logistic regression analysis (Table 5). The difference between the average charge in V2hv among x4 and r5 sequences was 0.56. All 30 sequence subsets attained a P value below 0.05, with a median P value of 0.008. The average difference in charge among x4 and r5 sequences in the subset corresponding to the median P value was 0.49. Frequency distributions of net charge of V2hv in the 30 subsets show that x4 sequences are consistently shifted to more positive values (Fig. 3A).
TABLE 5.
Regression analysis of covariation between V3 genotype and features of the V2 hypervariable regiona
| Set | Net chargeb
|
P | Length (amino acids)
|
P | ||||
|---|---|---|---|---|---|---|---|---|
| r5 | x4 | x4 − r5 | r5 | x4 | x4 − r5 | |||
| V1V3-set 1 | −2.45 | −1.89 | 0.56 | 0.0031 | 11.71 | 12.83 | 1.12 | 0.0384 |
| 30 subsets | ||||||||
| Min | −2.53 | −1.79 | 0.74 | 0.0002 | 11.71 | 13.13 | 1.42 | 0.0104 |
| Median | −2.45 | −1.96 | 0.50 | 0.0074 | 11.78 | 12.70 | 0.92 | 0.0718 |
| Max | −2.43 | −2.06 | 0.38 | 0.0351 | 11.89 | 12.37 | 0.48 | 0.3604 |
Sequences correspond to HXB residues 180 to 189. Average values for each subset corresponding to minimum (Min), median, and maximum (Max) P values among 30 subsets of sequences are shown. “Median” corresponds to the 15th highest P value.
(R + K) − (D + E).
FIG. 3.

Frequency distribution of characteristics of the V2 hypervariable region (HXB positions 180 to 189) among 30 subsets of x4 and r5 sequences. Each trace corresponds to either x4 (black dashed line) or r5 (gray line) sequences represented in one of the 30 subsets. (A) Frequency distribution of net charge. (B) Frequency distribution of the length of each sequence corresponding to HXB positions 180 to 189 in the alignment.
We also tested the correlation between V3 genotype and the length of V2hv. This association was more tenuous: the average length of the V2hv region of x4 sequences in V1V3-set 1 exceeded that of r5 sequences by 1.12 amino acids (P = 0.038), but the difference was significant (i.e., P < 0.05) in only 9 of the 30 subsets (median P value = 0.071; Table 5). The frequency distribution of V2hv length suggests that the linkage to V3 genotype is most pronounced when V2hv is 12 or more amino acids long (Fig. 3B).
We conclude that among the sequences examined in this study, there is good evidence that the x4 V3 genotype is associated with a net increase in positive charge in the V2hv region. There is a less well supported trend toward elongation of this region in x4 sequences. In addition, no correlation was found between V3 genotype and either charge or length in V1 or V4 (data not shown).
DISCUSSION
We have surveyed HIV-1 subtype B amino acid sequences spanning the first variable loop to the fourth constant region of gp120 for positions linked to entry phenotype-associated substitutions in V3. We used two different sequence sampling methods designed to include as many sequences as possible without confounding the results with close epidemiological relationships among isolates. Our analysis identified positions displaying differences both in the extent of variability and in the representation of specific amino acids among sequences with either X4/SI-like or R5/NSI-like V3 loop sequences. In addition, our results are consistent with previous studies suggesting that a switch to the X4 phenotype is accompanied by an increased net positive charge in the V1/V2 stem (16, 32, 75).
The vast majority of isolates corresponding to sequences available in public databases have not been assigned experimentally determined phenotypes; at most 15 sequences greater than 300 nucleotides in length representing isolates originating from different individuals were classified as X4 in the Los Alamos HIV database (http://hiv-web.lanl.gov) at the time of writing (data not shown). As a result, this analysis relied on the assignment of an inferred phenotype based on V3 sequence using a modification of the 11/25 rule, which assigns an x4 genotype to sequences with at least one basic substitution at V3 position 11 or 25 (HXB 306 and 322) (20, 29, 30, 48, 79). By eliminating sequences with arginine at V3 position 25, we improved the specificity of the phenotypic classification by reducing the number of R5 sequences misclassified as X4. The reliability of the test (or the probability that a sequence predicted to have a particular phenotype actually has that phenotype) gives an indication of the expected “purity” of the x4 and r5 sequence sets and depends on the test's specificity and sensitivity but also on the prevalence of X4 sequences in the data set (Table 2). The reliability of this test, called here the 11KR/25K rule, is high for the NSI and SI phenotypes and moderately good for coreceptor usage.
