

Abstract
Bacterial expression of β-lactamases is the most widespread resistance mechanism to β-lactam antibiotics, such as penicillins and cephalosporins. There is a pressing need for novel, non-β-lactam inhibitors of these enzymes. One previously discovered novel inhibitor of the β-lactamase AmpC, compound 1, has several favorable properties: it is chemically dissimilar to β-lactams and is a noncovalent, competitive inhibitor of the enzyme. However, at 26 μM its activity is modest. Using the X-ray structure of the AmpC/1 complex as a template, 14 analogues were designed and synthesized. The most active of these, compound 10, had a Ki of 1 μM, 26-fold better than the lead. To understand the origins of this improved activity, the structures of AmpC in complex with compound 10 and an analogue, compound 11, were determined by X-ray crystallography to 1.97 and 1.96 Å, respectively. Compound 10 was active in cell culture, reversing resistance to the third generation cephalosporin ceftazidime in bacterial pathogens expressing AmpC. In contrast to β-lactam-based inhibitors clavulanate and cefoxitin, compound 10 did not up-regulate β-lactamase expression in cell culture but simply inhibited the enzyme expressed by the resistant bacteria. Its escape from this resistance mechanism derives from its dissimilarity to β-lactam antibiotics.
Introduction
Microbial resistance to antibiotics is now a serious threat to public health.1,2 A pressing problem is resistance to the β-lactam antibiotics, including the penicillins and cephalosporins, which are among the most widely used class of antibiotics. Several mechanisms contribute to this resistance, including mutations in the target of these drugs, cell-wall biosynthesis transamidases called penicillin binding proteins, deletion and modification of the porin channels through which the drugs diffuse, and expression of pumps that export the drugs out of the bacterial cells.3–6 The most widespread resistance mechanism remains the expression of β-lactamase enzymes, which hydrolyze the lactam bond in the eponymous β-lactam ring of these drugs, inactivating them.7,8
To overcome these resistance enzymes, β-lactam molecules that inhibit (e.g. clavulanic acid) or evade (e.g. aztreonam) β-lactamases have been introduced. These molecules are themselves β-lactams and, like the penicillins, most are derivatives of microbial natural products that have been in the biosphere over evolutionary time. Consequently, resistance to them has evolved rapidly, often in the β-lactamases themselves.9 Mutant enzymes have arisen that can evade β-lactam-based β-lactamase inhibitors.10–12 Enzymes that are naturally resistant to current inhibitors, including class C β-lactamases such as AmpC, have become prominent in clinical settings.13 Mechanisms that alter the expression levels of the enzymes in the presence of the drugs or the inhibitors have appeared. There are strains of pathogenic bacteria that, recognizing the presence of a β-lactam-based inhibitor, will overexpress the β-lactamase that these drugs are meant to inhibit.14 There is thus a pressing need for novel β-lactamase inhibitors, not based on a β-lactam core structure. Such inhibitors would not be hydrolyzed by β-lactamases or mutant β-lactamases and would not be recognized by the suite of bacterial resistance mechanisms mobilized against β-lactams.15
Recently, we reported the structure-based discovery of a novel, noncovalent inhibitor of the widespread class C β-lac-tamase AmpC, compound 1 (Figure 1).16 This compound is dissimilar to penicillins and cephalosporins and binds to the enzymes noncovalently and reversibly, in contrast to the β-lactam substrates and inhibitors. Despite these differences, the X-ray crystal structure of the AmpC/1 complex revealed that 1 complements the core of the active site, interacting with key residues involved in β-lactam recognition and hydrolysis such as Ser64, Lys67, Asn152, and Tyr221. We concluded that the ligand recognition encoded by the AmpC structure was plastic enough to accommodate inhibitors genuinely dissimilar to β-lactams, allowing a new departure in the medicinal chemistry of their inhibitors.
Figure 1.

Characteristic β-lactam substrate and inhibitor of AmpC, and the lead compound for the novel inhibitor family discussed here.
The very novelty of this inhibitor posed several problems, ones that are perhaps shared by many genuinely new leads. First, the inhibition constant of compound 1 was, at 26 μM, weak, and its activity in vivo was poor. Moreover, its novelty confronted us with an unanticipated design problem. Whereas one can draw upon 60 years of medicinal chemistry in designing analogues of β-lactams,17 something we ourselves have done in the past,18,19 in a novel series this course is unavailable. Second, whereas β-lactams are evolved for cellular efficacy, neither the purely synthetic compound 1 nor its analogues need be active against bacteria. Having discovered a novel lead, the question became, could we improve its affinity and biological activity?
Here we describe our preliminary efforts to improve this series of inhibitors. We continue to use a structure-based approach. In the structure of the AmpC/1 complex, the inhibitor complements the core of the active site but leaves a distal region open. We sought derivatives to take advantage of this region that were relatively easy to synthesize and would not diminish the solubility and “leadlike” properties of the inhibitors.20 We therefore focused on derivates that made new interactions with polar residues in the distal part of the AmpC site, including Arg204, or that tested features of the ligands that appeared important for binding. Fourteen analogues were synthesized, the best of which bound to AmpC with 26-fold improved affinity over 1. To understand their improved affinity, X-ray crystal structures were determined for two of the new analogues in complex with AmpC. To explore their biological potential, several of the new inhibitors were tested in bacterial cell culture against clinical pathogens. We observe intriguing differences in their biological effects on resistant bacteria compared to classic, β-lactam-based inhibitors such as clavulanate and cefoxitin.
Results
Synthesis.
