

Abstract
Type II fatty acid biosynthesis represents an attractive target for the discovery of new antimalarial drugs. Previous studies have identified malarial ENR (enoyl acyl-carrier-protein reductase, or FabI) as the target for the antiseptic triclosan. In the present paper, we report the biochemical properties and 1.5 Å (1 Å=0.1 nm) crystal structure of OAR (3-oxoacyl acyl-carrier-protein reductase, or FabG), the second reductive step in fatty acid biosynthesis and its inhibition by hexachlorophene. Under optimal conditions of pH and ionic strength, Plasmodium falciparum OAR displays kinetic properties similar to those of OAR from bacteria or plants. Activity with NADH is <3% of that with NADPH. Fluorescence enhancement studies indicate that NADPH can bind to the free enzyme, consistent with kinetic and product inhibition studies suggesting a steady-state ordered mechanism. The crystal structure reveals a tetramer with a sulphate ion bound in the cofactor-binding site such that the side chains of the catalytic triad of serine, tyrosine and lysine are aligned in an active conformation, as previously observed in the Escherichia coli OAR–NADP+ complex. A cluster of positively charged residues is positioned at the entrance to the active site, consistent with the proposed recognition site for the physiological substrate (3-oxoacyl-acyl-carrier protein) in E. coli OAR. The antibacterial and anthelminthic agent hexachlorophene is a potent inhibitor of OAR (IC50 2.05 μM) displaying non-linear competitive inhibition with respect to NADPH. Hexachlorophene (EC50 6.2 μM) and analogues such as bithionol also have antimalarial activity in vitro, suggesting they might be useful leads for further development.
Keywords: acetoacetyl-CoA, fatty acid biosynthesis, hexachlorophene, malaria, NADPH, Plasmodium falciparum
Abbreviations: AcAcCoA, acetoacetyl-CoA; AcAcNAC, N-acetyl-S-acetoacetyl cysteamine; ACP, acyl-carrier protein; AUC, analytical ultracentrifugation; DTT, dithiothreitol; ENR, enoyl-ACP reductase; FAS, fatty acid synthase; KAS III, β-ketoacyl-ACP synthase III; MALDI–TOF, matrix-assisted laser-desorption ionization–time-of-flight; MCAT, malonyl-CoA:ACP transcylase; OAR, 3-oxoacyl-ACP reductase; R.M.S.D., root mean square deviation
INTRODUCTION
Plasmodium falciparum is the major causative agent of malaria, the world's most devastating parasitic disease. Five hundred million acute cases are reported each year [1], of which at least 1 million are fatal [2]. Despite extensive research efforts over the last 3 decades, an effective malaria vaccine remains elusive. Widespread drug resistance against the commonly used drugs such as chloroquine and pyrimethamine/sulfadoxine presents a serious threat to the management and treatment of this disease. If malaria is to be controlled, it is imperative that effective and affordable new drugs are developed.
The discovery of the apicoplast, a novel and essential chloroplast-like organelle found in all Plasmodium species and other apicomplexan parasites such as Toxoplasma gondii, holds great potential for drug development [3,4]. One of the metabolic processes that occurs within the apicoplast is fatty acid biosynthesis [5]. In vertebrates, yeast and mycobacteria, a single multifunctional polypeptide [type I FAS (fatty acid synthase)] containing several distinct domains catalyses the reactions that are involved in the elongation of the fatty acid chain [6,7]. In contrast, for plants and many bacteria, the individual metabolic transformations are catalysed by separate enzymes (type II FAS; reviewed in [8]). Despite the individual reaction steps of FAS I and FAS II being fundamentally similar, selective inhibition of malaria FAS II is possible with drugs such as thiolactomycin [5] and triclosan [9]. Although some apicomplexan parasites such as Cryptosporidium parvum lack type II FAS and others such as Toxoplasma gondii appear to posses both type I and type II systems, the malaria parasite only contains type II FAS, making it an attractive drug target (reviewed in [10]).
