

Abstract
The role of specific stromal-derived MMPs was analyzed in experimental metastasis assays in wildtype and either MMP9, MMP7, or MMP2 null mice. MMP9 null mice showed an 81% reduction in Lewis Lung Carcinoma tumor number, while MMP7 null mice showed a 42% increase in tumor number and there was no difference in tumor number in MMP2 null mice compared to wildtype controls. Similarly, in an orthotopic model of lung cancer, 50% fewer MMP9 null mice were able to establish tumors in the lung compared to control mice, although the size of the tumors was not different. The effect of MMP9 on lung tumor colonization was dependent on the expression of MMP9 from bone marrow-derived cells and is most likely contributed by neutrophils. To examine temporal effects of stromal MMP9, bioluminescence imaging from luciferase expressing human lung cancer-derived A549 cells revealed that there were fewer tumor cells in the lungs of MMP9 null mice as early as 19 hours after injection compared to control mice, with no difference in subsequent growth rates. Six hours after injection of tumor cells, MMP9 null mice showed a four-fold increase in the percent of tumor cells undergoing apoptosis compared to control mice. We conclude that MMP9 from the bone marrow contributes to the early survival and establishment of tumors in the lung and has no effect on subsequent growth. These results provide insights into the failure of MMP inhibitors in clinical trials in patients with late stage lung cancer.
Keywords: MMP9, metastasis, lung cancer, apoptosis, bone marrow-derived
Abbreviations: MMP, matrix metalloproteinases; ECM, extracellular matrix; Rag, recombinase activating gene; LLC, Lewis lung carcinoma; LUC, luciferase; TUNEL, terminal dUTP nicked end labeling; MMPI, matrix metalloproteinase inhibitor; TGFβ, transforming growth factor beta; VEGF, vascular endothelial growth factor
INTRODUCTION
Tumors develop and progress through a multi-step process involving the interaction of tumor cells and the host microenvironment. Communication between tumor cells and host components, such as immune cells, fibroblasts and endothelial cells contribute to the progression of cancer from its initial growth at a primary site in the body to its metastasis to distant organs (1). Matrix metalloproteinases (MMPs) are a family of zinc dependent endopeptidases that have been shown to contribute to tumor progression and metastasis (2). Both tumor and host-derived MMPs contribute to multiple stages of tumor progression through their ability to degrade the basement membrane and components of the extracellular matrix as well as non-matrix substrates (3,4).
MMPs are expressed by tumor cells as well as by the stromal component in several different tumor types (5). The importance of the role of host-derived MMPs in cancer has been examined in several tumor models (6–9). In a model of skin carcinogenesis, MMP9 from bone marrow-derived cells contributed to hyperproliferation and tumor incidence (6). In a model of pancreatic islet tumorigenesis, MMP2 and MMP9 contributed to tumor growth and stromal MMP9 had additional effects on the angiogenic switch (7). In addition, stromal fibroblast-contributed MMP-11 affects the growth of transplanted breast cancer cells (8). Taken together, these data indicate that host-derived MMPs remain an important contributor to different stages of tumor progression.
Despite the clear evidence that MMPs play a significant role in tumor progression, the results from the clinical trials of MMP inhibitors indicate that MMPIs are not a useful therapeutic approach (10). On evaluating the clinical trial results, it is clear that our understanding of MMP biology is incomplete. In order to better understand the role of host MMPs in the establishment of lung tumors, we have returned to experimental metastasis assays and devised an orthotopic model of lung cancer for use in MMP null mice, specifically focusing on the role of MMP9 in the establishment of lung tumors.
MATERIALS AND METHODS
Cell Culture
Lewis Lung carcinoma cells (LLC) were obtained from Dr. John Caterina (Bethel College, McKenzie, TN) from a stock at the NIH. Luciferase expressing human A549 cells (LUC-A549) were generously provided by Dr. Jerry Shay (University of Texas Southwestern Medical Center). A549 cells were obtained from ATCC. LLC were cultured in DMEM (Gibco BRL, Long Island, NY) supplemented with 10% FBS (Atlanta Biologicals, Norcross, GA) at 37°C, 5% CO2. LUC-A549 cells were cultured in F-12 Nutrient Mixture (HAM) (Gibco BRL, Long Island, NY) supplemented with 10% FBS (Atlanta Biologicals, Norcross, GA) at 37°C, 5% CO2. A549 cells were cultured in F12-K Nutrient Mixture (Kaighn’s modification) (Gibco BRL, Long Island, NY) supplemented with 10% FBS (Atlanta Biologicals, Norcross, GA) and 1.5g/L sodium bicarbonate at 37°C, 5% CO2.
Animal Models
Experimental metastasis assay
MMP2 (11), MMP7 (12), and MMP9 (13) null mice were generated as described previously and backcrossed into a C57Bl/6 background to N>10. C57Bl/6 mice (Harlan, Indianapolis, Indiana) were used as wildtype controls. Rag2 null mice maintained on a C57Bl/6 background were purchased from Taconic (Germantown, NY) and crossed into MMP9 null mice to obtain double Rag2/MMP9 knockout mice. At 6–8 weeks of age, tumor cells were injected into the tail vein of MMP null or C57Bl/6 WT mice or Rag2 null and Rag2/MMP9 null mice. After 2 weeks for the syngeneic model or 5 weeks for the immunocompromised model, the mice were sacrificed and surface lung tumors were counted and measured.
Orthotopic model
A549 cells were injected into the lung through the trachea of 6-week-old C57Bl/6 Rag2 null or MMP9/Rag2 null mice as a modification of the procedure described by McLemore et al, 1987. The intrabronchial injection is performed using a 1-inch piece of Tygon® microbore tubing (0.76 mm outer diameter) attached to a 2-inch 27 gauge blunt end needle (Popper, New Hyde Park, NY). A small incision is made in the neck of the mouse and in the trachea. The tubing is inserted and fed down the trachea. 1 x 106 tumor cells/100 μl PBS are injected into the bronchus. The incision in the skin is closed by wound clips. A primary tumor in the right lung of the mouse is established after 5 weeks in approximately 80% of the animals. Adaptation of this model to LLC cells was not successful due to the appearance of tracheal tumors that caused premature death of the mice.
