

Abstract
A critical barrier to entry into structure-based virtual screening is the lack of a suitable, easy to access database of purchasable compounds. We have therefore prepared a library of 727 842 molecules, each with 3D structure, using catalogs of compounds from vendors (the size of this library continues to grow). The molecules have been assigned biologically relevant protonation states and are annotated with properties such as molecular weight, calculated LogP, and number of rotatable bonds. Each molecule in the library contains vendor and purchasing information and is ready for docking using a number of popular docking programs. Within certain limits, the molecules are prepared in multiple protonation states and multiple tautomeric forms. In one format, multiple conformations are available for the molecules. This database is available for free download (http://zinc.docking.org) in several common file formats including SMILES, mol2, 3D SDF, and DOCK flexibase format. A Web-based query tool incorporating a molecular drawing interface enables the database to be searched and browsed and subsets to be created. Users can process their own molecules by uploading them to a server. Our hope is that this database will bring virtual screening libraries to a wide community of structural biologists and medicinal chemists.
INTRODUCTION
Structure-based virtual screening has had several important successes in recent years1–10 and is now a common technique in early stage drug discovery at most pharmaceutical companies as well as some university groups. Unfortunately, virtual screening techniques continue to require expert knowledge and extensive infrastructure and remain out of reach for many medicinally—and biologically—oriented investigators who might otherwise be able to exploit them. Among the steepest barriers to entry is the lack of a suitable database of small molecules with which to screen. These databases are either expensive to acquire or time-consuming and difficult to prepare and curate. To be useful for structure-based screening, 3D structures must be calculated for each available molecule. The structures must be linked to the supplier information, itself requiring some database design. More difficult are the problems of calculating multiple protonation, stereo- and regiochemical, tautomeric, and conformational states for the database molecules. Computing these multiple molecular states is challenging and is the focus of ongoing research.11–13 Finally, as supplier catalogs are often updated monthly, considerable curatorial work is required to remain current.
The “gold standard” for docking databases, at least in academic groups, has been the Available Chemicals Directory (ACD) from Molecular Design Limited (http://www.mdli.com, San Leandro, CA). This database contains about 250 000 purchasable compounds, while the screening compound analogue, the ACD-SC, has over 2.3 million compounds. The ACD has been extensively curated for chemical correctness, is compatible with corporate information systems using Oracle, and has molecules that are available as 3D models. Even the ACD, however, requires extensive post curation. For instance, correct protonation states for docking must be assigned. Many ACD molecules have counterions that must be removed prior to docking. Each molecule is present in only one tautomeric form, sometimes not the biologically relevant one. Only a single conformation of each molecule is available. Finally, the ACD is a commercial product that is often too expensive for nonspecialist labs to purchase and maintain. Similarly, the ChemNavigator database (http://www.chemnavigator.com) contains over 10 million unique purchasable drug-like compounds but is also neither entirely ready for docking nor free. Again, dealing with protonation, charge, tautomeric forms, and salts is left to the user.
Several free collections of small molecules are available, though none is entirely satisfactory for docking. The Ligand.Info database (http://Ligand.info)14 contains about 1 million compounds from various free databases. Although it contains 3D structures, there has been little effort so far to tautomerize, protonate, and charge them for docking. Furthermore, many of its compounds are not purchasable. The ChemBank project15 (http://chembank.med.harvard.edu) currently contains about 900 000 molecules, many annotated for function. As this is a 2D database, it is unsuitable for structure based screening as is.
In principle, a virtual screening library could be built from the 2D compound information provided by many compound suppliers. Indeed, this is what we have ultimately done with ZINC. To do so, a 3D structure must be generated, typically using the 2D molecular description supplied by the vendors; these often contain stereo- or regioisomeric ambiguities. The correct protonation state, charge, and tautomeric forms must be enumerated or chosen. To avoid wasted effort, insoluble, reactive, and aggregating compounds should be eliminated.