The precursors for type II FAS in P. falciparum are derived from the acetyl-CoA pool via malonyl-CoA [9]. The initiation of type II FAS requires ACP (acyl-carrier protein), MCAT (malonyl-CoA:ACP transcylase, or FabD) and KAS III [β-ketoacyl-ACP synthase III (3-oxoacyl-CoA synthase), or FabH]. MCAT produces malonyl-ACP from malonyl-CoA and ACP; subsequently KAS III catalyses the condensation of malonyl-ACP with either acetyl-CoA, or butyryl-CoA to form a 3-oxoacyl-ACP product [11,12]. This product then enters the elongation cycle and is reduced to 3-hydroxyacyl-ACP by the NADPH-dependent OAR (3-oxoacyl-ACP reductase, or FabG) [13]. Following dehydration to form enoyl-ACP, by HAD (β-hydroxyacyl-ACP dehydratase, or FabZ) [14], a second reductive step, catalysed by NADH-dependent ENR (enoyl-ACP reductase, or FabI) [9], takes place to complete the first round of the elongation cycle. Inhibition of ENR by the bactericidal drug triclosan has been shown to inhibit Plasmodium growth, validating this metabolic pathway as an antimalarial drug target [9,15]. However, bacteria readily develop resistance to triclosan in both laboratory and clinical settings via up-regulation [16] or point mutations in the active site of FabI [17]. Similar point mutations lead to reduced affinity for triclosan in P. falciparum ENR [18], raising concerns about the emergence of drug resistance in the malaria parasite. One approach to delay the emergence of resistance and enhance efficacy would be to target additional steps in this pathway. This strategy is already widely used in antifolate combination chemotherapy with pyrimethamine and sulfadoxine, which target dihydrofolate reductase and dihydropteroate synthase respectively [19].
OAR is an essential and validated antimicrobial target [20], but no specific inhibitors have been identified. Although (−)-epigallocatechin, the major component in green tea, inhibits bacterial OAR and ENR in the low micromolar range, inhibition of fatty acid biosynthesis is not the sole reason for antibacterial activity [21]. Indeed, catechins are known to be promiscuous inhibitors with multiple targets [22,23]. Structural studies on OAR from bacteria and plants have provided insight into the chemical mechanism of catalysis [24–27]; however, a detailed kinetic mechanism has not been established. Some preliminary properties of OAR from P. falciparum were reported recently [13]. However, our findings differ significantly in many respects, particularly with regard to the catalytic activity of the enzyme. In addition, we describe the structure and a kinetic mechanism for the enzyme and identify several potential lead inhibitors, including hexachlorophene, a well known antiseptic, as possible leads for further development.
EXPERIMENTAL
Materials
AcAcNAC (N-acetyl-S-acetoacetyl cysteamine) was obtained from Research Plus. AcAcCoA (acetoacetyl-CoA), β-hydroxybutyryl-CoA, fenticlor, bithionol, bithionol sulphoxide, clofoctol and di-resorcinol sulphide were obtained from Sigma. 2,2′-Thiobis (4-t-octylphenol), dichlorophene, hexachlorophene, isoniazid, triclocarban and triclosan were from Aldrich. Fluazifop-butyl, haloxyfop-methyl and quizalofop-ethyl were from Riedel-de Haen. Ponazuril and toltrazuril were gifts from Bayer. Bromochlorophen, cerulenin and diclazuril were from Ubichem, Calbiochem and LKT labs respectively. [3H]Hypoxanthine was from Amersham Biosciences. PCR-Blunt II TOPO cloning vector was from Invitrogen. The cDNA gametocyte plasmid library used to isolate the coding region of OAR was a gift from Dr D. Kaslow, National Institutes of Health, Bethesda, MD, U.S.A. The expression plasmid pJC40 was a gift from Dr J. Clos, University of Hamburg, Hamburg, Germany.