The average adjusted X4 and R5 reliabilities for this test are 0.65 and 0.91, respectively. As a result of these values, the x4 and r5 sets are expected to be asymmetrical in quality with respect to coreceptor usage, with the r5 set less likely to contain X4 sequences than the x4 set is to contain R5 sequences. Another factor leading to the asymmetrical quality of the r5 and x4 groups was the inclusion of dually tropic viruses in the x4 group; we would expect sequences with some R5-like characteristics to be found in the x4 set, to the extent that R5X4 and R5 have common determinants for CCR5 usage. Despite the mixed nature of the sequences classified as x4, X4/SI-associated changes are still apparent (and significant) compared to the much larger and relatively pure r5 sequence set. Indeed, for the reasons just stated, we expect that our results underestimate the true differences in amino acid composition and variability between X4 and R5 viruses. Thus, even an imperfect but asymmetric classification scheme is useful for discerning global changes over a large number of sequences. As an example, we have noted that patterns of variability among V3 sequences that are known to be either R5 or X4 closely resemble the patterns observed among V3 sequences classified as r5 or x4 using the less accurate 11/25 rule (54). It is reasonable to suppose that such global patterns should also be well reproduced outside of V3, with their detection further enhanced using the improved classification scheme.
Although the distinction between MT-2 tropism and coreceptor usage is not merely a semantic one, the two classifications are clearly closely related. We are not aware of any SI isolates that are not X4 or R5X4, and NSI isolates that can use CXCR4 are exceedingly rare (we identified a single example in the database [5]). It is therefore something of a paradox that the correlation between discrete changes in V3 sequence and MT-2 cell tropism is more robust than for coreceptor usage. This might be the result of selection bias in the sets of phenotyped V3 sequences used to test the rule or could reflect a difference in the sensitivity of the experimental assays used to test viral isolates for the two phenotypic classifications. Additionally, we have previously suggested that SI isolates may represent a later, more evolved stage of the X4 phenotype (54). The determinants for coreceptor usage might therefore be more complex or might be more likely to lie outside V3 than those for syncytium induction.
Despite these disparities, the 11KR/25K rule should exclude SI isolates from the r5 set approximately 97% percent of the time (based on the adjusted reliability for predicting the NSI phenotype), and the NSI sequences that remain are highly unlikely to use CXCR4. This should be taken as a strength of the approach for predicting coreceptor usage. And more generally, the connection between the SI/NSI phenotype and coreceptor usage seems sufficiently strong to lend additional support to the hypothesis that the observed V3-linked changes are associated with coreceptor preference.
What is the relationship between V3, the V1/V2 stem (containing positions 190 to 200), position 440, and the determination of coreceptor usage? A given position in gp120 could influence entry phenotype in a number of ways, most obviously by participating in a direct physical interaction between gp120 and the coreceptor. But the amino acid composition of a position could also hypothetically modify the tertiary structure of the gp120 subunit, the stability or rotational conformation of the Env oligomer, or the conformational responsiveness to CD4 binding, all of which could affect coreceptor preference or affinity. Changes in charge could also affect the ability of the virus to approach the cell-associated receptors or plasma membrane. In addition, X4 viruses tend to arise late in infection, are associated with a more advanced stage of disease, and are thus likely to be exposed to a different immunological environment as a group compared to R5 viruses. Thus, some changes linked to V3 genotype might not reflect a functional contribution to coreceptor usage. To assess these possibilities, we located positions identified by our statistical approach within the three-dimensional structure of a theoretical model of the HXB-2 gp120 trimer in a CD4-bound conformation (Fig. 4) (43).
FIG. 4.

Model of the gp120 trimer structure as published by Kwong et al. (43), detailing the physical location of positions linked to phenotype-associated changes in V3. Residues achieving significant linkage to V3 according to both sampling methods are filled in in red; others are colored blue (see Table 4). Positions located in the region of the V1/V2 loop for which the structure was not solved are also indicated. Critical CCR5 binding positions (57) are colored green and are surrounded by green dots; positions defining the CD4 binding site are orange (42). The V3 stem is colored purple (the structure of the rest of the V3 loop was not solved). Model coordinates were kindly provided by Peter Kwong. The illustration was created using Rasmol (60). (A) View of the trimer from the perspective of the target cell. (B) View of the trimer perpendicular to the plane of the viral membrane, with the coreceptor binding face directed toward the top of the page. Each subunit is colored a different shade for clarity.
Position 440 is located in the fourth constant region of gp120 (C4). We have previously identified a significant correlation between substitution of serine or glutamic acid at 440 and SI-associated changes in V3 (48). This position has also been shown to be under positive selection, based on the ratio of transitions to transversions at the corresponding codon (80). Although substitutions at position 440 in YU2 Env (an R5 primary isolate) reduce binding of purified gp120 to CCR5 (56, 57), there is evidence that the importance of that position lies in its interaction with the V3 loop rather than as a point of contact with the coreceptor. Position 440 is surface exposed in the gp120 monomer and located at the apex of the viral spike, but its side chain does not appear to project directly toward the coreceptor (42). Furthermore, in each of three hypothetical placements of V3 structures in a model of the gp120 trimer, the V3 loop is both closely associated with positions 440 and interposed between that position and the 17b antibody occupying the coreceptor binding site (43).