In an effort to make virtual screening more accessible to a broad community, we describe here a free database of purchasable molecules, many of them “drug-like” or “lead-like”, available in several 3D formats immediately usable by many popular docking programs. The salient criteria for our database, ZINC, an acronym for “ZINC is not commercial”, are as follows. Compounds should be purchasable for rapid testing of docking hypotheses. Subsets of molecules with variable properties such as functional groups, molecular weight, and calculated logP should be easy to create and manipulate. The database must support multiple protonation models, tautomeric forms, stereochemistries (e.g. racemic mixtures as well as stereochemically pure compounds), regioisomeric forms (E/Z isomerism), suppliers, and 3D conformational sampling. It should be possible to annotate molecules using both numeric and alphanumeric data. It should be easy to add new molecules, tag, or remove those that are no longer available and fix those that have errors. The database should be quick to search and download, and it should be straightforward to obtain regular updates.
METHODS
Compound Sources and Filters.
We use 10 vendor catalogs, most of which are updated monthly on the Web or CD-ROM (Table 1A). We filter-out molecules with formula weight greater than 700, calculated LogP greater than 6 and less than − 4, number of hydrogen-bond donors greater than 6, number of hydrogen-bond acceptors greater than 11, and number of rotatable bonds greater than 15. We also remove all molecules containing an atom other than H, C, N, O, F, S, P, Cl, Br, or I. We do make exceptions, for example, to include a number of actual drugs that violate these constraints; these rules are guidelines toward making the database loosely conform to current opinion in the field.
Table 1.
A. Compounds in ZINC by Vendor and B. Purchasable Compounds in ZINC by Compound Class
| A. | |||
|---|---|---|---|
| Supplier | in catalog | loaded into ZINC | exclusivea |
| ChemBridge | 324 536 | 50 116 | 30 319 |
| ChemDiv | 359 103 | 108 327 | 77 129 |
| Ryan | 303 939 | 78 791 | 67 943 |
| Asinex | 241 310 | 159 263 | 126 257 |
| Sigma-Aldrich | 149 390 | 101 942 | 59 042 |
| Maybridge | 45 253 | 26 099 | 13 526 |
| Specs | 174 245 | 61 778 | 59 310 |
| Comgenex | 166 132 | 20 221 | 20 214 |
| Otava | 94 912 | 75 692 | 54 129 |
| B. | |||
| molecule class | in database | mean number of rotatable bonds | mean molecular weight |
| Total | 727 842 | 5.8 | 387 |
| Lipinski-compliant | 494 915 | 4.8 | 339 |
| lead-like | 202 134 | 3.5 | 273 |
| fragment-like | 34 224 | 1.2 | 196 |
The number of molecules that are only sold by this vendor.
Molecule Preparation Protocol.
We obtain molecules from compound suppliers as 2D SDF files and convert them to isomeric SMILES using OpenEye’s convert.py tool (OpenEye Scientific Software, http://www.eyesopen.com). We use OpenEye’s filter.1.0.2 program to desalt the molecules and filter out undesirable molecules (a modified version of the filter_light.txt parameter file is included in the Supporting Information). Typically, over 70% of compounds are achiral and have no regioisomeric (E/Z) ambiguity. For the remainder, the information available from suppliers is often ambiguous. Fortunately, most of these have only one or two centers of ambiguity. Our choices include enumerating and processing the implied isomers or to make an educated guess at a single form. ZINC allows the user to make either choice. We enumerate up to four isomers corresponding to up to two centers. We also allow the user to obtain a single representation of each molecule, for example, for faster screening and a smaller database. It is also possible to select subsets of the database that have no ambiguities.
A single substance may be represented by more than one SMILES string. To ensure uniqueness in the database, we calculate a canonical representation with OpenEye’s OEchem library. OpenEye’s Omega program is then used to generate initial 3D models from unambiguous isomeric SMILES. Schrödinger’s ligprep program (Schrödinger, Inc., www.schrodinger.com) is employed to create relevant, correctly protonated forms of the molecule between pH 5 and 9.5. (Modified versions of some ligprep parameter files are included in the Supporting Information.) This includes deprotonating carboxylic acids and tetrazoles and protonating most aliphatic amines, for example. The semiempirical quantum mechanical program AMSOL16 calculates the partial atomic charges and atomic desolvation penalties for a single 3D conformation of each protonation state, stereoisomer, and tautomer.17 OpenEye’s program Omega generates 3D conformations, which are distilled into a flexibase format using our own program mol2db.18,19 We use Omega because it calculates accessible conformations relatively accurately and efficiently.20,21 We note that the calculation of small molecule conformations remains an active area of research in the field.