The synthesis of the methyl ester intermediates and their corresponding acids was accomplished by reacting the sulfonyl chloride with the appropriate amine in anhydrous pyridine (Scheme 1). The only exception was in the synthesis of compound 4A, where the amine reacted with the monomethylphthalate in the presence of diisopropylamine (DIEA) and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluoro-phosphate (PyBOP) to afford the desired methyl ester (Scheme 2). Each ester was subsequently hydrolyzed by suspending it in sodium hydroxide and isolating the final acids through precipitation with dilute HCl.
Scheme 1.
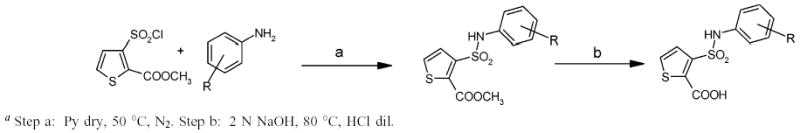
Synthesis of 3-((4-chloroanilin)sulfonyl)thiophene-2-carboxylic Acid Derivativesa
Scheme 2.
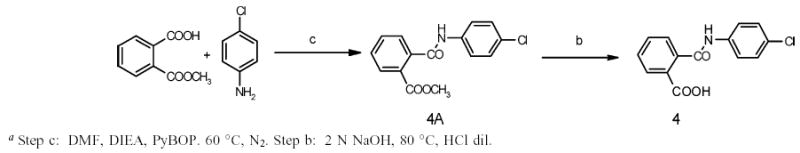
Synthesis of N-(4-Chlorophenyl)phthalamic Acid Derivativea
All intermediate esters (1A–14A) were characterized either through elementary analysis or monodimensional 1H NMR, or both, whereas all final carboxylic acids (1–14) were characterized through monodimensional 1H NMR and either mass spectroscopy or elementary analysis, or all three (see Materials and Methods and Supporting Information, Table S1). The structures of all compounds were consistent with their analytical and spectroscopic data.
Design, Enzymology, and Binding Affinity.
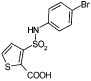
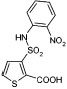
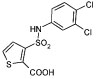
We wished to investigate which groups in the lead inhibitor 1 were critical for binding, and should be maintained, and where we could improve affinity through chemical elaboration. We first focused on the 3-(sulfonylamino)-2-carboxylic acid thiophene moiety of 1 (Figure 1), which in the AmpC complex structure hydrogen bonds with active site residues. For instance, the carboxylate of 1 binds in the “oxyanion” hole of AmpC, hydrogen bonding with main chain amide nitrogens, Ser64, and a conserved water molecule (Wat401), while the sulfonamide hydrogen bonds with the conserved Asn152 of AmpC. Derivates that replaced the thiophene carboxylate with a methyl ester substantially reduced affinity, consistent with the importance of the ion–dipole interactions made by this carboxylate (e.g., compound 1A, Table 1). Similarly, inverting the order of the sulfonyl–amino group reduced affinity 10–20-fold (compare compounds 1 and 2 or 11 and 14, Table 1), and replacing the sulfonamide with an amide reduced affinity 8-fold, consistent with the importance of the hydrogen bond between this group and Asn152 observed crystallographically. We also replaced the thiophene of 1 with a more synthetically tractable benzene. This substitution diminished activity, but by less than 2-fold (compound 4, Table 1).
Table 1.
Ki Values of Thiophene–Carboxy Derivatives vs AmpC
| CODE | STRUCTURE | Kia(μM) |
|---|---|---|
| 1 |

|
26 |
| 1A |
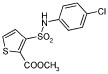
|
>>100b |
| 2 |

|
310 |
| 3 |

|
45 |
| 4 |
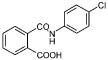
|
340 |
| 5 |

|
26 |
| 6 |
|
31 |
| 7 |

|
1.1 |
| 8 |

|
1.7 |
| 9 |

|
1.9 |
| 10 |
|
1.0 |
| 11 |

|
14 |
| 12 |
|
10 |
| 13 |
|
8.7 |
| 13A |

|
170 |
| 14 |
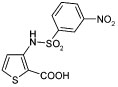
|
250 |
Estimated error margins on the Ki values are ±50%.
No detectable inhibition at 100 μM concentration of compound.
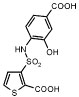
In an attempt to increase the affinity of compound 1, we considered derivates that improved either the apolar or polar complementarity in the distal chloro–phenyl ring of the inhibitor. This functionality is located in a more open region of the active site, flanked by the conserved Tyr221 on the “floor” of the active site, bulk solvent at the “top” of the inhibitor, and polar and apolar residues such as Arg204, Gly320, Thr319, and Val211 to its side. Introducing bulk or hydrophobicity, such as in 5, 6, and 13, affected affinity only modestly. More polar derivates, such as carboxylate and hydroxyl groups, improved affinity more substantially, up to 26-fold in compounds 7–10 (Table 1).
X-ray Crystallographic Structure Determination.
To investigate the structural bases for the higher affinities found for the new series, we determined the crystal structure of AmpC in complex with compound 10 and with compound 11 at 1.97 and 1.96 å resolution, respectively (Table 2). Proline and glycine residues excluded, 92.2% of the amino acids were in the most favored regions of the Ramachandran plot and 7.8% were in the additionally allowed regions for the crystal structure of AmpC/10. For AmpC/11, 92.5% of the nonproline and nonglycine amino acids were in the most favored regions of the Ramachandran plot and 7.5% were in the additionally allowed regions. The positions of both inhibitors in the active sites of each structure were unambiguously identified in the initial Fo–Fc difference map contoured at 3σ. In the AmpC/10 complex, the inhibitor was observed and modeled in both monomers in the asymmetric unit, whereas in the AmpC/11 complex, the inhibitor was only observed and modeled in one of the two monomers.