Cloning of P. falciparum OAR
A BLAST search of the P. falciparum genome database identified a sequence with high similarity to bacterial OARs. The entire coding region of the gene was amplified by PCR with Pfu polymerase (Hybaid) from a cDNA library using the sense oligonucleotide, 5′-GAATTCATGATCAAAATGAGTGTTCTAC-3′ (EcoRI site underlined), and the antisense oligonucleotide, 5′-GGATCCTTAAGGTGATAGTCCACCG-3′ (BamHI site underlined). PCR conditions were 94 °C for 7 min (1 cycle) followed by 30 cycles of 94 °C for 2 min, 45 °C for 2 min and 72 °C for 2 min, and a final elongation step at 72 °C for 7 min. The nucleotide sequence of the PCR fragment was determined by automated nucleotide sequencing on an automatic sequencer (ABI 377, Bio-Rad). Analyses of OAR sequences were performed with Vector NTI (Informax) and DNASTAR.
Expression and purification of P. falciparum OAR
OAR potentially resides in the apicoplast of P. falciparum and is therefore likely to possess a bipartite signal and transit peptide at its N-terminus for correct targeting to this unique organelle. Since the correct start site of mature P. falciparum OAR is unknown, an attempt was made to identify potential start sites by aligning the deduced amino acid sequence with other OAR sequences and searching for possible conserved residues. Four potential start sites were identified, and constructs were made for expression in Escherichia coli. These were amplified by PCR using either the full-length product obtained above or a P. falciparum 3D7 cDNA library as a template. The sense oligonucleotides used were 5′-GAATTCATGAAAGTTGCTTTAGTAACAGGTGCAGG-3′ (sense 1, for C1), 5′-GAATTCATGAATTTATTGTCTGAAAATAAGAAGG-3′ (sense 2, for C2), 5′-GAATTCATGGAAAATAAGAAGGAAAACTATTATTATT-3′ (sense 3, for C3) and 5′-GAATTCATGAACTATTATTATTGTGGGGAAAATAAAG-3′ (sense 4, for C4), all containing an artificial start codon (italics) and an EcoRI site (underlined) to aid in-frame and directional cloning into the expression plasmid pJC40 [28]. The antisense oligonucleotide used was 5′-GGATCCTTAAGGTGATAGTCCACCG-3′ which possesses a BamHI site for directional cloning into the expression plasmid pJC40. pJC40 leads to expression of an N-terminal decahistidine-tagged fusion protein which allows the purification of the recombinant protein using Ni2+-chelating chromatography (HiTrap™ chelating HP columns, Amersham Biosciences). PCR was performed using the same conditions as described above. In order to facilitate cloning into the expression plasmid, the PCR products were first subcloned into PCR-Blunt II TOPO, then isolated by digesting the TOPO clones with EcoRI and BamHI, followed by gel purification (Qiagen). The isolated inserts were ligated into pJC40 that had been previously digested with EcoRI and BamHI resulting in plasmids pJC40-C1 to pJC40-C4, whose sequences were confirmed before transformation into BL 21 Codon Plus RIL expression cell line (Stratagene). Transformed bacteria were grown on agar containing LB (Luria–Bertani) medium supplemented with 1 M sorbitol, 250 μM betaine and 50 μg·ml−1 ampicillin for 48 h at 37 °C. Single bacterial colonies containing pJC40-C1 to -C4 or pJC40 without an insert respectively were grown at 37 °C overnight in the above medium. Cultures were diluted 1:15 into fresh medium and grown at 37 °C for 16 h. Induction with IPTG (isopropyl β-D-thiogalactoside) was not required because the expression plasmid confers expression at high levels even without induction.
Cells were harvested by centrifugation at 3500 g for 45 min at 4 °C and were resuspended in buffer A [20 mM sodium phosphate, pH 6.8, 0.5 M NaCl and 1 mM tris-(2-carboxyethyl) phosphine], supplemented with protease inhibitor cocktail tablet (Roche), as per the manufacturer's instructions, and DNase type II (Sigma). Cells were flash-frozen and then stored at −80 °C. After rapid thawing, the cells were lysed using a French pressure cell and the extract clarified by centrifugation at 15000 g for 60 min at 4 °C. All subsequent steps were carried out at room temperature (22 °C) to minimize precipitation.