Molecules in ZINC are annotated by molecular property. These include molecular weight, number of rotatable bonds, calculated LogP, number of hydrogen-bond donors, number of hydrogen-bond acceptors, number of chiral centers, number of chiral double bonds (E/Z isomerism), polar and apolar desolvation energy (in kcal/mol), net charge, and number of rigid fragments.
The calculated octanol–water partition coefficient (calculated LogP) that is calculated for every molecule that is loaded into ZINC uses the fragment-based implementation by Molinspiration22 and agrees well with experimentally measured LogP23 for a diverse test set of molecules.23 This implementation, which at least partly draws on the xLogP algorithm of Wang,24 is robust, handling a broad range of chemistry. For the filtering step, in which we decide whether a molecule should be loaded into ZINC in the first place, we used OpenEye’s implementation of calculated LogP, which uses Wang’s algorithm,24 because it is an integral part of their filtering tools, which we use for this step in the processing.
Each molecule is also annotated with the vendor and original catalog number for each commercial source of that compound. Molecules may also be annotated for function or activity, when available. After molecules have been processed using this protocol, whether from a vendor’s catalog or as a result of a Web-originated request, they are loaded into the relational database using a Perl script (see Supporting Information Table S1).
Database Structure.
We have designed a database schema to organize data relationally, in a way that is compatible with our goals of efficient loading, incremental updates, querying, and data subsetting (Supporting Information Figure S1). Relational databases are fast, efficient, and, in the case of MySQL, free. With a relational-only structure however, there was some concern that exporting subsets of the database would be slow. To address this problem, molecule subsets are exported from the database into ready-to-download compressed files, and database-intensive work is scheduled in batch mode. Once prepared, subsets may be downloaded rapidly, completely bypassing the relational database. We use MySQL 4.0 and the Perl DBI/DBD toolkit. We use OE’s depict tool (part of the Ogham Suite) to render 2D depictions. We use the Cactvs suite25 and the software of Molinspiration (http://www.molinspiration.com) for proofreading, canonicalization, and property calculations.
RESULTS
ZINC is now available for download (http://zinc.docking.org). It is currently built from the catalogs of ten major compound vendors (Table 1A) and presently contains 727 842 purchasable compounds. The number of molecules in ZINC is growing, and the numbers reported here should be considered a representative snapshot; see the Web-page for up-to-date statistics. Of these 727842, 494 915 are Lipinski compliant,26 with the caveat that we have used Molinspiration’s LogP as a surrogate for cLogP. Of these, 202 134 are “lead-like”27–29 molecules, which we define here as having molecular weight between 150 and 350, calculated LogP less than four, number of hydrogen-bond donors less than or equal to three, and number of hydrogen-bond acceptors less than or equal to six. A total of 34 224 molecules are “fragment-like”,30 with calculated LogP values between − 2 and 3, less than three hydrogen-bond donors, less than six hydrogen-bond acceptors, less than three rotatable bonds, and molecular weight less than 250 (Table 1B, Figure 1).
Figure 1.
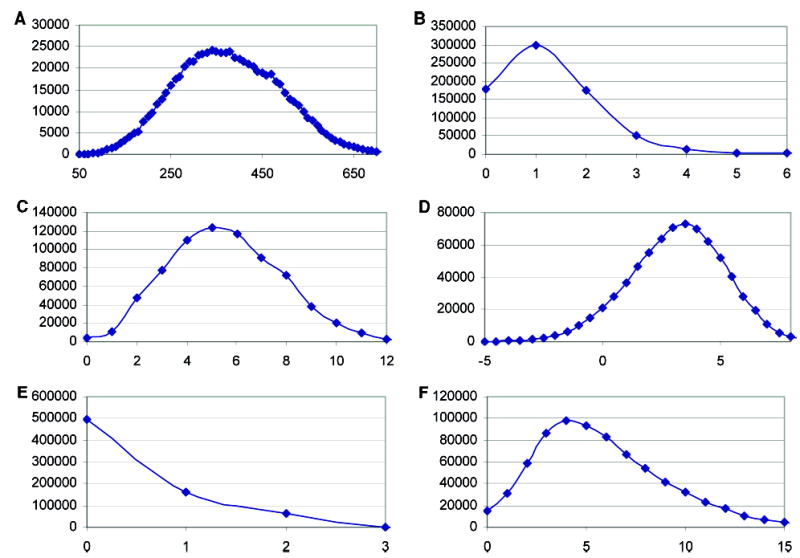
Molecular properties of compounds in ZINC. A. molecular weight, B. hydrogen-bond donors, C. hydrogen-bond acceptors, D. calculated LogP, E. violations of Lipinski’s rule-of-fives, and F. rotatable bonds.