Bioluminescent Imaging
Bioluminescent imaging was detected from luciferase expressing A549 cells after injection of the cells i.v. into mice. Beetle Luciferin (Promega) was used as the substrate for the luciferase expressing tumor cells and injected i.v. at a concentration of 150 mg/kg in PBS 1–2 minutes before imaging. Mice were anesthetized using 2% isofluorane and imaged at 2 hours, 19 hours, 43 hours post tumor cell injection and thereafter at weekly time points for 5 weeks using a cooled CCD camera (IVIS® system, Xenogen). Exposure times ranged from 1 min to 1 sec. Images were quantified as photons/sec using the Living Image® software from Xenogen.
Bone marrow transplantation studies
Bone marrow cells where collected from 6-week old C57Bl/6 wildtype or MMP9 null mice by flushing femurs and tibias with sterile PBS. Recipient mice were given 100 mg/l neomycin and 10mg/l polymyxin B sulfate in acidified water (Ph 2.7) 1 week before the transplantations and for the remainder of the study. Recipient mice, either C57Bl/6 wildtype or MMP9 null, were lethally irradiated (600 rads followed 3 hours later with 400 rads) using a cesium γ source. Four hours later, C57Bl/6 wildtype or MMP9 null mice were injected with 1x106 bone marrow cells/100 μl PBS by tail vein from wildtype or MMP9 null mice. 10 weeks after bone marrow transplantation, reconstitution of MMP9 in the bone marrow was confirmed by gelatin zymography.
Histological Analysis
Formalin-fixed paraffin-embedded tissues were dewaxed, hydrated through graded alcohols, stained with hematoxylin and eosin, dehydrated and cover slipped.
Immunohistochemistry
Five-micrometer formalin-fixed paraffin-embedded sections were dewaxed, hydrated through graded ethanols, treated with 0.6% hydrogen peroxide in methanol (to destroy endogenous peroxidases), micro waved in 10mM sodium citrate for 1 min at high power and 9 min at medium power for antigen retrieval. Sections were exposed to blocking solution (10% rabbit serum/TBS) for 1 hr. Sections were incubated overnight in blocking solution with rat monoclonal anti-neutrophil (1:100 dilution; Serotec Inc., Raleigh, NC), rabbit anti-von willebrand factor (Dako Corporation, Carpinteria, CA) or appropriate control IgG (Pharmingen, Mississauga, ON Canada). Sections were washed with TBST (150mM NaCl, 10mM Tris, and 0.05% Tween-20) and incubated with biotinylated anti-rat IgG (1:1000; Vector Laboratories, Burlingame, CA) for 1 hour. Labeled cells were visualized using an avidin-biotin peroxidase complex (Vectastain ABC kit, Vector Laboratories) and 3,3′ –Diaminobenzidine tetrahydrochloride substrate (Sigma, St. Louis, MO). Sections were counterstained with hematoxylin. For neutrophil quantitation five 10X fields/lung were analyzed in 3 control and 4 MMP9 null mice. Von willebrand factor staining was assessed by counting multiple areas defined by Metamorph Imaging System (Universal Imaging Corporation, Downingtown, PA) in seven mice per experimental group.
Zymography
Bone marrow cells were washed 1X with PBS and lysed in lysis buffer (1% Triton-X, 0.1% SDS, 1% Deoxycholic acid, 50mM Tris pH 7.5, 0.15M NaCl, 5 Mm EDTA, supplemented with 20 μg/ml leupeptin, 20μg/ml aprotinin, 20μg/ml PMSF) on ice for 10 minutes. Protein concentrations from bone marrow lysates were determined using the BCA assay (Pierce). Equal amounts of protein were loaded on 10% SDS-polyacrylamide gels containing 0.1 mg/ml gelatin and run at 100 volts for 4 hours in non-reducing conditions. After electrophoresis, gels were washed with 2.5 % Triton-X twice for 15 minutes and incubated at 37°C in substrate buffer (50mM Tris-HCL, pH 7.5, 10 mM CaCl2) overnight. Gels were stained with 0.5% Coomassie blue, 50% methanol, 10% acetic acid and destained in 50% methanol, 10% acetic acid.
Clodronate Treatment
Liposomal clodronate was prepared as described previously (14). 80 μl of the liposomal clodronate or liposomal PBS solution was injected i.t. into C57Bl/6 wildtype mice. 24 hours later LLC cells were injected i.v. Bronchoalveolar lavage (BAL) fluid was collected 48 hours after liposomal clodronate administration. Mouse tracheas were cannulated with a 20-gauge blunt end needle attached to a 1-ml syringe, and the lungs were instilled with sterile PBS until a total lavage volume of 3ml was collected. The BAL was centrifuged at 400 rpm for 5min, and the cell pellet resuspended in 1ml of 3% FBS/PBS. Total cell counts were determined using a grid hemocytometer. Differential cell counts were obtained by staining cytocentrifuge slides with a modified Wright's stain (Richard Allan-Scientific, Kalamazoo, MI).
Apoptosis Analysis
TUNEL was performed on frozen sections using the ApoTag Fluorescein In Situ Apoptosis Detection Kit (Chemicon International, Temecula, CA) according to manufacturer’s directions. Nuclei were counterstained with Hoechst 33258.
Labeling of tumor cells
CellTracker™ probe (CellTracker Red CMPTX) (Molecular Probes, Eugene, OR) (10μM) was added to the cell culture medium at a dilution of 1:1000. Cells were labeled for 30 min. Labeled cells were visualized using Axioplan 2 imaging microscope (Zeiss) and Openlab 4.0.2 software.
Statistical Analysis
All data generated using the experimental metastasis assays were analyzed using a non-parametrical (Mann-Whitney) method. Data generated using the orthotopic model were analyzed using Fisher’s exact test. (Statview software, SAS Institute).