Table 2.
X-ray Data Collection and Refinement Statistics for AmpC/10 and AmpC/11
| AmpC/10 | AmpC/11 | |
|---|---|---|
| cell constants | ||
| a (Å) | 117.75 | 118.469 |
| b (Å) | 76.77 | 76.279 |
| c (Å) | 97.90 | 97.674 |
| β (deg) | 116.63 | 116.37 |
| space group | C2 | C2 |
| resolution (Å) | 1.97 | 1.96 |
| unique reflcns | 55,165 | 53,587 |
| total obs | 105,106 | 154,202 |
| Rmerge (%) | 9.1 (25.8)a | 5.4 (39.3)a |
| completenessb (%) | 93.7 (94.6) | 95.4 (90.3) |
| resolution range for refinement (Å) | 1.97-20 (1.97-2.04) | 1.96-20 (1.96-2.03) |
| 〈I〉/〈σI;〉 | 9.43 (2.14) | 11.07 (2.09) |
| no. of protein residues | 709 | 700 |
| no. of water molecules | 516 | 313 |
| rmsd bond lengths (Å) | 0.0148 | 0.0085 |
| rmsd bond angles (deg) | 1.7413 | 1.472 |
| Rcryst (%) | 16.84 | 19.81 |
| Rfree (%)c | 21.53 | 22.65 |
| av B factor, protein atoms(Å2; molecules 1 and 2) | 17.71 | 28.57 |
| av B factor, inhibitor atoms(Å2; molecule 1) | 39.78 | |
Highest resolution shell in parentheses. Subsequent values in parentheses are for that shell.
Fraction of theoretically possible reflections observed.
Rfree was calculated with 5% of reflections, randomly selected, set aside.
Overall, the complexes with 10 and 11 resemble that of the AmpC/1 complex, with a few interesting differences. Key hydrogen bond interactions in the conserved parts of all three structures, the thiophene–carboxylate and the sulfonamide functionalities, are maintained (Tables S2 and S3, Supporting Information; Figures 2 and 3). The thiophene carboxylate in both 10 and 11 hydrogen bonds with both the Oγ and the main-chain NH of the hydrolytic Ser64, with a third hydrogen bond formed with the main-chain nitrogen of Ala318 and a fourth with the conserved Wat401. There are two enzyme monomers in the asymmetric unit in the AmpC structures, and in one of them, monomer 2, a fifth hydrogen bonds is formed to Wat481. As with the lead compound 1, the thiophene ring is within van der Waals distance of residues Leu119 and Leu293, which form a hydrophobic patch on AmpC. A sulfonamide oxygen interacts with the Oγof Ser64 and the Nζ of Lys67; the other hydrogen bonds with the Nδ2 of Asn152. As in the AmpC/1 complex, the nitrogen atom of the sulfonamide interacts with the main-chain oxygen of Ala318.
Figure 2.
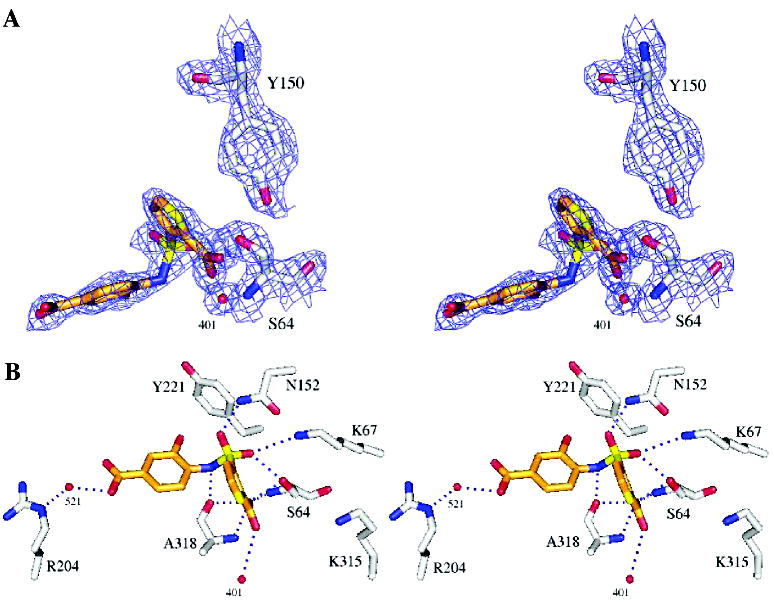
X-ray crystal structure of the AmpC/10 complex. (A) Stereoview of 2Fo – Fc electron density maps contoured at 1σ of the refined model. Carbon atoms are gray for the protein and orange for the ligand; oxygen atoms, red; nitrogen atoms, blue; sulfur atoms, yellow. (B) AmpC/10 complex in the active site. Dashed blue lines represent hydrogen bonds; red spheres represent water molecules. Interaction distances are listed in Table S2, Supporting Information. Figures 2 and 3 were generated with Pymol (http://pymol.sourceforge.net/).
Figure 3.