The molecular properties of the molecules in ZINC loosely conform to current opinion in the field about eligible compounds for screening (Figure 1). As it is easy to make subsets using stricter criteria, we have deliberately relaxed our compound filters to include a number of molecules at the periphery of what many investigators might consider desirable. The molecular weight (Figure 1A), number of hydrogen-bond donors (Figure 1B), number of hydrogen-bond acceptors (Figure 1C), and calculated LogP (Figure 1D) are widely used parameters of “drug-likeness”. The number of single violations of the Lipinski rules26 (Figure 1E) in ZINC is largely due to high calculated LogP values. We have tolerated higher levels of calculated LogP than proscribed by the Lipinski rules because of the uncertainties in the calculated values. Another widely followed metric of suitability for screening are the number of rotatable bonds (Figure 1F); more than half of the molecules in the ZINC database have five or fewer.
A Web server has been established to distribute the ZINC database, allowing investigators to search, browse, subset, and download some or all of the molecules in SMILES, mol2, SDF, and DOCK flexibase18,19 formats. Users may also upload and process molecules on the server. We have used the relational database program MySQL to implement ZINC because it is fast, robust, and free. The current MySQL implementation of ZINC occupies about 4 GB of disk space (Supporting Information Table S1). The molecule files in mol2 and DOCK flexibase format use an additional 150GBof disk space when compressed. The Web server requires 350 GB of temporary storage for database subsets and other files, which are derived from the database and help to optimize download performance. The ZINC Web server runs on a dual processor Xeon 2.4GHz server, has a similar machine dedicated solely to running MySQL, and can draw on a 50-CPU 2.4 GHz Xeon Linux cluster for processing.
Searching ZINC.
Users may search ZINC based on several criteria (Figure 2A). Limits on molecular properties such as net charge and molecular weight may be specified on the left-hand side of the search Web page (Figure 2A). On the bottom left, individual ZINC database registration codes, the unique serial number assigned to each substance in ZINC, may be specified, either by typing them or choosing a text file of codes to upload from the browsing computer. Molecules matching any of the ZINC codes specified will be found. A constraint on the compound vendor may also be specified. On the right, molecular substructures may be drawn using the Java Molecular Editor (JME).31 A list of SMILES32 strings in a text file may also be uploaded and used to search.
Figure 2.
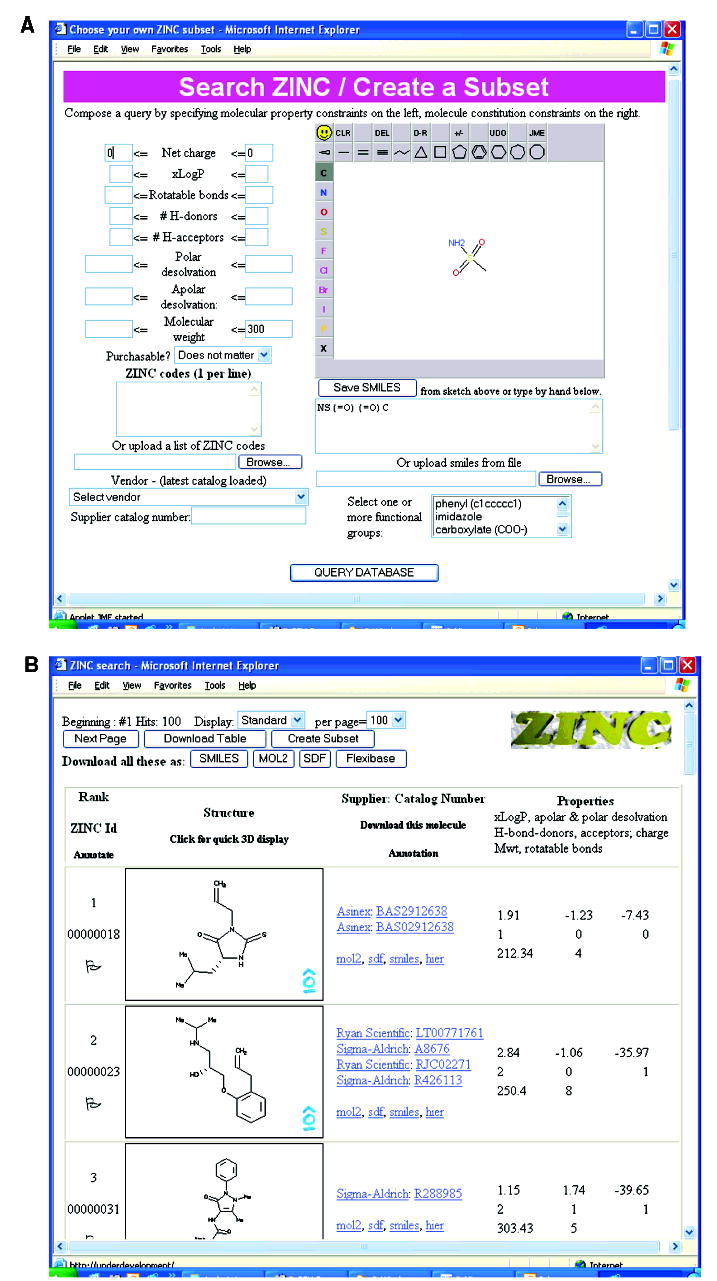
A. The ZINC search tool and B. the ZINC database browser.
Database Browser.