A physical and functional relationship between V3 and C4 has been established by monoclonal antibody epitope mapping (49, 50, 78) as well as by mutational analysis in both HIV and simian immunodeficiency virus (SIV) (9, 51). The pattern of substitutions at position 440 reported here also suggests an interaction between that position and the V3 loop: the consensus arginine is much more likely to be replaced by an acidic or uncharged amino acid among isolates with basic substitutions in V3 (Fig. 2). These results thus support the previously suggested possibility that the nonbasic substitutions at position 440 may compensate for the accumulation of positive charges in V3 by minimizing the electrostatic repulsion between that position and the V3 loop (9, 48). Such compensatory changes might be necessary to maintain a stable conformation of gp120 or to aid in the formation of epitopes required for coreceptor interaction.
Position 424 also lies within C4, and unlike most other positions identified, 424 is not surface exposed. It is, however, near amino acids making contact with either CCR5 (419 to 422) or CD4 (425 to 430) (42, 57). The transition from isoleucine to the less bulky valine among x4 sequences may influence the conformation or flexibility of regions of gp120 involved in interaction with either CD4 or the coreceptor. Another buried, hydrophobic position was found to be linked to V3 genotype, this one located at the conserved, N-terminal end of V4: position 382 is always F among r5 sequences but experiences a low level of substitution in an x4 background, although the difference was significant only in set 1. The side chains of residues 424 and 382 are close to one another in the gp120 crystal structure, suggesting a possible interaction between these positions.
One provocative finding was the cluster of five positions linked to V3 genotype between positions 190 and 200. This region forms parts of the V1/V2 stem and bridging sheet and lies at the oligomer interface (42, 43). It has been suggested that positions in the V1/V2 stem may contribute to the interaction between gp120 and CCR5 (56). Although positions 195, 198, and 200 are well exposed on the surface of gp120 (fractional solvent accessibility, >0.4) (42), only the side chains of positions 198 and 200 point toward the coreceptor target; the side chain of 195 projects in the opposite direction. Structural information is not available for V2 positions N-terminal of 195, but by extrapolating from the alternating pattern of side chain orientation of positions 195 to 202, we speculate that 190 is oriented toward the target cell. V3-associated changes at each of these positions were significant in sets of sequences assembled using both selection methods.
Several of these positions are strong candidates for experimental verification. For example, the consensus sequence at positions 188 to 190 (NTS) forms a motif predicted to direct N-linked glycosylation of position 188. Position 190 is more frequently serine or threonine among r5 sequences (86 versus 61%); most of the substitutions among x4 sequences result in both the disruption of the glycosylation motif and the accumulation of a positive charge (Fig. 2). The S190R mutation was observed among clones of an HIV-1 isolate selected for syncytium formation in microglia by passage in vitro (65). It is also possible that the loss of glycosylation at 190 among x4 viruses is due to a release from antibody selection late in infection, when X4 isolates are most likely to arise. Like position 440, positions 195 and 200 are under significant positive selection, which is consistent with a role in determining Env function (80). Positions 198 and 200 lie in a beta sheet in the V1/V2 stem directly across from positions 123 and 121, respectively, both of which have been shown to be critical for CCR5 binding (56, 57) (Fig. 4).
According to structural models, the V1/V2 stem is separated from V3 within a single gp120 monomer in the CD4-bound state, but it may be close to the V3 loop of a neighboring subunit in the Env oligomer (Fig. 4) (43). There is also experimental evidence of a physical and functional interaction between V3 and V1/V2: neutralizing antibodies in serum from monkeys infected with HIV-1/SIV chimeric viruses recognize discontinuous epitopes that are either composed of or influenced by changes at both residue 13 (HXB gp160 308) in V3 and positions 187 and 190 in the V1/V2 stem (26). Several studies have demonstrated a cooperative interaction between V3 and positions in the V1/V2 stem to the extent that changes in both regions are required to confer a particular coreceptor usage, cell tropism, or cytopathic effect (13, 33, 37, 59). A critical unifying feature of these studies, however, is the context dependence of the changes described; that is, specific substitutions at a particular position often influence phenotype only in a very restricted set of isolates. In this light, the fact that global patterns of V3-linked variation were apparent at all in the V1/V2 stem is significant and calls for experimental confirmation of the overall role of this region in gp120 function.
The linkage of other positions to V3 is less easily interpreted due to a lack of structural information, because linkage to V3 is only marginally significant or because location in the three-dimensional structure of gp120 does not suggest an obvious involvement in coreceptor binding or oligomer assembly. Like position 190, a mutation at position 166 in V2 (R166G) was involved in the acquisition of syncytium induction in microglia in vitro (65); in this study, R166K was overrepresented in x4 sequences. Position 177 lies in a region of V2 shown to be involved in gp41-independent dimerization of gp120 (10), and positions 204 and 211 are located at the oligomer interface (43). Any of these positions could hypothetically influence the assembly or conformation of the gp120 oligomer. Rizzuto and Sodroski (56) identified position R117, whose side chain lies within 5 Å of that of 204 (204 is situated between 117 and the coreceptor binding face) and also projects into the oligomer interface, as being critical for CCR5 binding; the authors of this study also noted that CCR5-associated positions tended to lie close to the trimer axis.