The resulting supernatant was diluted 3-fold with buffer A, filtered (0.2 μm pore-size filter; Nalgene) and loaded on to a 5 ml HiTrap chelating HP column pre-equilibrated with Ni2+. The column was washed with 10 vol. of buffer A, followed by a gradient of 0–0.5 M imidazole in buffer A (30 column vol.) to remove contaminants, before eluting OAR with 1 M imidazole in buffer A. Fractions containing OAR were pooled and immediately dialysed overnight against three changes of buffer B [20 mM sodium phosphate, pH 6.8, 500 mM NaCl, 1 mM DTT (dithiothreitol), 0.5 mM EDTA and 0.01% sodium azide]. Subsequently the dialysed sample was centrifuged at 100000 g for 1 h at 24 °C to remove aggregated protein. The protein concentration was determined using Coomassie Blue (Bio-Rad) with BSA as standard, and purity was assessed by SDS/PAGE (4–12% gradient gels).
In order to determine whether the His10-tag affected the specific activity or physical properties of OAR, 250 μg of protein was digested with 50 μg of Factor Xa (Roche/New England Biolabs) at 37 °C overnight, followed by gel filtration using a Superdex 75 column (26/60; Amersham Biosciences). Subsequently, the specific activity of OAR was determined in the collected fractions using AcAcNAC as the substrate (see below).
Molecular size and oligomeric state of P. falciparum OAR
The subunit molecular size of OAR with and without a His10-tag was determined by SDS/PAGE and MS. The oligomeric state of both protein species was assessed by AUC (analytical ultracentifugation) and gel filtration. AUC was carried out at various protein concentrations in a Beckman Coulter XL-I analytical ultracentrifuge with an AN50Ti rotor at 35000 rev./min and 24 °C. Gel filtration (0.37 mg, 0.2 ml) was carried out at 0.5 ml·min−1 on a Superdex S-200 column (1 cm×30 cm) previously equilibrated with buffer B. The column was calibrated with a mixture of thyroglobulin (670 kDa), bovine γ-globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17 kDa) and vitamin B12 (1.35 kDa). All fractions were assayed for enzyme activity using AcAcNAC as the substrate. Molecular mass was also determined by MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight) spectroscopy in linear mode, with sinapinic acid as the matrix, on a Voyager-DE STR mass spectrometer from PerSeptive Biosystems.
Western blotting
Supernatant and pellet fractions of bacteria expressing P. falciparum OAR of varying length (pJC40-C1 to -C4) were separated on SDS/PAGE and subsequently blotted on to nitrocellulose. After blocking with 5% skimmed milk in PBS at 4 °C overnight, the blots were incubated with the primary anti-His antibody (Clontech) at room temperature for 1 h, washed in PBS containing 0.1% (v/v) Tween 20 and then incubated with the secondary anti-(mouse IgG) antibody before they were developed using the ECL® (enhanced chemiluminescence) system from Amersham Biosciences.
Enzyme assays
The optimal ionic strength for the reaction was determined in 20 mM Hepes, pH 7.4, 200 μM NADPH, 50 mM AcAcNAC and various concentrations of NaCl (0–750 mM) using 4.5 μg of OAR. The pH optimum for the reaction was determined using a mixed buffer system consisting of 50 mM Mes, 50 mM Ches [2-(N-cyclohexylamino)ethanesulphonic acid] and 50 mM Hepes at a pH range of 5–10, 200 μM NADPH, 50 mM AcAcNAC and 2.4 μg of OAR. The ionic strength was maintained at 0.325 M with NaCl.