Results of the search may be reviewed using the Database Browser (Figure 2B). Whereas most Web queries can be answered in half a minute or less, complex queries or multiple simultaneous requests may take longer. The Database Browser displays molecules in a table containing ZINC registration code, a 2D sketch, purchasing information, and molecular properties such as calculated LogP and number of rotatable bonds (Figure 2B). Clicking on a vendor’s catalog number links to the vendor’s e-commerce Website, if available. The following options are also available: (a) download individual molecules or the set of all molecules matched in SMILES, mol2, SDF, and DOCK flexibase formats, (b) download a table of molecular properties including purchasing information for analysis in a spreadsheet, and (c) create a subset for docking or download.
Database Subsets.
Many users may only be interested in some of the molecules in ZINC. The ZINC Web pages allow the download of subsets by vendor and other criteria such as Lipinski-compliant,26 “lead-like”,27–29 and “fragment-like”30 compounds. The search page may be used to download small subsets immediately or to create user-defined subsets using arbitrary criteria, including functional groups and molecular properties. Once prepared, each subset is available in SMILES, mol2, SDF, and DOCK flexibase format. Large files are broken into slices of approximately 20 to 100MB for easier download. In the limit, the entire ZINC database may be downloaded.
Process your Own Molecules.
Users may upload their own molecules to the ZINC server in SMILES, SDF, or mol2 formats and have them processed using the same protocol we use to build ZINC. The uploaded molecules subsequently appear as a subset for download in the usual way and disappear from the server after a week.
Sample Uses of ZINC.
Some possible uses of ZINC may be illustrated by example.
Search Example 1: Build a Library of Small Neutral Compounds Containing a Sulfonamide.
In the ZINC search page (Figure 2A), the user draws a sulfonamide group into the JME editor and clicks “Save SMILES” or simply types the SMARTS pattern “NS(=O)(=O))[#6]” directly into the SMILES field. Using the molecular properties fields (middle left, Figure 2A), a maximum molecular weight of 300 is specified. A minimum and a maximum molecular charge of zero is input (left top, Figure 2A). If a search is conducted using these criteria, a browser displaying the 2548 purchasable compounds in ZINC satisfying these constraints will appear (this search takes about 20 s when conducted locally). With this list, further analyses are possible, for instance, to inquire what the distribution of calculated LogP values in this subset are. This can be obtained by clicking on “Download table” to bring up a spreadsheet in Excel, from which calculated LogP vs molecular weight may be graphed. If the range of values of this subset is satisfactory, the user may then return to the ZINC Database Browser and download this subset in mol2, SDF, SMILES, or flexibase format by clicking on the appropriate button at the top of the page. Vendor information is included with this subset.
Search Example 2: Obtain Purchasing Information for Top Scoring Hits.