It has not been well established whether the increase in net positive charge in V2 accompanies a switch to the X4 or SI phenotype. Groenink et al. (32) first reported a significantly higher positive charge in the hypervariable V2 locus of SI and “switch NSI” isolates (i.e., those with an NSI phenotype isolated from individuals who also harbored SI virus) than in NSI isolates. Other groups have also noted a trend toward higher positive charge in V2 in SI isolates (16, 75). Here, we report a significant correlation between X4/SI-associated changes in V3 and an increase in net positive charge of gp120 sequence fragments that include V2hv (HXB positions 180 to 189). Although the average difference in charge, approximately +0.5, is not large, this charge accumulation occurs in conjunction with an additional x4-associated basic substitution at position 190 (see Fig. 2 and 3).
Modeling of the coreceptors suggests that the extracellular surface of CXCR4 is more negatively charged than that of CCR5 (22), and others have shown that that the surface components of CXCR4 used by HIV-1 are more acidic than the corresponding regions of CCR5 (7, 46). In addition, substitution of alanine for certain acidic residues in CXCR4 not only resulted in the loss of coreceptor activity but also allowed R5 viruses to infect cells using the mutant receptors (11). This charge difference between CCR5 and CXCR4 fits well with the observation that basic substitutions in V3 are associated with CXCR4 usage. Because of the location of V2hv near the coreceptor binding face in the gp120 trimer, it is tempting to speculate that the increase in positive charge in the V1/V2 stem among X4 isolates further enhances a direct interaction with the negatively charged surface of CXCR4.
We have noted an increase in V2hv length among x4 sequences, although the significance of this correlation is marginal. It is a matter of some controversy whether the accumulation of insertions in V2hv (HXB positions 185 to 189) is associated with either changes in phenotype or disease progression. Groenink et al. (32) observed extension of V2 among SI and switch NSI sequences, although the same group failed to confirm this result using a more extensive data set (61). Another recent study showed an increased length of V2 in both SI isolates and NSI isolates obtained shortly before a phenotypic switch; the same group demonstrated that X4, R5X4, and R3R5X4 viruses were indistinguishable from one another, but all had longer average V2 lengths than R5 viruses (36). Two other studies failed to find a relationship between V2 extension and entry phenotype (34, 75). An extensive longitudinal study of Env sequence evolution in 12 subjects demonstrated a positive correlation between V2 extension and slow disease progression and showed that insertions in V2 reduced virus replication in macrophages (67). Further elucidation of the connection between V2hv extension and entry phenotype will probably have to wait for the availability of a large set of isolates with experimentally determined coreceptor usage.
Our analysis has identified positions in gp120 that are linked to entry phenotype-associated changes in V3. The cluster of such positions in V2 complements experimental evidence showing an often context-dependent functional association between V2, V3, and coreceptor usage. We have also confirmed and extended observations that CXCR4 usage is associated with increased positive charge in the V2 hypervariable region. Using a statistical approach, we have provided specific predictions about the functional role of regions of gp120 that can now be tested experimentally.
Acknowledgments
We thank Brian Foley and the Los Alamos HIV Sequence Database for the classification of gp120 sequences by patient and Peter Kwong for providing coordinates for the gp120 trimer model. We also thank Brenda Temple for help manipulating protein structures and Wolfgang Resch for useful discussion and programming expertise.
This work was supported by NIH grant R01-AI44667 to R.S. N.G.H. was supported in part by NIH Training Grants T32-AI07419 and T32-AI07001.
REFERENCES
- 1.Alkhatib, G., C. Combadiere, C. C. Broder, Y. Feng, P. E. Kennedy, P. M. Murphy, and E. A. Berger. 1996. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955-1958. [DOI] [PubMed] [Google Scholar]
- 2.Berger, E. A., R. W. Doms, E. M. Fenyo, B. T. Korber, D. R. Littman, J. P. Moore, Q. J. Sattentau, H. Schuitemaker, J. Sodroski, and R. A. Weiss. 1998. A new classification for HIV-1. Nature 391:240.. [DOI] [PubMed] [Google Scholar]
- 3.Bickel, P. J., P. C. Cosman, R. A. Olshen, P. C. Spector, A. G. Rodrigo, and J. I. Mullins. 1996. Covariability of V3 loop amino acids. AIDS Res. Hum. Retrovir. 12:1401-1411. [DOI] [PubMed] [Google Scholar]