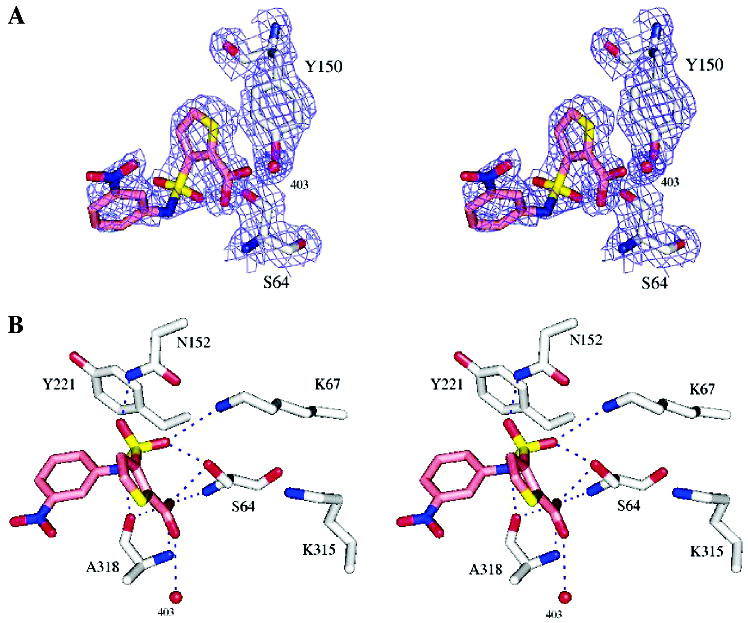
X-ray crystal structure of the AmpC/11 complex. (A) Stereoview of 2Fo – Fc electron density maps (blue, contoured at 1σ) of the refined model. Carbon atoms are colored gray for the protein and pink for the ligand; oxygen atoms, red; nitrogen atoms, blue; sulfur atoms, yellow. (B) AmpC/11 complex in the active site. Dashed blue lines represent key hydrogen bonds. Red spheres represent water molecules. Interaction distances are listed in Table S3, Supporting Information.
Differences emerge in the 4′-carboxylate-2′-hydroxy benzene ring, which is the point of substitution for compound 10. The ring itself appears to form edge-to-face quadrupole interactions with Tyr221 via the introduced 2′-hydroxyl of 10; the distance between this hydroxyl and Cδ1 of Tyr221 is 3.43 Å. This interaction is also present in the structure of the lead 1, but in this case it appears to be a ring–ring or quadrupolar interaction, whereas with 10 the intercession of the hydroxyl makes it a dipole–quadrupolar interaction. The presence of the 2′-hydroxyl also displaces 10 slightly “up” in the site, moving the center of the ring away from Tyr221 and toward polar residues such Arg204 and Thr319, by about 0.2 Å in monomer 2. In both crystallographic monomers 1 and 2 of AmpC the 4′-carboxylate points toward the solvent accessible entry of the binding site. In monomer 1, this carboxylate hydrogen bonds with an ordered water molecule, Wat521 (distance 2.9 Å), which in turn hydrogen bonds with the Nε of Arg204 (distance, 2.8 Å). In monomer 2, Arg204 adopts a different conformation and these interactions are not observed. Instead, a new interaction is observed between Gln120 and the sulfonamide of 10, with the Gln120 in this monomer pointing toward the inhibitors rather than out of the site, as it does in monomer 1. Also in monomer 2, difference electron density suggests that a second conformation of the inhibitor may exist, with the distal phenyl ring rotated by 180°, allowing the 2′-hydroxyl group to hydrogen bond with the side chain of Asn343 through an ordered water molecule.
To investigate how important these polar and quadrupolar interactions might be, we also determined the structure of the AmpC/11 complex by X-ray crystallography. Compound 11 replaces the 4′-carboxylate of 10 with a 3′-nitro and correspondingly loses an order of magnitude in affinity (Table 1). In the structure of the AmpC/11 complex, the inhibitor is moved slightly down in the site, keeping the quadrupole edge-to-face stacking with Tyr221, but this time without the intercession of the 2′-hydroxyl, which is no longer present. The 3′-nitro is relatively distant from Arg204, and we observe no interaction with an ordered water; the loss of these polar interactions may be consistent with the diminished affinity for this inhibitor.
Microbiology.
To investigate the potential of these compounds to reverse antibiotic resistance, we undertook antimicrobial activity studies in bacterial cell culture. Compounds 10 and 11 were tested for synergy with the third generation cephalosporin ceftazidime (CAZ) against pathogenic bacteria from clinical isolates from the Clermont-Ferrand University Hospital (France), and the results were compared with those of the lead compound 1. Strains tested were E. aerogenes 298, E. cloacae P99, C. freundii 63, K. pneumoniae Kus, K. pneumoniae 704, and P aeruginosa CF34. All were resistant to CAZ because of the expression of class C β-lactamases. The minimum inhibitory concentration (MIC) of CAZ against these pathogens ranged from 256 to 64 μg/mL (Table 3; Figure 4).
Table 3.
Synergy of Compounds 10, 11, and 1 with Ceftazidime against Clinical Strains Producing AmpC-type β-lactamase
| MICa (μg/mL)
|
||||
|---|---|---|---|---|
| strains | CAZb | CAZ/10c | CAZ/11c | CAZ/1c |
| E. aerogenes 298 | 128 | 16 | 64 | 32 |
| E. cloacae P99 | 128 | 16 | 128 | 64 |
| C. freundii 63 | 128 | 16 | 64 | 64 |
| K. pneumoniae Kus | 256 | 32 | 256 | 256 |
| K. pneumoniae 704 | 64 | 16 | 64 | 64 |
| P. aeruginosa CF34 | 64 | 64 | 64 | 64 |
Minimum inhibitory concentration.
CAZ = ceftazidime.
The inhibitor:CAZ ratio was 1:1, and the concentration of DMSO was always less than 1%.
Figure 4.

Activity in cell culture of the lead compound 1 and compound 10 vs bacteria from clinical isolates that express AmpC β-lactamase.