The standard assay for OAR contained 50 mM sodium phosphate buffer, pH 6.8, containing 0.25 M NaCl, 200 μM NADPH, 50 mM AcAcNAC or 0.5 mM AcAcCoA and 0.2–0.8 μg of OAR in a final volume of 0.5 ml. The assay mixture was pre-incubated for 5 min at 25 °C before the reaction was initiated by the addition of substrate or enzyme. The initial rate of NADPH oxidation was determined spectrophotometrically, by monitoring the reduction in absorbance at 340 nm (Beckman DU 640 or Shimadzu UV-1601 spectrometer). Under these standard assay conditions, activity was proportional to enzyme added between 0.09 and 14 μg of OAR.
Kinetic analyses
Initial Km (app) values for AcAcNAC, AcAcCoA and NADPH were established under the standard assay conditions by varying the concentration of one substrate while maintaining the other substrate at a fixed saturating level (5×Km). Each assay was performed in triplicate, and the linear rates obtained over the first 5–20 s were analysed by non-linear methods using the software program GraFit (Erithacus), fitting to all the individual values. For clarity, only the mean values are presented in the Figures.
To investigate the enzyme mechanism initially, a matrix of kinetic data was taken where NADPH was varied at five fixed concentrations of AcAcCoA. Individual data sets were analysed using the Michaelis–Menten equation (eqn 1) in order to establish whether the double-reciprocal plots are parallel or convergent:
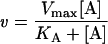 |
(1) |
To investigate further the mechanism and the order of substrate binding, assays were performed where each substrate was varied in turn with a fixed concentration of one of the products, either NADP+ or β-hydroxybutyryl-CoA. The second substrate was kept constant at a subsaturating concentration. The data were analysed by fitting each set for a given product concentration to the Michaelis–Menten equation (eqn 1). The pattern of the corresponding Lineweaver–Burk transformation was analysed and assigned as competitive, mixed or uncompetitive inhibition. The combination inhibition of these four graphs is indicative of a particular mechanism.
Finally, the kinetic data in the absence of products were globally fitted to the equation for the steady-state ordered mechanism (eqn 2), and its goodness of fit was established.
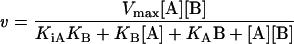 |
(2) |
where v is the rate, Vmax is the maximum rate, [A] and [B] are the concentrations of substrates A and B, KA and KB are the Michaelis constants for A and B respectively, and KiA is the dissociation constant for A.
Substrate-binding studies
The ability of NADPH to bind to OAR was measured by ultrafiltration and fluorescence titration. In the ultrafiltration method, a 1 ml mixture containing 50 μM NADPH and 12.5 μM OAR in buffer B was allowed to equilibrate for 10 min at room temperature. An aliquot (0.5 ml) of this solution was transferred to a Vivaspin concentrator (10 kDa molecular mass cutoff) and centrifuged at 1500 g for 1 min. The concentration of NADPH in the flowthrough was measured spectrophotometrically at 340 nm and compared with a control where no OAR had been included.
For the fluorescence method, NADPH was titrated in 1 μl aliquots into 1 ml of buffer B, and the fluorescence emitted at 461 nm was determined when excited at 341 nm. This was repeated with 3.57 μM OAR diluted in buffer B which had been allowed to equilibrate for 1 h. The difference in fluorescence was calculated (ΔF, corrected to zero), which corresponds to the amount of NADPH bound, and fitted as a function of total NADPH concentration ([N]). Since a significant part of the free NADPH was bound to OAR, the data were fitted by non-linear regression to the quadratic equation (eqn 3):
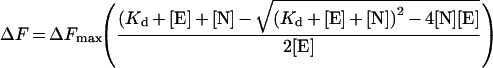 |
(3) |
where ΔFmax is the maximum fluorescence change, Kd is the dissociation constant and [E] is the OAR concentration.
Inhibitor studies
Potential inhibitors were screened at 20 μM using AcAcNAC as the substrate (see above), and IC50 values were determined using AcAcCoA as the substrate, where appropriate. The inhibition mechanism was examined for hexachlorophene by varying the AcAcCoA or NADPH concentrations at different fixed concentrations of hexachlorophene. Individual data sets were fitted to the Michaelis–Menten equation (eqn 1), and the resulting slopes were used to plot Km/Vmax against hexachlorophene concentration.