- 4.Bieniasz, P. D., R. A. Fridell, I. Aramori, S. S. Ferguson, M. G. Caron, and B. R. Cullen. 1997. HIV-1-induced cell fusion is mediated by multiple regions within both the viral envelope and the CCR-5 coreceptor. EMBO J. 16:2599-2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjorndal, A., H. Deng, M. Jansson, J. R. Fiore, C. Colognesi, A. Karlsson, J. Albert, G. Scarlatti, D. R. Littman, and E. M. Fenyo. 1997. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 71:7478-7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyd, M. T., G. R. Simpson, A. J. Cann, M. A. Johnson, and R. A. Weiss. 1993. A single amino acid substitution in the V1 loop of human immunodeficiency virus type 1 gp120 alters cellular tropism. J. Virol. 67:3649-3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brelot, A., N. Heveker, O. Pleskoff, N. Sol, and M. Alizon. 1997. Role of the first and third extracellular domains of CXCR-4 in human immunodeficiency virus coreceptor activity. J. Virol. 71:4744-4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cann, A. J., M. J. Churcher, M. Boyd, W. O'Brien, J. Q. Zhao, J. Zack, and I. S. Chen. 1992. The region of the envelope gene of human immunodeficiency virus type 1 responsible for determination of cell tropism. J. Virol. 66:305-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrillo, A., and L. Ratner. 1996. Human immunodeficiency virus type 1 tropism for T-lymphoid cell lines: role of the V3 loop and C4 envelope determinants. J. Virol. 70:1301-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Center, R. J., P. L. Earl, J. Lebowitz, P. Schuck, and B. Moss. 2000. The human immunodeficiency virus type 1 gp120 V2 domain mediates gp41-independent intersubunit contacts. J. Virol. 74:4448-4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chabot, D. J., P. F. Zhang, G. V. Quinnan, and C. C. Broder. 1999. Mutagenesis of CXCR4 identifies important domains for human immunodeficiency virus type 1 X4 isolate envelope-mediated membrane fusion and virus entry and reveals cryptic coreceptor activity for R5 isolates. J. Virol. 73:6598-6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chesebro, B., J. Nishio, S. Perryman, A. Cann, W. O'Brien, I. S. Chen, and K. Wehrly. 1991. Identification of human immunodeficiency virus envelope gene sequences influencing viral entry into CD4-positive HeLa cells, T-leukemia cells, and macrophages. J. Virol. 65:5782-5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho, M. W., M. K. Lee, M. C. Carney, J. F. Berson, R. W. Doms, and M. A. Martin. 1998. Identification of determinants on a dualtropic human immunodeficiency virus type 1 envelope glycoprotein that confer usage of CXCR4. J. Virol. 72:2509-2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choe, H., M. Farzan, Y. Sun, N. Sullivan, B. Rollins, P. D. Ponath, L. Wu, C. R. Mackay, G. LaRosa, W. Newman, N. Gerard, C. Gerard, and J. Sodroski. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135-1148. [DOI] [PubMed] [Google Scholar]
- 15.Connor, R. I., and D. D. Ho. 1994. Human immunodeficiency virus type 1 variants with increased replicative capacity develop during the asymptomatic stage before disease progression. J. Virol. 68:4400-4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cornelissen, M., E. Hogervorst, F. Zorgdrager, S. Hartman, and J. Goudsmit. 1995. Maintenance of syncytium-inducing phenotype of HIV type 1 is associated with positively charged residues in the HIV type 1 gp120 V2 domain without fixed positions, elongation, or relocated N-linked glycosylation sites. AIDS Res. Hum. Retrovir. 11:1169-1175. [DOI] [PubMed] [Google Scholar]
- 17.Cornelissen, M., G. Mulder-Kampinga, J. Veenstra, F. Zorgdrager, C. Kuiken, S. Hartman, J. Dekker, L. van der Hoek, C. Sol, and R. Coutinho. 1995. Syncytium-inducing (SI) phenotype suppression at seroconversion after intramuscular inoculation of a non-syncytium-inducing/SI phenotypically mixed human immunodeficiency virus population. J. Virol. 69:1810-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Jong, J. J., A. de Ronde, W. Keulen, M. Tersmette, and J. Goudsmit. 1992. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J. Virol. 66:6777-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Jong, J. J., J. Goudsmit, W. Keulen, B. Klaver, W. Krone, M. Tersmette, and A. de Ronde. 1992. Human immunodeficiency virus type 1 clones chimeric for the envelope V3 domain differ in syncytium formation and replication capacity. J. Virol. 66:757-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delwart, E. L., J. I. Mullins, P. Gupta, G. H. Learn, Jr., M. Holodniy, D. Katzenstein, B. D. Walker, and M. K. Singh. 1998. Human immunodeficiency virus type 1 populations in blood and semen. J. Virol. 72:617-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng, H., R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. Di Marzio, S. Marmon, R. E. Sutton, C. M. Hill, C. B. Davis, S. C. Peiper, T. J. Schall, D. R. Littman, and N. R. Landau. 1996. Identification of a major coreceptor for primary isolates of HIV-1. Nature 381:661-666. [DOI] [PubMed] [Google Scholar]
- 22.Dimitrov, D. S., X. Xiao, D. J. Chabot, and C. C. Broder. 1998. HIV coreceptors. J. Membr. Biol. 166:75-90. [DOI] [PubMed] [Google Scholar]
- 23.Doranz, B. J., J. Rucker, Y. Yi, R. J. Smyth, M. Samson, S. C. Peiper, M. Parmentier, R. G. Collman, and R. W. Doms. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149-1158. [DOI] [PubMed] [Google Scholar]
- 24.Dragic, T., V. Litwin, G. P. Allaway, S. R. Martin, Y. Huang, K. A. Nagashima, C. Cayanan, P. J. Maddon, R. A. Koup, J. P. Moore, and W. A. Paxton. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667-673. [DOI] [PubMed] [Google Scholar]
- 25.Eddy, S. R. 2001. HMMER: profile hidden Markov models for biological sequence analysis. http://hmmer.wustl.edu.