Compound 10 increased bacterial susceptibility to CAZ, improving its MIC values by 4- to 8-fold. This was sufficient to partially restore clinically relevant susceptibility for E. Aerogenes 298, E. cloacae P99, C. freundii 63, and K. pneumoniae 704. Conversely, the lead compound 1 and compound 11, both of which have lower affinities than does 10, had relatively little effect on the potency of CAZ. Consistent with this result, 10 synergized the activity of CAZ and the penicillin piperacillin (PIP) in disk diffusion assays, as can be seen by the characteristic tear-drop shapes of the zones of bacterial growth inhibition (Figure 5). This contrasts dramatically with the same experiment performed with the β-lactam-based inhibitors clavulanate and cefoxitin (IC50 of 340 nM vs AmpC; not shown), both of which induce AmpC expression.15,21,22 Although clavulanate and cefoxitin inhibit the AmpC- type β-lactamase expressed by these cells, they nevertheless antagonize the activity of CAZ and PIP in the disk diffusion studies. This results in flattened zones of bacterial grown inhibition, reflecting the up-regulation of β-lactamase production in the E. cloacae bacteria.
Figure 5.

Effects on up-regulation of a class C β-lactamase in E. cloacae by β-lactamase inhibitors, each with piperacillin (PIP) and ceftazidime (CAZ): (A) β-lactam-based inhibitor clavulanate (center); (B) β-lactam-based inhibitor cefoxitin (FOX); (C) non-β-lactam inhibitor 10.
Discussion
There is a pressing need for novel inhibitors to combat antibiotic resistance, but the discovery of such confronts one with new problems. In the case of lead compound 1, a non-β-lactam inhibitor of β-lactamases, the challenges were to improve the binding affinity in a series where one could not simply apply 60 years worth of β-lactam structure–activity relationships and to improve the biological efficacy in a scaffold that, unlike β-lactams, had not been evolved for in vivo activity. Three important results emerge from our preliminary efforts to improve this series of compounds. First, a cycle of structure-based design and testing led to small, soluble inhibitors whose improved affinities could be explained on the basis of the X-ray structures subsequently determined for their complexes. Second, the improved affinity is mirrored by the improved activity in cell culture. Third, new biology follows from the new chemical structures in this series, which, not being β-lactams, simply inhibit β-lactamase and do not up-regulate its expression in cell culture, as do classic β-lactam-based inhibitors such as clavulanate (Figure 5).7
It is encouraging that modest changes in this series can improve inhibition significantly (compare the lead 1 with 10, Table 1). Also, the resulting analogues remain relatively soluble and small, leaving further room for chemical elaboration. Indeed, an encouraging aspect of this series is that increased affinity comes from polar groups that act to maintain the overall solubility of the compounds, most particularly the addition of a carboxylate on the distal phenyl ring. Exactly what interactions are responsible for the improved affinity is less certain. The crystal structures of the 1.0 μM inhibitor 10 and the 14 μM inhibitor 11 in complex with AmpC suggest that water-mediated interactions with Arg204 are responsible for the improved affinity of 10. This conclusion must be seen as quite tentative at this point, since this interaction is only seen in one of the two monomers of the AmpC/10 complex. From a molecular recognition standpoint, it is surprising that a water-mediated hydrogen bond between the carboxylate and Arg204, in a relatively solvent-exposed region, can significantly improve affinity, though this sort of effect is not unprecedented in AmpC.23 Other explanations are possible; for instance the improved affinity may derive from an overall improved complementarity to the highly electropositive active site of AmpC. Detailed conclusions as to the importance of these interactions must remain tentative pending more rigorous analyses, such as double-perturbation thermodynamic cycles;23,24 what we can say is that the X-ray structures of these complexes provide templates for designing and understanding such experiments.
What might justify this effort is that the improved enzyme affinity of inhibitor 10 results in 4- to 8-fold improvement in antibiotic efficacy. In bacterial cell culture, a 1:1 ration of 10 to CAZ synergizes the activity of the latter against widespread hospital pathogens such as E. cloacae and C. freundii (Table 3 and Figure 4) and is significantly more potent than the lead compound 1 or compound 11. This antibiotic effect suggests that this series of inhibitors, whose Ki values remain relatively modest, may be useful leads for further optimization. The X-ray crystal structures of compound 10 in complex with AmpC provide atomic resolution templates for future design efforts.
Perhaps the most striking result to emerge from this study, however, comes from comparing the effects of the classic β-lactam-based inhibitors clavulanate and cefoxitin with those of compound 10 on an inducible strain of E. cloacae. These bacteria up-regulate the expression of class C β-lactamase in the presence of β-lactams such as clavulanate, thereby overwhelming these β-lactams with the very enzyme they are meant to inhibit (Figure 5A,B). Conversely, compound 10, which is not a β-lactam, simply inhibits the β-lactamase expressed by these bacteria, it does not lead to its up-regulation. Thus, the novel chemical structures represented by this series of inhibitors leads to novel, therapeutically relevant biological effects.
Materials and Methods
Chemistry.
All reagents were purchased from Sigma-Aldrich or Fluka and were reagent grade. Progress of the reaction was monitored by thin-layer chromatography (TLC) on silica gel plates (Riedel-de-Haen, Art. 37341). Merck silica gel (Kieselgel 60) was used for flash chromatography (230–400 mesh) when required. Extracts were dried over MgSO4, and solvents were removed under reduced pressure.