RESULTS
Host MMPs contribute to the establishment of tumors in the lung
In order to determine if host-derived MMPs contribute to lung tumor colonization, experimental metastasis assays were performed by injecting 3 x 105 Lewis lung carcinoma cells (LLC) into the tail vein of syngeneic C57Bl/6 wildtype mice or MMP2, MMP7 or MMP9 null mice. Two weeks after tail vein injection, the surface lung tumors were counted and measured. In contrast to previous reports (15), we saw no difference in tumor number comparing control and MMP2 null mice (Fig. 1A). Surprisingly, MMP7 null mice showed a 42% increase in tumor number compared to control mice (p=0.01), suggesting a protective effect of MMP7 on lung colonization (Fig. 1C). Most interestingly, MMP9 null mice showed an 81% reduction in tumor number compared to wildtype mice (p=0.0002) (Fig. 1E). The results from the MMP9 null mice are in agreement with those previously reported (16) although the effect of MMP9 ablation on tumor number was greater in our study. There was no significant difference in tumor size in any of the MMP null mice compared to wildtype mice (Fig. 1B,D,F), suggesting that these MMPs are not having an effect on the growth of tumors that successfully establish in the lung. These experiments show that different host-derived MMPs can have different effects on metastasis to the lung. Because MMP9 null mice showed the most dramatic phenotype, we chose to pursue this observation to understand the mechanism by which MMP9 contributes to lung tumor development.
Figure 1.
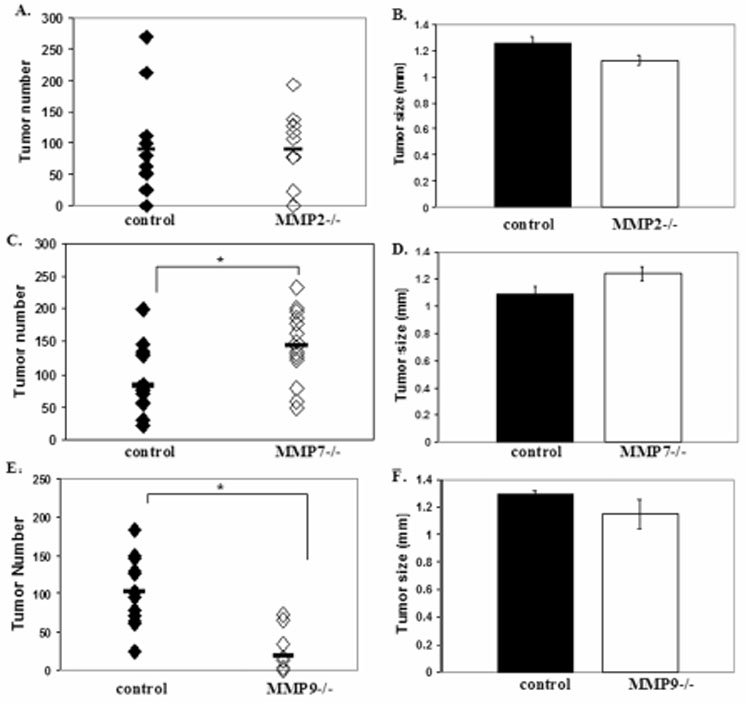
Host MMP contributions to lung tumor formation. (A) Number of surface lung tumors in control (n=12) and MMP2 null (n=12) mice 2 weeks after injection of 3 x 105 LLC cells i.v. (p=NS). (B) Tumor size in control and MMP2 null mice (p=NS). (C) Number of surface lung tumors in control (n=14) and MMP7 null (n=14) mice 2 weeks after injection of 3 x 105 LLC cells i.v. (*p=0.01). (D) Tumor size in control and MMP7 null mice (p=NS). (E) Number of surface lung tumors in control (n=13) and MMP9 null (n=11) mice 2 weeks after injection of 3 x 105 LLC cells i.v. (*p=0.0002). (F) Tumor size in control and MMP9 null mice (p=NS).
MMP9 contributes to the establishment of a primary tumor in the lung
To test if host MMP9 also has an effect on the establishment of primary tumors in the lung, an orthotopic model of lung cancer was established by injecting human lung carcinoma A549 cells directly into the lung via the trachea. This procedure produces distinct primary tumors in the lung in 5 weeks (Fig. 2A). 1x106 A549 cells were injected into either immunocompromised Rag2 null mice or immunocompromised Rag2/MMP9 null mice and tumor growth was examined after 5 weeks. Tumors develop in 82% of wildtype mice compared to 44% of MMP9 null mice, suggesting a contribution of host MMP9 to initial tumor take (Table. 2B). However, when comparing tumors that develop in either the wildtype or MMP9 null animals, approximately 50% in each case can be classified as microscopic (< 0.1mm diameter) and approximately 50% are macroscopic with sizes ranging from 0.5 – 87 mm3 (Table 2C). This suggests that the presence or absence of MMP9 is not the prime determinant in tumor progression from microscopic to macroscopic tumors. These data are similar to those obtained with experimental metastasis assays in which the initial tumor ‘take’ or colonization is dependent on MMP9 expression, while subsequent growth is not influenced by MMP9.
Figure 2.

Host MMP9 contributes to establishment of a primary tumor in the lung. (A) Histology of lung 5 weeks after orthotopic injection of 1 x 106 A549 cells (H/E). T= tumor. Table B: Percent of control (n=17) and MMP9 null mice (n=18) that establish a primary tumor in the lung after orthotopic injection of A549 cells (*p=0.02). Table C: Percentage of tumors in control and MMP9−/− mice that were classified as microscopic (micro) (<0.1 mm3) or macroscopic (macro) (0.5–87.5 mm3). *Fisher’s exact test-2-tailed.
MMP9 from the bone marrow contributes to lung tumor formation
MMP9 is expressed in the stromal component in human lung tumors including localization in inflammatory cells (17,18), and inflammatory cells have been shown to be a source of MMP9 in several cancer models (6,7,19,20). To determine if MMP9 from bone marrow-derived cells plays a role in lung metastasis, bone marrow transplantation assays were performed. Lethally irradiated mice received either 1 x 106 wildtype or MMP9 null bone marrow cells i.v. into the tail vein. Ten weeks after transplantation, 1 x 105 LLC cells were injected i.v. into wildtype mice receiving either wildtype or MMP9 null bone marrow cells or MMP9 null mice receiving either wildtype or MMP9 null bone marrow cells. Two weeks later, lungs were harvested and surface lung tumors were counted. Bone marrow reconstitution was determined by gelatin zymography to detect MMP9 from bone marrow cell lysates in transplanted mice (Fig. 3A). MMP9 was present in wildtype mice that received wildtype bone marrow and absent in MMP9 null mice receiving MMP9 null bone marrow. Two of the five wildtype mice receiving MMP9 null bone marrow showed complete reconstitution and three showed partial reconstitution. MMP9 was detected in the bone marrow of all MMP9 null mice that received wildtype bone marrow but at different levels. The number of lung nodules that developed in MMP9 null mice reconstituted with MMP9 null bone marrow was 14% of those that developed in wildtype mice reconstituted with wildtype bone marrow (Fig. 3B), similar to the results obtained with intact wildtype or MMP9 null mice (Fig. 1E). In mice reconstituted with bone marrow of a different genotype, the number of lung nodules was directly proportional to the level of MMP9 in the bone marrow (Fig. 3B). Thus, the effect of MMP9 in the experimental metastasis assay was the result of MMP9 contributed by bone marrow-derived cells.