- 26.Etemad-Moghadam, B., G. B. Karlsson, M. Halloran, Y. Sun, D. Schenten, M. Fernandes, N. L. Letvin, and J. Sodroski. 1998. Characterization of simian-human immunodeficiency virus envelope glycoprotein epitopes recognized by neutralizing antibodies from infected monkeys. J. Virol. 72:8437-8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felsenstein, J. 2000. PHYLIP (Phylogeny Inference Package), 3.6(alpha) ed. J. Felsenstein, Seattle, Wash.
- 28.Feng, Y., C. C. Broder, P. E. Kennedy, and E. A. Berger. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872-877. [DOI] [PubMed] [Google Scholar]
- 29.Fouchier, R. A., M. Brouwer, S. M. Broersen, and H. Schuitemaker. 1995. Simple determination of human immunodeficiency virus type 1 syncytium-inducing V3 genotype by PCR. J. Clin. Microbiol. 33:906-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fouchier, R. A., M. Groenink, N. A. Kootstra, M. Tersmette, H. G. Huisman, F. Miedema, and H. Schuitemaker. 1992. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 66:3183-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groenink, M., A. C. Andeweg, R. A. Fouchier, S. Broersen, R. C. van der Jagt, H. Schuitemaker, R. E. de Goede, M. L. Bosch, H. G. Huisman, and M. Tersmette. 1992. Phenotype-associated env gene variation among eight related human immunodeficiency virus type 1 clones: evidence for in vivo recombination and determinants of cytotropism outside the V3 domain. J. Virol. 66:6175-6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Groenink, M., R. A. Fouchier, S. Broersen, C. H. Baker, M. Koot, A. B. van't Wout, H. G. Huisman, F. Miedema, M. Tersmette, and H. Schuitemaker. 1993. Relation of phenotype evolution of HIV-1 to envelope V2 configuration. Science 260:1513-1516. [DOI] [PubMed] [Google Scholar]
- 33.Hoffman, T. L., E. B. Stephens, O. Narayan, and R. W. Doms. 1998. HIV type I envelope determinants for use of the CCR2b, CCR3, STRL33, and APJ coreceptors. Proc. Natl. Acad. Sci. USA 95:11360-11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes, E. S., J. E. Bell, and P. Simmonds. 1997. Investigation of population diversity of human immunodeficiency virus type 1 in vivo by nucleotide sequencing and length polymorphism analysis of the V1/V2 hypervariable region of env. J. Gen. Virol. 78:2871-2882. [DOI] [PubMed] [Google Scholar]
- 35.Hwang, S. S., T. J. Boyle, H. K. Lyerly, and B. R. Cullen. 1991. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science 253:71-74. [DOI] [PubMed] [Google Scholar]
- 36.Jansson, M., E. Backstrom, G. Scarlatti, A. A. Bjorndal, S. Matsuda, P. Rossi, J. Albert, and H. Wigzell. 2001. Length variation of glycoprotein 120 V2 region in relation to biological phenotypes and coreceptor usage of primary HIV type 1 isolates. AIDS Res. Hum. Retrovir. 17:1405-1414. [DOI] [PubMed] [Google Scholar]
- 37.Koito, A., G. Harrowe, J. A. Levy, and C. Cheng-Mayer. 1994. Functional role of the V1/V2 region of human immunodeficiency virus type 1 envelope glycoprotein gp120 in infection of primary macrophages and soluble CD4 neutralization. J. Virol. 68:2253-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koito, A., L. Stamatatos, and C. Cheng-Mayer. 1995. Small amino acid sequence changes within the V2 domain can affect the function of a T-cell line-tropic human immunodeficiency virus type 1 envelope gp120. Virology 206:878-884. [DOI] [PubMed] [Google Scholar]
- 39.Koot, M., I. P. Keet, A. H. Vos, R. E. de Goede, M. T. Roos, R. A. Coutinho, F. Miedema, P. T. Schellekens, and M. Tersmette. 1993. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann. Intern. Med. 118:681-688. [DOI] [PubMed] [Google Scholar]
- 40.Korber, B. T., R. M. Farber, D. H. Wolpert, and A. S. Lapedes. 1993. Covariation of mutations in the V3 loop of human immunodeficiency virus type 1 envelope protein: an information theoretic analysis. Proc. Natl. Acad. Sci. USA 90:7176-7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kornfeld, R., and S. Kornfeld. 1985. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54:631-664. [DOI] [PubMed] [Google Scholar]
- 42.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwong, P. D., R. Wyatt, Q. J. Sattentau, J. Sodroski, and W. A. Hendrickson. 2000. Oligomeric modeling and electrostatic analysis of the gp120 envelope glycoprotein of human immunodeficiency virus. J. Virol. 74:1961-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leitner, T., S. Kumar, and J. Albert. 1997. Tempo and mode of nucleotide substitutions in gag and env gene fragments in human immunodeficiency virus type 1 populations with a known transmission history. J. Virol. 71:4761-4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leonard, C. K., M. W. Spellman, L. Riddle, R. J. Harris, J. N. Thomas, and T. J. Gregory. 1990. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 265:10373-10382. [PubMed] [Google Scholar]