Melting points were determined on a Büchi 510 capillary melting point apparatus and are uncorrected. Elemental analyses were performed on a Perkin-Elmer Elemental Analyzer 240C. 1H NMR spectra were recorded on a Bruker DPX 200 MHz spectrometer (Centro Interdipar-timentale Grandi Strumenti Università di Modena) with trimethylsilane (TMS) as internal standard; the values of the chemical shifts (δ) are given in parts per million and coupling constants (J) in hertz. Dimethyl sulfoxide-d6 (DMSO-d6) was used as the solvent. Mass spectra were determined on a Finnigan MAT SSQ 710 A mass spectrometer (EI, 70 eV).
3-((4-Chlorophenyl)sulfamoyl)thiophene-2-carboxylic Acid Methyl Ester (1A).
The starting amine, 4-chlorophenylamine (35 mg, 0.28 mmol) was dissolved in 10 mL of anhydrous pyridine, and the 3-(chlorosulfonyl)thiophene-2-carboxylic acid methyl ester (100 mg, 0.42 mmol) was added dropwise to the solution under stirring. The reaction was stirred for 24 h, at 50 °C, under nitrogen. The resulting solution was treated with water and ice; the crude product, corresponding to the 3-((4-chlorophenyl)sulfamoyl)thiophene-2-carboxylic acid methyl ester (1A) was formed and isolated through filtration. Yield: 73 mg, 80%; mp 87–90 °C. Anal. Calcd for C12H10ClNO4S2: C, 43.44;H, 3.04; N, 4.22. Found: C, 43.51; H, 3.10; N, 4.28.
3-((4-Chlorophenyl)sulfamoyl)thiophene-2-carboxylic Acid (1).
Compound 1A (60 mg, 0.18 mmol) was suspended in 10 mL of 2 N NaOH, and the reaction was stirred for 1 h at 80 °C, until the starting material was completely dissolved. Thereafter, the solution was cooled, and dilute HCl was added until the solution reached pH 2. A precipitate, corresponding to 3-((4-chlorophenyl)sulfamoyl)thiophene-2-carboxylic acid (1) was formed and isolated through filtration. Yield: 42 mg, 73%; mp 195–198 °C. 1H NMR: 7.97 (H1, d, J 5.4 Hz); 7.49 (H2, d, J 5.4 Hz); 7.21 (H3, d/o, J 8.8 Hz); 7.40 (H4, d/o, J 9 Hz); 7.40 (H5, d/o, J 9 Hz); 7.21 (H6, d/o, J 8.8 Hz); 10.20 (H7, s). Anal. Calcd for C11H8ClNO4S2: C, 41.58; H, 2.54; N, 4.41. Found: C, 41.45; H, 2.48; N,4.51.
Intermediate ester derivatives (2A–14A) and final carboxylic acids (2–14) were synthesized according to the method followed for 1A and 1.Details are reported in the Supporting Information.
Enzymology.
Inhibitors were dissolved in DMSO at a concentration of 50 mM; more dilute stocks were subsequently prepared as necessary. Kinetic measurements were performed using nitrocefin as substrate in 50 mM Tris buffer, pH 7.0, and monitored in an HP8453 UV–vis spectrophotometer. The Km of nitrocefin for AmpC in this buffer was 127 μM. The concentration of AmpC was determined spectrophotometrically in concentrated stock solutions made from lyophilized powder and subsequently diluted; this enzyme had been previously expressed and purified, as described.15 The concentration of enzyme in all reactions was 1.75 nM. Inhibition Ki values were obtained from IC50 plots assuming competitive inhibition, an assumption consistent with both previous inhibition patterns in this series16 and with experiments investigating the effect of increasing substrate concentrations (not shown). For compound 10, the Ki value was also obtained by comparison of progress curves in the presence and in the absence of inhibitor;18,25,26 the result was consistent with the value determined from the IC50 plots.
Crystal Growth and Structure Determination.
Cocrystals of AmpC in complex with compounds 10 and 11 were grown by vapor diffusion in hanging drops equilibrated over 1.8 M potassium phosphate buffer (pH 8.7) using microseeding techniques. The initial concentration of the protein in the drop was 3.8 mg/mL, and the concentrations of compounds 10 and 11 were 0.8 mM. The compounds were added to the crystallization drops in a 1.2% DMSO, 1 M potassium phosphate buffer (pH 8.7) solution. Crystals appeared a few weeks after equilibration at 21 °C. Before data collection, crystals were immersed in a cryoprotectant solution of 20% sucrose, 1.8 M potassium phosphate, pH 8.7, for about 30 s, and were flash-cooled in liquid nitrogen.
Diffraction data were collected on frozen crystals at the Advance Light Source (ALS, Lawrence Berkeley Laboratory, CA). Both data sets were measured from single crystals. Reflections were indexed, integrated, and scaled using the HKL software package.27 For both structures, the space group was C2, with two molecules in the asymmetric unit. For AmpC/10, molecule 1 of the asymmetric unit was modeled with 352 residues, including the inhibitor (7 residues, 284–290, were left out due to poor density in this region, which is often disordered in AmpC structures), and molecule 2 was modeled with 359 residues, including the inhibitor. For AmpC/ 11, molecule 1 was modeled with 342 residues (16 residues were left out), and molecule 2 of the asymmetric unit was modeled 359 residues, including the inhibitor. The initial phasing models were an apo AmpC structure (PDB entry 1KE4) for AmpC/10 and an AmpC/boronic acid complex (PDB entry 1MY8) for AmpC/11, with inhibitor, water molecules, and ions removed. The models were positioned using rigid body refinement and refined using the maximum likelihood target in CNS including: simulated annealing, positional minimization, and individual B-factor refinement, with a bulk solvent correction.28 σ A-weighted electron density maps were calculated using CNS and used in further steps of manual model rebuilding and placement of water molecules with the programs XtalView and O.29,30 The inhibitors were built into the 2Fo – Fc and Fo – Fc electron density maps in each active site of the asymmetric unit. Subsequent refinement cycles consisted of positional minimization and B-factor refinement in CNS.