Figure 3.
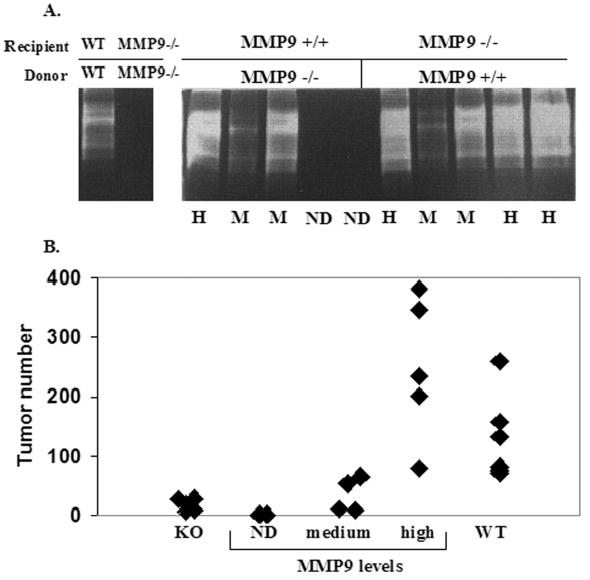
MMP9 levels from bone marrow-derived cells correlate with lung tumor formation. (A) Gelatin zymography of bone marrow cell lysates showing MMP levels in representative samples of wildtype (WT) and MMP9 null (MMP−/−) mice after reconstitution with either MMP9 positive or MMP9 null bone marrow cells. ND=not detected, M=medium, H=high levels of bone marrow MMP9. Duplicate zymograms in the presence of EDTA confirms metal-dependent enzymatic activity. (B) Quantitation of surface lung tumors in control or MMP9 null mice reconstituted with MMP9 positive or MMP9 null bone marrow cells.
MMP9 contributes to the early establishment of tumors in the lung and has no effect on tumor growth
To further examine the effect of host MMP9 on tumor growth and establishment in the lung, the experimental metastasis assay was performed using luciferase expressing human A549 cells (LUC-A549) in immunocompromised Rag2 null and Rag2/MMP9 null mice. Since the tumor cells express luciferase, tumor growth rates in the lung could be measured over time by measuring luciferase expression by bioluminescence. 2x106 LUC-A549 cells were injected i.v. into the tail vein of Rag2 null and Rag2/MMP9 null mice. Bioluminescent images were obtained 2 hours, 19 hours, 43 hours, and weekly up to 5 weeks after injection of tumor cells (Fig. 4A). Lung-localized luciferase activity was identical in wildtype and MMP9-null mice at the two hour time point, but as early as 19 hours and continuing throughout the experiment there was a statistically significant decrease in the bioluminescent signal in MMP9 null compared to wildtype mice (Fig. 4B). After 5 weeks, mice were sacrificed and surface lung tumors were counted and measured. MMP9 expression was confirmed by immunohistochemistry in the A549 cells and in stromal cells in the lungs of control but not MMP9 null mice (data not shown). MMP9 null mice showed a 97% reduction in tumor number compared to wildtype mice (Fig. 4C). Of the tumors that developed, there was no significant difference in tumor size between control and MMP9 null mice (Fig. 4D).
Figure 4.
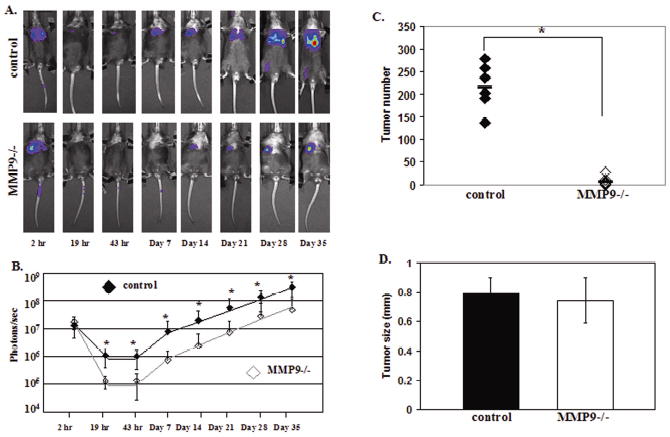
MMP9 contributes to the early establishment of tumors in the lung. (A) In vivo bioluminescent images of representative control and MMP9 null mice injected with 2x106 luciferase expressing A549 cells (LUC-A549) i.v. into the tail vein. (B) Mean ± SD tumor growth rates in control (n=10) and MMP9 null (n=10) mice at the time points indicated in (A) determined by bioluminescence (photons/sec) from the LUC-A549 cells (*p <0.05). (C) Number of surface lung tumors in control (n=6) and MMP9 null (n=10) mice 5 weeks after injection of 2 x 106 LUC-A549 cells i.v. (* p=0.0007) (D) Surface lung tumor size (mm) in control and MMP9 null mice (p=NS).
To further examine the early stages of the experimental metastasis assay, Rag2 null and Rag2/MMP9 null mice were sacrificed 24 hours after injection of A549 cells i.v. into the tail vein and the lungs examined at the histological level. Clusters of tumor cells could be seen in the MMP9 null mice and the wildtype mice (Fig. 5A,B). Quantitation of the tumor cells in the Rag2 null and Rag2/MMP9 null lungs shows a 72% reduction in the number of tumor cells in the Rag2/MMP9 null mice at the 24-hour time point compared to the control mice (p=0.02) (Fig. 5C). This confirms the bioluminescence data where decreased luciferase activity was seen in the lungs of MMP9 null mice 19 hours after injection of tumor cells compared to control mice. Thus, the data obtained by bioluminescence measurements, gross, and histological examination supports the conclusion that the lack of MMP9 affects the initial steps of tumor formation in the lung and not subsequent tumor growth.