- 46.Lu, Z., J. F. Berson, Y. Chen, J. D. Turner, T. Zhang, M. Sharron, M. H. Jenks, Z. Wang, J. Kim, J. Rucker, J. A. Hoxie, S. C. Peiper, and R. W. Doms. 1997. Evolution of HIV-1 coreceptor usage through interactions with distinct CCR5 and CXCR4 domains. Proc. Natl. Acad. Sci. USA 94:6426-6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milich, L., B. Margolin, and R. Swanstrom. 1993. V3 loop of the human immunodeficiency virus type 1 Env protein: interpreting sequence variability. J. Virol. 67:5623-5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milich, L., B. H. Margolin, and R. Swanstrom. 1997. Patterns of amino acid variability in NSI-like and SI-like V3 sequences and a linked change in the CD4-binding domain of the HIV-1 Env protein. Virology 239:108-118. [DOI] [PubMed] [Google Scholar]
- 49.Moore, J. P., Q. J. Sattentau, R. Wyatt, and J. Sodroski. 1994. Probing the structure of the human immunodeficiency virus surface glycoprotein gp120 with a panel of monoclonal antibodies. J. Virol. 68:469-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moore, J. P., M. Thali, B. A. Jameson, F. Vignaux, G. K. Lewis, S. W. Poon, M. Charles, M. S. Fung, B. Sun, and P. J. Durda. 1993. Immunochemical analysis of the gp120 surface glycoprotein of human immunodeficiency virus type 1: probing the structure of the C4 and V4 domains and the interaction of the C4 domain with the V3 loop. J. Virol. 67:4785-4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morrison, H. G., F. Kirchhoff, and R. C. Desrosiers. 1993. Evidence for the cooperation of gp120 amino acids 322 and 448 in SIVmac entry. Virology 195:167-174. [DOI] [PubMed] [Google Scholar]
- 52.O'Brien, W. A., Y. Koyanagi, A. Namazie, J. Q. Zhao, A. Diagne, K. Idler, J. A. Zack, and I. S. Chen. 1990. HIV-1 tropism for mononuclear phagocytes can be determined by regions of gp120 outside the CD4-binding domain. Nature 348:69-73. [DOI] [PubMed] [Google Scholar]
- 53.Palmer, C., P. Balfe, D. Fox, J. C. May, R. Frederiksson, E. M. Fenyo, and J. A. McKeating. 1996. Functional characterization of the V1V2 region of human immunodeficiency virus type 1. Virology 220:436-449. [DOI] [PubMed] [Google Scholar]
- 54.Resch, W., N. Hoffman, and R. Swanstrom. 2001. Improved success of phenotype prediction of the human immunodeficiency virus type 1 from envelope variable loop 3 sequence using neural networks. Virology 288:51-62. [DOI] [PubMed] [Google Scholar]
- 55.Richman, D. D., and S. A. Bozzette. 1994. The impact of the syncytium-inducing phenotype of human immunodeficiency virus on disease progression. J. Infect. Dis. 169:968-974. [DOI] [PubMed] [Google Scholar]
- 56.Rizzuto, C., and J. Sodroski. 2000. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res. Hum. Retrovir. 16:741-749. [DOI] [PubMed] [Google Scholar]
- 57.Rizzuto, C. D., R. Wyatt, N. Hernandez-Ramos, Y. Sun, P. D. Kwong, W. A. Hendrickson, and J. Sodroski. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280:1949-1953. [DOI] [PubMed] [Google Scholar]
- 58.Roos, M. T., J. M. Lange, R. E. de Goede, R. A. Coutinho, P. T. Schellekens, F. Miedema, and M. Tersmette. 1992. Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. J. Infect. Dis. 165:427-432. [DOI] [PubMed] [Google Scholar]
- 59.Ross, T. M., and B. R. Cullen. 1998. The ability of HIV type 1 to use CCR-3 as a coreceptor is controlled by envelope V1/V2 sequences acting in conjunction with a CCR-5 tropic V3 loop. Proc. Natl. Acad. Sci. USA 95:7682-7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sayle, R. A., and E. J. Milner-White. 1995. RASMOL: biomolecular graphics for all. Trends Biochem. Sci. 20:374.. [DOI] [PubMed] [Google Scholar]
- 61.Schuitemaker, H., R. A. Fouchier, S. Broersen, M. Groenink, M. Koot, A. B. van 't Wout, H. G. Huisman, M. Tersmette, and F. Miedema. 1995. Envelope V2 configuration and HIV-1 phenotype: clarification. Science 268:115.. [DOI] [PubMed] [Google Scholar]
- 62.Schuitemaker, H., M. Koot, N. A. Kootstra, M. W. Dercksen, R. E. de Goede, R. P. van Steenwijk, J. M. Lange, J. K. Schattenkerk, F. Miedema, and M. Tersmette. 1992. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 66:1354-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X. L. Huang, and J. I. Mullins. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489-10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shannon, C. E. 1948. A mathematical theory of communication. Bell Syst. Technol. J. 27:379-423. [Google Scholar]