The user has already downloaded the “drug-like” subset of the ZINC database and has docked it against a target of interest using his own docking program. The user would now like to purchase two dozen compounds that have been hand picked from among the top scoring ligands. Having prepared the ZINC codes in a text file, the user goes to the ZINC search Web page (Figure 2A) and, in the lower left, chooses his file of ZINC codes to upload as a search constraint. Clicking “Search”, the user obtains a browser of his top scoring compounds (Figure 2B). Each molecule may be downloaded separately by clicking on the SMILES, mol2, SDF, or flexibase hyperlinks to the right of the 2D depiction. The user clicks on “Download Table” to obtain a spreadsheet containing purchasing information.
Search Example 3: Find Compounds Similar to the 42 Known Ligands of a Target.
From the search page, the user uploads a file containing SMILES of 42 ligands for a target of interest. By clicking “Search”, the ZINC database is searched for molecules matching these SMILES. A trial 3D geometry of each molecule may be inspected in the Java applet J-mol by clicking on the 2D depiction. If the results in the Database Browser look interesting, the molecules may be downloaded in SMILES, mol2, SDF, or flexibase formats.
DISCUSSION
A key barrier to entry in virtual screening is the inaccessibility of a database to screen. We present here a new research tool suitable for both novices and experts to address this deficiency. The ZINC database provides 3D molecules in several formats compatible with most docking programs. The Web-based interface is fast and supports moderately complex queries. We have made it easy to prepare subsets, as we ourselves frequently only want to screen a subset of the database against a particular target. To accelerate experimental testing, we have made it straightforward to purchase compounds online, by supporting direct links to e-commerce systems where available. The interface allows tables of data to be downloaded to a spreadsheet, to enable users to graph properties, and to spot trends within the database. The ZINC server enables users to upload and process their own compounds, as we ourselves often have molecules such as positive and negative controls that we wish to dock that are not part of the existing database. We hope ZINC will be useful for virtual screening by experts and nonspecialists alike and enable more investigators to attempt computational ligand discovery.
Acknowledgments
This work is supported by NIH GM71896 (to B.K.S. and J.J.I.). We thank OpenEye Scientific Software (SantaFe, NM) for the use of Omega, OEChem, filter1.0.2, Vida, QuacPAC, Ogham, and other tools. We thank Schrödinger, Inc. for the use of ligprep. We are grateful to Dr. Peter Ertl for the Java Molecular Editor (JME), Dr. W. D. Ihlenfeldt for Cactvs, and Molinspiration for the mitools toolkit, including calculated LogP. We thank Dr. Niu Huang and Austin Kirchner for scripts, tools, and thoughtful comments and Dr. Ruth Brenk, Veena Thomas, and Alan Graves for reading this manuscript.
Footnotes
Supporting Information Available: Current size of the MySQL tables used to hold the ZINC database (Table S1), schema of the ZINC database (Figure S1), our version of the filter_light.txt parameter file for OpenEye’s filter.1.0.2 (Figure S2), our version of the services/data/ionizer.ini file for Schrodinger’s LigPrep (Figure S3), and our version of the macro-model/data/tautomer_list file for Schrodinger’s LigPrep (Figure S4). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J Med Chem. 2003;46:3045–3059. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- 2.Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 3.Carlson HA. Protein flexibility and drug design: how to hit a moving target. Curr Opin Chem Biol. 2002;6:447–452. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- 4.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337:209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 6.Zavodszky MI, Sanschagrin PC, Korde RS, Kuhn LA. Distilling the essential features of a protein surface for improving protein-ligand docking, scoring, and virtual screening. J Comput-Aided Mol Des. 2002;16:883–902. doi: 10.1023/a:1023866311551. [DOI] [PubMed] [Google Scholar]