Figure 5.
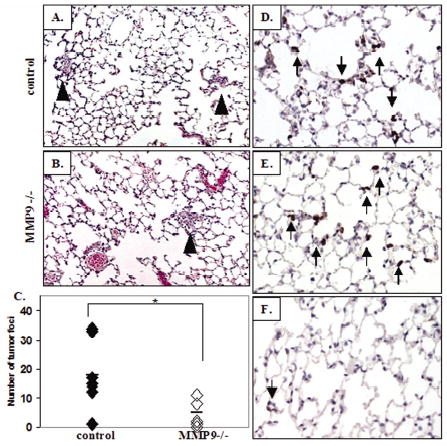
MMP9 null mice show fewer tumor cells in the lung 24 hours after tumor cell inoculation compared to control mice and both groups show neutrophil infiltration. Histological (H/E) analysis of lungs in control (A) and MMP9 null (B) mice 24 hours after i.v. injection of A549 cells into the tail vein. Clusters of tumor cells are indicated by arrow heads. (C) Quantitation of tumor cell clusters in the lungs of control (n=7) and MMP9 null mice (n=6) 24 hours after tumor cell inoculation (*p=0.018). Immunohistochemical analysis of neutrophils 24 hours after tumor cell inoculation in control (D) and MMP9 null (E) mice. Arrows indicate neutrophils in the lungs. (F) Neutrophils in the lungs of non-injected Rag2 null mice.
Cellular source of MMP-9
MMP9 in the lung tumor microenvironment is contributed by bone marrow-derived cells (Fig. 3). The absence of lymphocytes in the Rag2 null mice, which are deficient in mature B and T cells, did not appear to influence the effect of MMP9 ablation on lung tumor formation since we observed similar results in the experimental metastasis assay performed with immunocompetent and immunodeficient mice (Fig. 1E, 4C). To examine other bone marrow-derived cells that may be contributing MMP-9, we treated mice with liposomal clodronate to eliminate alveolar macrophages from the lung. Liposomal clodronate reduced alveolar macrophages by an average of 32% but we saw no reduction in tumor number in clodronate treated mice compared to control mice treated with liposomal PBS (data not shown). However, we did observe an abundant influx of neutrophils in tumor-bearing lungs which have been demonstrated to be a predominant source of MMP9 in inflamed lung tissue (21). Immunostaining for a neutrophil marker revealed a marked elevation in neutrophils in tumor-bearing wildtype and MMP9 null mice (Fig. 5D, E) compared to non-injected Rag2 null mice where few neutrophils were observed in the lung (Fig. 5F). The neutrophils were clustered adjacent to tumor cells. There was no difference in the number of neutrophils in wildtype vs. MMP9 null mice 6 hours after tumor inoculation (28.2 ± 7.5 vs. 28.5 ± 3.8 neutrophils/10X field, respectively), demonstrating that the lack of MMP9 did not alter neutrophil influx at this early timepoint. These data suggest that rapid influx of bone marrow-derived neutrophils after tumor cell inoculation contributes the majority of host-derived MMP9 in our model.
Tumor cell survival in the lung 6 hours after inoculation is dependent on MMP9
To determine if the absence of MMP9 leads to reduced cell survival in the lung at the early stages of tumor cell colonization in the experimental metastasis assay, apoptosis was measured by TUNEL 6 hours and 20 hours after i.v. injection of CellTracker-labeled A549 cells. Using fluorescence, the percentage of tumor cells that were undergoing apoptosis could be measured in sections from control and MMP9 null mice (Fig. 6A–F). Five random fields from each mouse lung were examined to determine the percentage of tumor cells undergoing apoptosis. When apoptosis was examined at 6 hours, 8% of tumor cells in MMP9 null mice were under going apoptosis compared to only 2% of tumor cells in control mice (p=0.01) (Fig. 6G). However, at 20 hours, more tumor cells in the control mice (8%) were undergoing apoptosis compared to MMP9 null mice (1%) (p=0.04) (Fig. 6H). These data confirm that very early after injection of tumor cells (6 hours), the cells in the MMP9 null mice are undergoing more apoptosis, resulting in fewer tumors in the MMP9 null mice at later time points compared to control mice.
Figure 6.
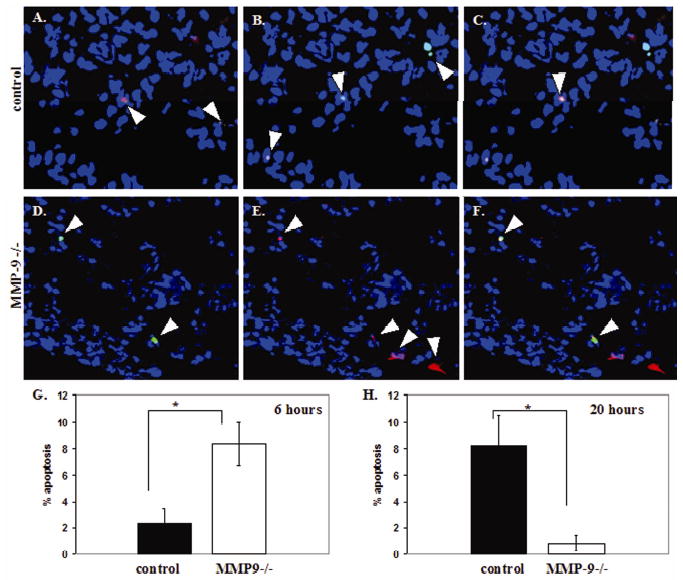
MMP9 null mice show significantly more tumor cells undergoing apoptosis 6 hours after injection of tumor cells. (A–G) TUNEL analysis at 6 hours after injection of CellTracker labeled A549 cells in control (A–C) and MMP9 null (D–F) mice. Apoptotic cells (green, A and D). CellTracker labeled tumor cells (red, B and E). C and F are the localization of apoptotic tumor cells shown in yellow by a merge of TUNEL staining (green) and CellTracker staining (red). Nuclei are blue (Hoechst). (G) Percentage of tumor cells in control (n=5) and MMP9 null (n=6) mice undergoing apoptosis 6 hours after injection of A549 cells i.v. (*p=0.01). (H) Percentage of tumor cells in control (n=5) and MMP9 null (n=4) mice undergoing apoptosis 20 hours after injection of A549 cells i.v. (*p=0.04).