- 65.Shieh, J. T., J. Martin, G. Baltuch, M. H. Malim, and F. Gonzalez-Scarano. 2000. Determinants of syncytium formation in microglia by human immunodeficiency virus type 1: role of the V1/V2 domains. J. Virol. 74:693-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shioda, T., J. A. Levy, and C. Cheng-Mayer. 1992. Small amino acid changes in the V3 hypervariable region of gp120 can affect the T-cell-line and macrophage tropism of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 89:9434-9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shioda, T., S. Oka, X. Xin, H. Liu, R. Harukuni, A. Kurotani, M. Fukushima, M. K. Hasan, T. Shiino, Y. Takebe, A. Iwamoto, and Y. Nagai. 1997. In vivo sequence variability of human immunodeficiency virus type 1 envelope gp120: association of V2 extension with slow disease progression. J. Virol. 71:4871-4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh, A., G. Besson, A. Mobasher, and R. G. Collman. 1999. Patterns of chemokine receptor fusion cofactor utilization by human immunodeficiency virus type 1 variants from the lungs and blood. J. Virol. 73:6680-6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Speck, R. F., K. Wehrly, E. J. Platt, R. E. Atchison, I. F. Charo, D. Kabat, B. Chesebro, and M. A. Goldsmith. 1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J. Virol. 71:7136-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Starcich, B. R., B. H. Hahn, G. M. Shaw, P. D. McNeely, S. Modrow, H. Wolf, E. S. Parks, W. P. Parks, S. F. Josephs, and R. C. Gallo. 1986. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell 45:637-648. [DOI] [PubMed] [Google Scholar]
- 71.Tersmette, M., R. E. de Goede, B. J. Al, I. N. Winkel, R. A. Gruters, H. T. Cuypers, H. G. Huisman, and F. Miedema. 1988. Differential syncytium-inducing capacity of human immunodeficiency virus isolates: frequent detection of syncytium-inducing isolates in patients with acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. J. Virol. 62:2026-2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trkola, A., T. Dragic, J. Arthos, J. M. Binley, W. C. Olson, G. P. Allaway, C. Cheng-Mayer, J. Robinson, P. J. Maddon, and J. P. Moore. 1996. CD4-dependent, antibody-sensitive interactions between HIV-1 and its coreceptor CCR-5. Nature 384:184-187. [DOI] [PubMed] [Google Scholar]
- 74.Trujillo, J. R., W. K. Wang, T. H. Lee, and M. Essex. 1996. Identification of the envelope V3 loop as a determinant of a CD4-negative neuronal cell tropism for HIV-1. Virology 217:613-617. [DOI] [PubMed] [Google Scholar]
- 75.Wang, N., T. Zhu, and D. D. Ho. 1995. Sequence diversity of V1 and V2 domains of gp120 from human immunodeficiency virus type 1: lack of correlation with viral phenotype. J. Virol. 69:2708-2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Westervelt, P., H. E. Gendelman, and L. Ratner. 1991. Identification of a determinant within the human immunodeficiency virus 1 surface envelope glycoprotein critical for productive infection of primary monocytes. Proc. Natl. Acad. Sci. USA 88:3097-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu, L., N. P. Gerard, R. Wyatt, H. Choe, C. Parolin, N. Ruffing, A. Borsetti, A. A. Cardoso, E. Desjardin, W. Newman, C. Gerard, and J. Sodroski. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179-183. [DOI] [PubMed] [Google Scholar]
- 78.Wyatt, R., M. Thali, S. Tilley, A. Pinter, M. Posner, D. Ho, J. Robinson, and J. Sodroski. 1992. Relationship of the human immunodeficiency virus type 1 gp120 third variable loop to a component of the CD4 binding site in the fourth conserved region. J. Virol. 66:6997-7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiao, L., S. M. Owen, I. Goldman, A. A. Lal, J. J. deJong, J. Goudsmit, and R. B. Lal. 1998. CCR5 coreceptor usage of non-syncytium-inducing primary HIV-1 is independent of phylogenetically distinct global HIV-1 isolates: delineation of consensus motif in the V3 domain that predicts CCR-5 usage. Virology 240:83-92. [DOI] [PubMed] [Google Scholar]
- 80.Yamaguchi-Kabata, Y., and T. Gojobori. 2000. Reevaluation of amino acid variability of the human immunodeficiency virus type 1 gp120 envelope glycoprotein and prediction of new discontinuous epitopes. J. Virol. 74:4335-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang, Y. J., T. Dragic, Y. Cao, L. Kostrikis, D. S. Kwon, D. R. Littman, V. N. KewalRamani, and J. P. Moore. 1998. Use of coreceptors other than CCR5 by non-syncytium-inducing adult and pediatric isolates of human immunodeficiency virus type 1 is rare in vitro. J. Virol. 72:9337-9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu, T., H. Mo, N. Wang, D. S. Nam, Y. Cao, R. A. Koup, and D. D. Ho. 1993. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261:1179-1181. [DOI] [PubMed] [Google Scholar]