Effect of OAR inhibitors on P. falciparum in vitro
P. falciparum 3D7 (a clone isolated from a patient in The Netherlands) was cultivated essentially as described by Trager and Jensen [29] in RPMI 1640 medium with human A+ erythrocytes and 0.5% (w/v) Albumax II (Life Technologies). The growth effects of inhibitors on asynchronous cultures of P. falciparum were determined by incorporation of [3H]hypoxanthine in 96-well microtitre plates essentially as described in [30], except the initial haematocrit and parasitaemia were 5% and 0.25% respectively. Drug stock solutions were prepared in DMSO and were diluted in growth medium as appropriate. Each drug was added in a single dose, and the final dilutions contained less than 0.1% DMSO, which had no measurable effect on the parasites in this system. [3H]Hypoxanthine was added to the culture after 24 h, and incorporation into DNA was measured at 48 h. The antimalarial drug chloroquine was included as a positive control.
Crystallization and structure solution
Following gel filtration using a Superdex 75 column, fractions containing the His10-tagged OAR were pooled and dialysed into 20 mM Hepes, pH 6.8, 0.5 M NaCl, 1 mM DTT and 0.5 mM EDTA. Crystals were then grown by vapour diffusion using 0.5 μl of protein solution (at 10 mg·ml−1) and an equal volume of mother liquor consisting of 100 mM Mes, pH 6.0, 35% (v/v) 2-methyl-2,4-pentanediol and 200 mM Li2SO4, equilibrated against a reservoir containing 1 ml of mother liquor incubated at room temperature. A single crystal appeared after less than 6 weeks and was approx. 0.1 mm×0.1 mm×0.1 mm in size. The crystal was frozen directly in a nitrogen cryostream for data collection. Data were collected on beamline BM14 at the European Synchrotron Radiation Facility (Grenoble, France) and processed with the HKL suite of programs [31]. The data between 20 and 1.5 Å (1 Å=0.1 nm) were scaled in P42212 (a=b=73.12, c=100.18 Å; one molecule per asymmetric unit) with an overall Rmerge of 0.055 (0.583 for the last shell), 99.9% completeness (99.8% for the last shell) and 5.9-fold redundancy (5.2-fold for the last shell). The structure was solved by molecular replacement with AMoRe [32], using the E. coli OAR structure complexed with NADP+ (PDB entry 1Q7B) as a search model, giving a distinct solution with an R-factor of 0.293 against 15–3-Å data. The molecular replacement phases were used in warpNtrace [33], which built 241 residues. Refinement was performed with CNS [34] interspersed with model building in O [35]. The first seven N-terminal residues of OAR (residues 47–53) and residues 248–252 did not have well-defined electron density and were not included in the model. Figures were generated using the PyMOL Molecular Graphics System, DeLano Scientific (http://www.pymol.org).
RESULTS
Cloning of P. falciparum OAR
A putative sequence of P. falciparum OAR was constructed by compiling segments found on several contigs of chromosome 9, identified from the P. falciparum genome database (PlasmoDB; http://plasmodb.org/) by BLAST searches against bacterial OARs. The first methionine within the open reading frame was chosen as the start site of the full-length clone, and specific oligonucleotides were designed to amplify the entire coding region of the gene from cDNA of P. falciparum 3D7. Sequencing of this clone revealed that the gene encompasses five exons and four introns, with conserved exon–intron boundaries, when compared with the genomic sequence. The coding region excluding the introns is 915 bp long and encodes a polypeptide of 305 amino acids (Figure 1). The deduced amino acid sequence is 38% and 39% identical with OAR from Brassica napus and E. coli respectively. As expected, the deduced amino acid sequence of P. falciparum OAR possesses an N-terminal extension of 58 amino acids relative to E. coli OAR, which corresponds to the typical apicoplast bipartite hydrophobic signal peptide (residues 1–22) as predicted by SignalP 3.0 [36] followed by an asparagine- and lysine-rich stretch of approx. 32 amino acids (residues 23–54) coding for the potential transit peptide that diverts import of the protein into the apicoplast of the parasite [37]. Since the exact cleavage site for the apicoplast transit peptide is unknown, four different constructs (pJC40-C1 to -C4) were generated for recombinant expression trials in E. coli (see Figure 1).