DISCUSSION
The failure of MMP inhibitor clinical trials prompted us to analyze the discrepancy between the clinical results and the numerous preclinical reports demonstrating an advantage of MMP inhibition. We reproduced the results of Itoh et al, (16) demonstrating that the lack of host MMP9 reduced LLC lung nodules, and demonstrated that this result was consistent across systems with an 81% inhibition with LLC cells in syngeneic mice and a 97% reduction in A549 cells in immunocompromised mice. However, of the three MMPs examined, MMP9 was the only one to show this effect, and MMP7 demonstrated an opposing protective role in the LCC experimental metastasis assay. Recently, protective roles for MMP-3 and MMP-8 in carcinogenesis models have been demonstrated (22,23), and MMP inhibitors have been shown to have negative effects in some specific animal models (24,25) and clinical trials1. These results underscore the complexity of MMP biology and the importance of understanding the specificity of a targeted cancer therapy.
In addition to demonstrating selectivity in the effect of MMP9 on lung colonization, we demonstrated that MMP9 contributes to lung metastasis specifically at the earliest stages of colonization in the lung and has no effect on subsequent tumor growth. Bioluminescence measurements indicated that the LUC-A549 cells reached the lungs of wildtype and MMP9 null mice in equal proportions, but within 19 hours there was an 88% reduction in photons/sec in the MMP9 null mice compared to controls. Histological analysis confirmed the reduction in the number of A549 tumor cells in the lungs of MMP9 null mice 24 hours after injection compared to wildtype mice (72%). Despite the difference in tumor number, there was no difference in tumor size between control and MMP9 null mice in either of the experimental metastasis models. The LUC-A549 cells clearly demonstrated that the increase in bioluminescence with time, an indication of the rate of tumor growth, was virtually identical between 48 hours and 5 weeks in wildtype and MMP9-null mice. These results are consistent with results reported from an experimental metastasis model of i.v. injected T-cell lymphoma cells and liver metastasis, where administration of an inhibitor specific to the gelatinases reduced liver metastasis only when the gelatinase inhibitor was given 1-hour before tumor cell inoculation with no effect when it was given 1-day after injection of tumor cells (26). We conclude that host MMP9 contributes to the earliest stages of establishment of tumors in the lung and has no effect on subsequent tumor growth.
It is intriguing that host MMP9 influences tumor establishment in tumor cells that express MMP9 (A549 cells) as well as those that do not (LLC cells). Explanation for this include the possibility that A549 cells express MMP9 that is not activated, or that they do not express CD44, a known receptor for MMP9 (27). However, A549 cells have been reported to express CD44 (28), and we have demonstrated in preliminary studies that A549 cells produce active MMP9 as measured using a proteolytic beacon (29) designed to be selective for MMP9 (data not shown). We conclude that tumor-produced MMP9 does not compensate for the lack of host-derived MMP9 in this model system.
The lack of an effect of MMP9 on tumor growth differs with previous reports of both positive and negative effects of MMP9 on tumor angiogenesis. MMP9 can promote tumor angiogenesis and subsequent tumor growth in several systems (7,18), most likely through a mechanism of releasing VEGF from the matrix allowing increased accessibility to its receptor (7). However, MMP9 can also inhibit angiogenesis by generating endogenous angiogenesis inhibitors, angiostatin and tumstatin, and consequently, MMP9 has been shown to be associated with reduced tumor growth and angiogenesis (30,31). In our studies, the tumors formed with either the LLC or A549 cells were less than 2 mm in diameter, a size at which angiogenesis is not required (32). We found no difference in angiogenesis as measured by von willebrand factor staining in the tumors of control and MMP9 null mice (Figure 1E) (data not shown). Therefore, the absence of a difference in tumor size between wildtype and MMP9 null mice in our model may be due to the tumors not growing large enough to undergo an angiogenic switch.
Bone marrow transplantation studies revealed that MMP9 from bone marrow-derived cells contributes to the difference in tumor number that was observed in the experimental metastasis assay. Inflammatory cells derived from the bone marrow are a source of MMP9 in human tumors, including lung tumors (16,17,33,34), and we observed MMP9 immunoreactivity in stromal cells in our model. We speculated that T cells may be required for the MMP9 effect. T-lymphocytes release MMP9 (35,36), and T-lymphocyte membranes in contact with interstitial macrophages, but not alveolar macrophages, stimulated the release of MMP9 from the macrophages (37). However, we saw that the absence of MMP9 has a similar effect on tumor number in the lung in wildtype and Rag2null mice, which are deficient in mature B and T lymphocytes. Ablating alveolar macrophages by administration of liposomal chlodronate also did not affect tumor number in the lung, suggesting that macrophages may not be the predominant source of MMP9 in the experimental metastasis assay. We note that the administration of liposomal chlodronate reduced the number of alveolar macrophages by 32% rather than causing complete ablation, but the zymography data of MMP9 expression in the bone marrow cell lysates indicates that a small reduction in MMP9 from the bone marrow is sufficient to cause a concomitant reduction in tumor number. It is likely that MMP9 from neutrophils is contributing to the phenotype in the experimental metastasis assay. We observed an influx of neutrophils in the lung within 6 hours and at 24 hours after injection of tumor cells, and neutrophils are a predominant source of MMP9 (21). In addition, there is evidence that neutrophils contribute to mammary adenocarcinoma metastasis (38,39). The lack of MMP9 did not alter neutrophil influx, since we observed equivalent numbers of neutrophils in both MMP9-null and wildtype mice after tumor cell inoculation. We speculate that as metastatic tumors cells enter the lung, neutrophils are recruited and perform protumorigenic functions by releasing MMP9 and allowing tumor cells to establish and initiate growth.