Microbiology.
Compounds 10 and 11 were tested for synergy with the β-lactam ceftazidime against pathogenic bacteria from clinical isolates at the Clermont-Ferrant Hospital (France); these bacteria were resistant to β-lactams because of the expression of class C β-lactamases. Strains of bacteria tested were E. aerogenes 298, E. cloacae P99, C. freundii 63, K. pneumoniae Kus, K. pneumoniae 704, and P. aeruginosa CF34.31–33 Minimum inhibitor concentration values were determined with Mueller-Hinton broth using the microdilution method according to NCCLS guidelines.34 To test the inhibitory activity, compounds were dissolved in DMSO and dilutions were performed using growth medium. An adequate final concentration in which to determine the MIC was obtained where the concentration of DMSO was <1%. The ratio Inhib/CAZ was 1. A control done with DMSO showed that DMSO did not have an effect in the bacteria growth.
For the β-lactamase induction experiments, plates of Mueller-Hinton agar were inoculated with a clinical strain of E. cloacae in which production of AmpC is inducible by β-lactams (Clermont-Ferrand hospital, unpublished strain). Inhibitors were added to blank disks, and the final content of inhibitor per disk was 128 μg for compound 10 and 8 μg for clavulanic acid. Disks of ceftazidime, cefoxitin, and piperacillin contained 30, 30, and 75 μg, respectively.35
Data Deposition.
The coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 1XGJ for AmpC in complex with compound 10 and under accession code 1XGI for AmpC in complex with 11.
Acknowledgments
This work was supported by Grant GM63815 (to B.K.S.). We thank Piergiorgio Pecorari for his valuable suggestions and Alberto Venturelli for conversations, Yu Chen and Veena Thomas for reading this manuscript, and Jesus Blazquez for introducing some of us to synergy and antagonism tests for β-lactamase inhibitors.
Footnotes
Supporting Information Available: Detailed information related to the synthesis and chemical characterization of compounds 2-14 and a table listing all compounds characterized through 1H NMR and their numbering (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Rice LB, Bonomo RA. beta-Lactamases: Which Ones Are Clinically Important? Drug Resist Updates. 2000;3(3):178–189. doi: 10.1054/drup.2000.0144. [DOI] [PubMed] [Google Scholar]
- 2.Cosgrove S, Carmeli Y. The Impact of Antimicrobial Resistance on Health and Economic Outcomes. Clin Infect Dis. 2003;36:1433–1437. doi: 10.1086/375081. [DOI] [PubMed] [Google Scholar]
- 3.Chambers HF. Penicillin-Binding Protein-Mediated Resistance in Pneu-mococci and Staphylococci. J Infect Dis. 1999;179(Suppl 2):353– 359. doi: 10.1086/513854. [DOI] [PubMed] [Google Scholar]
- 4.Li XZ, Nikaido H. Efflux-Mediated Drug Resistance in Bacteria. Drugs. 2004;64(2):159–204. doi: 10.2165/00003495-200464020-00004. [DOI] [PubMed] [Google Scholar]
- 5.Li XZ, Zhang L, Nikaido H. Efflux Pump-Mediated Intrinsic Drug Resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother. 2004;48(7):2415–2423. doi: 10.1128/AAC.48.7.2415-2423.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiolas A, Bornet C, Davin-Regli A, Pages JM, Bollet C. Resistance to Imipenem, Cefepime, and Cefpirome Associated with Mutation in Omp36 Osmoporin of Enterobacter Aerogenes. Biochem Biophys Res Commun. 2004;317(3):851–856. doi: 10.1016/j.bbrc.2004.03.130. [DOI] [PubMed] [Google Scholar]
- 7.Bennett PM, Chopra I. Molecular Basis of beta-Lactamase Induction in Bacteria. Antimicrob Agents Chemother. 1993;37(2):153–158. doi: 10.1128/aac.37.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Essack SY. The Development of beta-Lactam Antibiotics in Response to the Evolution of beta-Lactamases. Pharm Res. 2001;18(10):1391–1399. doi: 10.1023/a:1012272403776. [DOI] [PubMed] [Google Scholar]
- 9.Medeiros AA. Cooperative Evolution of Mechanisms of beta-Lactam Resistance. Clin Microbiol Infect. 2000;6(Suppl 3):27–33. doi: 10.1111/j.1469-0691.2000.tb02037.x. [DOI] [PubMed] [Google Scholar]
- 10.Meroueh SO, Roblin P, Golemi D, Maveyraud L, Vakulenko SB, Zhang Y, Samama JP, Mobashery S. Molecular Dynamics at the Root of Expansion of Function in the M69L Inhibitor-Resistant TEM β-Lacta-mase from Escherichia coli. J Am Chem Soc. 2002;124:9422–9430. doi: 10.1021/ja026547q. [DOI] [PubMed] [Google Scholar]
- 11.Sun T, Bethel CR, Bonomo RA, Knox JR. Inhibitor-Resistant Class A β-Lactamases: Consequences of the Ser130-to-Gly Mutation Seen in Apo and Tazobactam Structures of the SHV-1 Variant. Biochemistry. 2004;43:14111–14117. doi: 10.1021/bi0487903. [DOI] [PubMed] [Google Scholar]
- 12.Bush K. beta-Lactamases of Increasing Clinical Importance. Curr Pharm Des. 1999;5(11):839–845. [PubMed] [Google Scholar]