Figure 1. Alignment of OAR from P. falciparum, E. coli and B. napus.

P. falciparum OAR (Pf; Swiss-Prot accession number AAO32669) was aligned with B. napus OAR (Bn; Swiss-Prot accession number Q93X62) and E. coli OAR (Ec; Swiss-Prot accession number P25716) using ClustalW. The potential signal peptide of P. falciparum OAR is underlined, and the positions of the four introns are marked with grey arrows. The chloroplast transit peptide, which is removed to form the mature protein in B. napus OAR, is indicated in lower case. The start of the four expression constructs used in the present study are marked C1–C4; X indicates the start site of P. falciparum OAR of Pillai et al. [13]. Active-site residues that are conserved in all bacterial and plant OARs are indicated as white on black. Grey triangles indicate the two arginine residues that are central to ACP binding. Secondary-structure elements of P. falciparum OAR are shown below the alignment. The disordered region of the P. falciparum OAR corresponding to the α6 helix is represented by a feint ‘disordered’ helix. Connected black arrows show the β4–α4 and β5–α5 loops, which are the main regions that show conformational change, as well as the active-site residues. The consensus sequence was generated by aligning an additional 22 OAR sequences from the Swiss-Prot database using T-Coffee. Residues are marked as identical (*), conservative (:) or semi-conservative (.) substitutions.
Expression and purification of P. falciparum OAR
Expression of the two shortest constructs (pJC40-C1 and -C2) resulted in the production of recombinant OAR. However, the protein was found almost exclusively in the insoluble pellet (Figure 2). Constructs pJC40-C3 and -C4 were also expressed at equally high yields, and an appreciable amount of active enzyme was found to be in the soluble fraction (Figure 2). The cleavage site of the potential bipartite signal/transit peptide is unknown in the native enzyme, so we decided to carry out all subsequent analyses using pJC40-C4, the longest of the four expression constructs. During the purification of the protein, it became apparent that recombinant OAR tended to precipitate at temperatures below 20 °C, especially at protein concentrations above 0.5 mg·ml−1. Therefore OAR was stored at room temperature in the presence of azide to prevent bacterial growth, where it maintained 100% of its activity for at least 12 weeks. Overall yield was 0.8 mg·l−1 of bacterial culture. The purified protein was ∼98% homogeneous after Ni2+-affinity chromatography, although there appeared to be faint bands where the protein was either degraded or mRNA translation was terminated prematurely (Figure 3A). The subunit size of OAR by SDS/PAGE was estimated to be 31 kDa, consistent with the predicted size of the His10-tagged protein (31486 Da) and the results obtained from MALDI–TOF analysis (31332 Da).
Figure 2. Recombinant expression of P. falciparum OAR constructs C1–C4.

Soluble (S) and pellet (P) fractions of lysates from E. coli cultures transformed with plasmids pJC40-C1 to -C4 were prepared as described in the Experimental section. Recombinant protein was separated by SDS/PAGE and detected by Western blotting using antibody that detected the N-terminal His10-tag. Molecular-mass sizes are given in kDa.
Figure 3. Purification and physical properties of recombinant OAR.