In exploring cellular mechanisms by which MMP9 affects lung tumor establishment, we observed significantly more apoptosis in the tumor cells in MMP9 null lungs (8%) compared to control lungs (2%) 6 hours after injection, providing an explanation for the decrease in tumor cells in MMP9 null mice at all subsequent times. It has been shown that blocking MMP9 in vitro using an anti-MMP9 monoclonal antibody induces apoptosis in B-cell chronic lymphocytic leukemia cells, but only when these cells are in contact with bone marrow stromal cells (40). This data is interesting in light of our finding that MMP9 from the bone marrow is contributing to tumor cell survival in the lung. Interestingly, at 20 hours post injection the effect was reversed and significantly more tumor cells in the control mice were undergoing apoptosis compared to MMP9 null mice. The reason for this difference is unclear, but could be explained if one considers that apoptosis of cells when they reach an ectopic environment is probably a normal event, but the timing is accelerated in the absence of MMP9. A proportion of the tumor cells are capable of surviving long enough to reach the next stage of initiating proliferation, but the number of surviving cells capable of initiating proliferation is reduced in MMP9 null mice as a result of this premature apoptosis. Sustained growth of the colonies that manage to initiate proliferation is unaffected by MMP9 since we observed similar growth rates and tumor sizes in control and MMP9 null mice.
How MMP9 contributes to survival or the absence of MMP9 contributes to cell death remains unknown. MMP9 may contribute to the apoptotic process by modifying cell surface molecules or processing matrix to release soluble factors. MMP9 has been shown to process latent TGFβ to its active form at the cell surface by complexing with CD44 (41). When this complex of MMP9 and CD44 is disrupted, tumor cells injected into the lung undergo apoptosis. It is therefore possible that the absence of MMP9 is inducing apoptosis by allowing less active TGFβ to be present to induce tumor cell survival. As well, apoptosis of tumor cells in MMP9 null mice may be related to tumor cell attachment to the lung endothelium. It has been shown that rat lung endothelium has exposed areas of basement membrane where laminin-5 in these areas is important in binding to integrin α3β1 on tumor cells to facilitate tumor cell arrest in the lung vasculature (42). Therefore, an absence of MMP9 in the lung may prevent tumor cells from attaching to the lung vasculature because MMP9 is not available to expose basement membrane on the lung endothelium to facilitate adhesion.
Additional evidence for MMP9 contributing to tumor establishment is observed from the results in the orthotopic model. 50% fewer MMP9 null mice had primary tumors in the lung compared to control mice after orthotopic injection of A549 cells. However, of the tumors that did develop in the lung, tumors in MMP9 null mice were able to convert to macroscopic size in numbers equivalent to control mice. MMP9 affected early tumor take in the lung, and did not contribute to subsequent growth of tumors in the lung. Therefore, in the orthotopic model, as was observed in the experimental metastasis assay, MMP9 contributed to tumor cell survival and the absence of MMP9 caused fewer primary tumors to form in the lung.
MMPs have multiple roles in different stages of tumor progression, and we have demonstrated that host MMP9 can contribute to the early stages of metastasis in the lung as well as establishment of transplanted primary lung tumors. Treatment of patients with late-stage lung cancer with several synthetic MMPIs, all of which targeted MMP9, was not successful (10). Although our data indicate a contribution of MMP9 to metastasis, it is only for a narrow window of time following the arrival of the tumor cells in the lung microenvironment, after which point, ablation of MMP9 has no effect on subsequent tumor growth. Based on this information, MMP9 inhibition with MMPIs would be expected to have little or no effect on patients in which metastasis had already occurred, such as those enrolled in the MMPI clinical trials. Effective treatment based on MMP9 inhibition would require identifying patients before metastatic seeding occurs, and maintaining constant levels of MMP inhibition. These results demonstrate the importance of understanding the specific stage(s) of tumor progression recapitulated in animal models, and the necessity of a thorough understanding of mechanism of action of targeted therapies.
Acknowledgments
The authors wish to thank Drs. David Johnson and David Carbone for their continual support and encouragement. The authors would also like to thank Oliver McIntyre for his help with the in vivo imaging studies.
This work was supported by NIH NCI P50 CA90949. H.A. was a predoctoral trainee supported by T32 CA009385.
Footnotes
Abstract: M. Moore et al., Proc Am Soc Clin Oncol 19, 240a (2001).
References
- 1.Fidler IJ. The organ microenvironment and cancer metastasis. Differentiation. 2002;70:498–505. doi: 10.1046/j.1432-0436.2002.700904.x. [DOI] [PubMed] [Google Scholar]
- 2.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 3.Stamenkovic I. Matrix Metalloproteinases in tumor invasion and metastasis. Seminars in Cancer Biology. 2000;10:415–33. doi: 10.1006/scbi.2000.0379. [DOI] [PubMed] [Google Scholar]
- 4.Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation. 2002;70:561–73. doi: 10.1046/j.1432-0436.2002.700909.x. [DOI] [PubMed] [Google Scholar]
- 5.DeClerk YA. Interactions between tumour cells and stromal cells and proteolytic modification of the extracellular matrix by metalloproteases in cancer. Eur J Cancer. 2000;36:1258–68. doi: 10.1016/s0959-8049(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 6.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–90. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Bio. 2000;2:737–44. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masson R, Lefebvre O, Noël A, et al. In vivo evidence that stromelysin-3 metalloproteinase contributes in a paracrine manner to epithelial cell malignancy. J Cell Biol. 1998;140:1535–41. doi: 10.1083/jcb.140.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang S, Van Arsdall M, Tedjarati S, et al. Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J Natl Cancer Inst. 2002;94:1134–42. doi: 10.1093/jnci/94.15.1134. [DOI] [PubMed] [Google Scholar]