- 13.Philippon A, Arlet G, Jacoby G. Plasmid-Determined AmpC-Type beta-Lactamases. Antimicrob Agents Chemother. 2002;46(1):1–11. doi: 10.1128/AAC.46.1.1-11.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanson ND, Sanders CC. Regulation of Inducible AmpC beta-Lactamase Expression among Enterobacteriaceae. Curr Pharm Des. 1999;5(11):881–894. [PubMed] [Google Scholar]
- 15.Powers RA, Blazquez J, Weston GS, Morosini MI, Baquero F, Shoichet BK. The Complexed Structure and Antimicrobial Activity of a Non-beta-lactam Inhibitor of AmpC beta-Lactamase. Protein Sci. 1999;8(11):2330–2337. doi: 10.1110/ps.8.11.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powers RA, Morandi F, Shoichet BK. Structure-Based Discovery of a Novel, Noncovalent Inhibitor of AmpC beta-Lactamase. Structure (London) 2002;10(7):1013–1023. doi: 10.1016/s0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 17.Konaklieva MI. Lactams as Inhibitors of Serine Enzymes. Curr Med Chem: Anti-Infect Agents. 2002;1(3):215–238. [Google Scholar]
- 18.Caselli E, Powers RA, Blasczcak LC, Wu CY, Prati F, Shoichet BK. Energetic, Structural, and Antimicrobial Analyses of beta-Lactam Side Chain Recognition by beta-Lactamases. Chem Biol. 2001;8(1):17–31. doi: 10.1016/s1074-5521(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 19.Trehan I, Morandi F, Blaszczak LC, Shoichet BK. Using Steric Hindrance To Design New Inhibitors of Class C beta-Lactamases. Chem Biol. 2002;9(9):971–980. doi: 10.1016/s1074-5521(02)00211-9. [DOI] [PubMed] [Google Scholar]
- 20.Oprea TI. Current Trends in Lead Discovery: Are We Looking for the Appropriate Properties? Mol Diversity. 2002;5(4):199–208. doi: 10.1023/a:1021368007777. [DOI] [PubMed] [Google Scholar]
- 21.Kadima TA, Weiner JH. Mechanism of Suppression of Piperacillin Resistance in Enterobacteria by Tazobactam. Antimicrob Agents Chemother. 1997;41(10):2177–2183. doi: 10.1128/aac.41.10.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders CC, Bradford PA, Sanders WEJ. Penicillin-Binding Proteins and Induction of AmpC β-Lactamase. Antimicrob Agents Chemother. 1997;41:2013–2015. doi: 10.1128/aac.41.9.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roth T, Minasov G, Shoichet B. Thermodynamic Cycle Analysis and Inhibitor Design against β-Lactamase. Biochemistry. 2003;42:14483–14491. doi: 10.1021/bi035054a. [DOI] [PubMed] [Google Scholar]
- 24.Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MMY, Winter G. Hydrogen Bonding and Biological Specificity Analysed by Protein Engineering. Nature. 1985;314:235. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 25.Waley GS. A Quick Method for the Determination of Inhibition Constants. Biochem J. 1982;205:631–633. doi: 10.1042/bj2050631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weston GS, Blazquez J, Baquero F, Shoichet BK. Structure-Based Enhancement of Boronic Acid Inhibitors of AmpC β-Lactamase. J Med Chem. 1998;41:4577–4586. doi: 10.1021/jm980343w. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol, Macromol Crystallogr, Part A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.McRee DE. XtalView/Xfit–A Versatile Program for Manipulating Atomic Coordinates and Electron Density. J Struct Biol. 1999;125(2–3):156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 30.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved Methods for Building Protein Models in Electron Density Maps and the Location of Errors in These Models. Acta Crystallogr, Sect A: Found Crystallogr. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 31.Bauernfeind A, Schneider I, Jungwirth R, Sahly H, Ullmann U. A Novel Type of AmpC beta-Lactamase, ACC-1, Produced by a Klebsiella Pneumoniae Strain Causing Nosocomial Pneumonia. Antimicrob Agents Chemother. 1999;43(8):1924–1931. doi: 10.1128/aac.43.8.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Champs C, Poirel L, Bonnet R, Sirot D, Chanal C, Sirot J, Nordmann P. Prospective Survey of beta-Lactamases Produced by Ceftazi-dime-Resistant Pseudomonas aeruginosa Isolated in a French Hospital in 2000. Antimicrob Agents Chemother. 2002;46(9):3031–3034. doi: 10.1128/AAC.46.9.3031-3034.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fleming PC, Goldner M, Glass DG. Observations on the Nature, Distribution, and Significance of Cephalosporinase. Lancet. 1963;29(1):1399–1401. doi: 10.1016/s0140-6736(63)92051-8. [DOI] [PubMed] [Google Scholar]
- 34.NCCLS, National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Tests for Bacteria That Grow Aerobically Approved Standard, 4th ed.; NCCLS document M7-A5; NCCLS: Wayne, PA, 2000.
- 35.Jarlier V, Nicolas M, Fournier G, Philippon A. Extended Broad-Spectrum beta-Lactamases Conferring Transferable Resistance to Newer beta-Lactam Agents in Enterobacteriaceae: Hospital Prevalence and Susceptibility Patterns. Rev Infect Dis. 1988;10(4):867–878. doi: 10.1093/clinids/10.4.867. [DOI] [PubMed] [Google Scholar]