- 7.Lyne PD. Structure-based virtual screening: an overview. Drug Discov Today. 2002;7:1047–1055. doi: 10.1016/s1359-6446(02)02483-2. [DOI] [PubMed] [Google Scholar]
- 8.Taylor RD, Jewsbury PJ, Essex JW. A review of protein-small molecule docking methods. J Comput-Aided Mol Des. 2002;16:151–166. doi: 10.1023/a:1020155510718. [DOI] [PubMed] [Google Scholar]
- 9.Gohlke H, Klebe G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew Chem Int Ed Engl. 2002;41:2644–2676. doi: 10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 10.Shoichet BK, McGovern SL, Wei B, Irwin JJ. Lead discovery using molecular docking. Curr Opin Chem Biol. 2002;6:439–446. doi: 10.1016/s1367-5931(02)00339-3. [DOI] [PubMed] [Google Scholar]
- 11.Pospisil P, Ballmer P, Scapozza L, Folkers G. Tautomerism in computer-aided drug design. J Recept Signal Transduct Res. 2003;23:361–371. doi: 10.1081/rrs-120026975. [DOI] [PubMed] [Google Scholar]
- 12.Fornabaio M, Cozzini P, Mozzarelli A, Abraham DJ, Kellogg GE. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 2. Computational titration and pH effects in molecular models of neuraminidase-inhibitor complexes. J Med Chem. 2003;46:4487–4500. doi: 10.1021/jm0302593. [DOI] [PubMed] [Google Scholar]
- 13.Duarte HA, Carvalho S, Paniago EB, Simas AM. Importance of tautomers in the chemical behavior of tetracyclines. J Pharm Sci. 1999;88:111–120. doi: 10.1021/js980181r. [DOI] [PubMed] [Google Scholar]
- 14.von Grotthuss M, Pas J, Rychlewski L. Ligand-Info, searching for similar small compounds using index profiles. Bioinformatics. 2003;19:1041–1042. doi: 10.1093/bioinformatics/btg117. [DOI] [PubMed] [Google Scholar]
- 15.Strausberg RL, Schreiber SL. From knowing to controlling: a path from genomics to drugs using small molecule probes. Science. 2003;300:294–295. doi: 10.1126/science.1083395. [DOI] [PubMed] [Google Scholar]
- 16.Cramer CJ, Truhlar DG. AM1-SM2 and PM3-SM3 parametrized SCF solvation models for free energies in aqueous solution. J Comput-Aided Mol Des. 1992;6:629–666. doi: 10.1007/BF00126219. [DOI] [PubMed] [Google Scholar]
- 17.Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 18.Lorber DM, Shoichet BK. Flexible ligand docking using conformational ensembles. Protein Sci. 1998;7:938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorber DM, Udo MK, Shoichet BK. Protein-protein docking with multiple residue conformations and residue substitutions. Protein Sci. 2002;11:1393–1408. doi: 10.1110/ps.2830102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bostrom J, Greenwood JR, Gottfries J. Assessing the performance of OMEGA with respect to retrieving bioactive conformations. J Mol Graph Model. 2003;21:449–462. doi: 10.1016/s1093-3263(02)00204-8. [DOI] [PubMed] [Google Scholar]
- 21.Bostrom J. Reproducing the conformations of protein-bound ligands: a critical evaluation of several popular conformational searching tools. J Comput-Aided Mol Des. 2001;15:1137–1152. doi: 10.1023/a:1015930826903. [DOI] [PubMed] [Google Scholar]
- 22.Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 23.Molinspiration.com www.molinspiration.com 2004.
- 24.Wang R, Fu Y, Lai L. A New Atom-Additive Method for Calculating Partition Coefficients. J Chem Inf Comput Sci. 1997;37:615–621. [Google Scholar]
- 25.Ihlenfeldt WD, Voigt JH, Bienfait B, Oellien F, Nicklaus MC. Enhanced CACTVS browser of the Open NCI Database. J Chem Inf Comput Sci. 2002;42:46–57. doi: 10.1021/ci010056s. [DOI] [PubMed] [Google Scholar]
- 26.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 27.Schneider G. Trends in virtual combinatorial library design. Curr Med Chem. 2002;9:2095–2101. doi: 10.2174/0929867023368755. [DOI] [PubMed] [Google Scholar]
- 28.Oprea TI. Current trends in lead discovery: are we looking for the appropriate properties? J Comput-Aided Mol Des. 2002;16:325–334. doi: 10.1023/a:1020877402759. [DOI] [PubMed] [Google Scholar]
- 29.Teague SJ, Davis AM, Leeson PD, Oprea T. The Design of Leadlike Combinatorial Libraries. Angew Chem Int Ed Engl. 1999;38:3743–3748. doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 30.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 31.Ertl P, Jacob O. WWW-Based Chemical Information System. J Mol Struct. 1997;419:113–120. [Google Scholar]
- 32.Weininger D. SMILES: Introduction and Encoding Rules. J Chem Inf Comput Sci. 1988;28:31. [Google Scholar]