- 10.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–92. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 11.Vu TH, Shipley JM, Bergers G, et al. MMP9/Gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chrondrocytes. Cell. 1998;93:411–22. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson CL, Heppner KJ, Labosky PA, Hogan BLM, Matrisian LM. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci. 1997;94:1402–07. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of β-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–92. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- 14.Koay MA, Gao X, Washington MK, et al. Macrophages are necessary for maximal nuclear factor-κB activation in response to endotoxin. Am J Respir Cell Mol Biol. 2002;26:572–78. doi: 10.1165/ajrcmb.26.5.4748. [DOI] [PubMed] [Google Scholar]
- 15.Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H, Itohara S. Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res. 1998;58:1048–51. [PubMed] [Google Scholar]
- 16.Itoh T, Tanioka M, Matsuda H, et al. Experimental metastasis is suppressed in MMP9 deficient mice. Clin Exp Metastasis. 1999;17:177–81. doi: 10.1023/a:1006603723759. [DOI] [PubMed] [Google Scholar]
- 17.Cox G, Jones JL, O’Byrne KJ. Matrix metalloproteinase 9 and epidermal growth factor signal in operable non-small cell lung cancer. Clin Cancer Res. 2000;6:2349–55. [PubMed] [Google Scholar]
- 18.Cox G, Jones JL, Andi A, Walker DA, O’Byrne KJ. A biological staging model for operable non-small cell lung cancer. Thorax. 2001;56:561–6. doi: 10.1136/thorax.56.7.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Debusk LM, Fukada K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 20.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP9 expressing macrophages and angiogensis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623–33. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atkinson JJ, Senior RM. Translational Review: Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol. 2003;28:12–24. doi: 10.1165/rcmb.2002-0166TR. [DOI] [PubMed] [Google Scholar]
- 22.McCawley LJ, Crawford HC, King LE, Mudgett J, Matrisian LM. A protective role for matrix metalloproteinase-3 in squamous cell carcinoma. Cancer Res. 2004;64:6965–72. doi: 10.1158/0008-5472.CAN-04-0910. [DOI] [PubMed] [Google Scholar]
- 23.Balbin M, Fueyo A, Tester AM, et al. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–7. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 24.Krüger A, Soeltl R, Sopov I, et al. Hydroxamate-type matrix metalloproteinase inhibitor batimastat promotes liver metastasis. Cancer Res. 2001;61:1272–75. [PubMed] [Google Scholar]
- 25.Chen X, Su Y, Fingleton B, et al. Increased plasma MMP9 in integrin a1-null mice enhances lung metastasis of colon carcinoma cells. Int J Cancer. 2005;116:52–61. doi: 10.1002/ijc.20997. [DOI] [PubMed] [Google Scholar]
- 26.Krüger A, Arlt M, Gerg M, et al. Antimetastatic activity of a novel mechanism-based gelatinase inhibitor. Cancer Res. 2005;65:3523–26. doi: 10.1158/0008-5472.CAN-04-3570. [DOI] [PubMed] [Google Scholar]
- 27.Yu Q, Stamenkovix I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koslowski R, Fichtner F, Barth K, Roehlecke C, Seidel D, Kasper M. Apoptosis and release of CD44s in bleomycin-treated L132 cells. J Cell Biochem. 2005;95:1146–56. doi: 10.1002/jcb.20483. [DOI] [PubMed] [Google Scholar]
- 29.McIntyre JO, Fingleton B, Wells KS. Development of a novel fluorogenic proteolytic beacon for in vivo detection and imaging of tumour-associated matrix metalloproteinase-7 activity. Biochem J. 2004;377:617–28. doi: 10.1042/BJ20030582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pozzi A, Lefine WF, Gardner HA. Low plasma levels of matrix metalloproteinase 9 permit increased tumor angiogenesis. Oncogene. 2002;21:272–81. doi: 10.1038/sj.onc.1205045. [DOI] [PubMed] [Google Scholar]
- 31.Hamano Y, Zeisberg M, Sugimoto H, et al. Physiological levels of tumstatin, a fragment of collagen IV α3 chain, are generated by MMP9 proteolysis and suppress angiogenesis via αVβ3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3:65–71. [PubMed] [Google Scholar]
- 33.Jones JL, Glynn P, Walker RA. Expression of MMP-2 and MMP-9, their inhibitors, and the activator MT1-MMP in primary breast carcinomas. J Pathology. 1999;189:161–8. doi: 10.1002/(SICI)1096-9896(199910)189:2<161::AID-PATH406>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 34.Saito K, Takeha S, Shiiba K, et al. Clinicopathologic significance of urokinase receptor- and MMP-9-positive stromal cells in human colorectal cancer: functional multiplicity of matrix degradation on hematogenous metastasis. Int J Cancer. 2000;86:24–9. doi: 10.1002/(sici)1097-0215(20000401)86:1<24::aid-ijc4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 35.Weeks BS, Schnaper W, Handy M, Holloway E, Kleinman HK. Human T lymphocytes synthesize the 92 kDa type IV collagenase (gelatinase B) J Cell Physiol. 1993;157:644–9. doi: 10.1002/jcp.1041570326. [DOI] [PubMed] [Google Scholar]
- 36.Leppert D, Waubant E, Galardy R, Bunnett NW, Hauser SL. T cell gelatinases mediate basement membrane transmigration in vitro. J Immunol. 1995;154:4379–89. [PubMed] [Google Scholar]
- 37.Ferrari-Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer J. Human lung tissue macrophages, but not alveolar macrophages, express matrix metalloproteinase after direct contact with activated T lymphocytes. Am J Respir Cell Mol Biol. 2001;24:442–51. doi: 10.1165/ajrcmb.24.4.4008. [DOI] [PubMed] [Google Scholar]
- 38.Aeed PA, Nakajima M, Welch DR. The role of polymorphonuclear leukocytes (PMN) on the growth and metastatic potential of 13762NF mammary adenocarcinoma cells. Int J Cancer. 1988;42:748–59. doi: 10.1002/ijc.2910420521. [DOI] [PubMed] [Google Scholar]
- 39.Welch DR, Schissel DJ, Howery RP. Aeed PA. Tumor-elicited polymorphonuclear cells, in contrast to “normal” circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells . Proc Natl Acad Sci. 1989;86:5859–63. doi: 10.1073/pnas.86.15.5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ringshausen I, Dechow T, Schneller F, et al. Constitutive activation of the MAPkinase p38 is critical for MMP-9 production and survival of B-CLL cell on bone marrow cells. Leukemia. 2004;18:1964–70. doi: 10.1038/sj.leu.2403544. [DOI] [PubMed] [Google Scholar]
- 41.Yu Q, Stamenkovic I. Transforming growth factor-beta facilitates breast carcinoma metastasis by promoting tumor cell survival. Clin Exp Met. 2004;21:235–42. doi: 10.1023/b:clin.0000037705.25256.d3. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, Fu W, Im J, et al. Tumor cell α3β1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J Cell Biol. 2004;164:935–41. doi: 10.1083/jcb.200309112. [DOI] [PMC free article] [PubMed] [Google Scholar]