(A) Purification of OAR-C4. Purity was assessed by SDS/PAGE followed by Coomassie Blue staining. Lane 1, lysate from pJC40-OAR-C4; lane 2, flowthrough from Ni2+ HiTrap chelation column; lane 3, pooled active fractions from the affinity column; lane 4, pooled fractions after dialysis and ultracentrifugation to remove precipitate. Molecular-mass sizes are given in kDa. (B) Native molecular mass by size-exclusion chromatography. Solid line, absorbance at 280 nm; broken line, OAR activity. The inset shows Ve (eluted volume)/Vo (void volume) against log Mr for protein standards (○), His10-tagged OAR (●) and OAR after removal of the His10-tag (□). mAU, milli-absorbance units; AU, absorbance units. (C) Determination of optimum ionic strength. OAR was assayed as described in the Experimental section in 20 mM Hepes buffer at pH 7.4 supplemented with NaCl. (D) Determination of optimum pH. OAR activity was determined over a range of pH using a Mes/Ches [2-(N-cyclohexylamino)ethanesulphonic acid]/Hepes buffer mixture with ionic strength maintained constant at 325 mM with NaCl.
Physical and steady-state kinetic properties of P. falciparum OAR
Gel filtration on Superdex S-200 gave a single peak of activity and protein corresponding to a size of 78.7 kDa, suggesting that the native enzyme is either a dimer or a trimer (Figure 3B). Removal of the N-terminal His10-tag, by digestion with Factor Xa also suggested a dimeric or trimeric native structure using gel filtration. However, AUC of samples containing 1.5 or 0.15 mg·ml−1 revealed a native molecular mass of 121.6 kDa, which would correspond to a tetramer. In addition, a smaller peak (2.2% of the total) at 214 kDa was present at the higher concentration, suggesting that aggregation into a higher oligomeric state can occur.
Cleavage of the His10-tag marginally increased the specific activity of OAR by 21%, but was accompanied by poor recovery of protein (approx. 20% after size-exclusion chromatography). Accordingly, all subsequent steady-state kinetic parameters were determined for the His10-tagged enzyme.
The ionic strength optimum and pH optimum at constant ionic strength for the enzymatic reaction with AcAcNAC and NADPH were determined to be 0.325 M and pH 6.8 (Figures 3C and 3D). Steady-state kinetic parameters of OAR using these optimal conditions of pH and ionic strength yielded Km (app) values and specific activity in the same range as those reported for other OARs (Table 1). P. falciparum OAR clearly prefers NADPH as a cofactor to NADH, since the enzyme has only 3% of the activity with NADH compared with NADPH, with AcAcCoA as co-substrate.
Table 1. Properties of P. falciparum OAR compared with B. napus and E. coli enzymes.
Data for P. falciparum are from the present study; data for B. napus are from [25,56] and Q93X62 (TrEMBL accession code); data for partially purified E. coli enzyme are from [43,57], E. coli enzyme quaternary structure is from [26] and P25716 (Swiss-Prot accession number). N.D., not determined.
| Property | Unit | P. falciparum | B. napus | E. coli |
|---|---|---|---|---|
| Signal peptide | – | Yes | Yes | No |
| Predicted mass for mature protein | Da | N.D. | 27050 | 22560 |
| Oligomeric state: | ||||
| Gel filtration | – | Dimer/trimer | Tetramer | N.D. |
| Ultracentrifugation | – | Tetramer | N.D. | N.D. |
| Crystallography | – | Tetramer | Tetramer | Tetramer |
| pH optimum | – | 6.8 | N.D. | 6.0–7.0 |
| Ionic strength optimum | M | 0.325 | N.D. | N.D. |
| Specific activity (with AcAcCoA) | units·mg−1 | 62.5 | 136 | ∼17 |
| Km (app) AcAcNAC | mM | 48.0±5.8 | 35 | 43 |
| Km (app) AcAcCoA | mM | 0.51±0.05* | 0.26 | 2.5 |
| Km (app) NADPH | μM | 47.5±4.5* | 23 | 21 |
| NADH | – | Not active | N.D. | Not active |
| kcat | s−1 | 265±35* | N.D. | N.D. |
* Absolute values for the kinetic constants are KiA=10.2±5.3 μM; KmA=179±37 μM (NADPH); KmB=2.6±0.5 mM (AcAcCoA).
The enzyme is stable at room temperature at concentrations >200 μg·ml−1. However, stock solutions diluted to approx. 10 μg·ml−1 for kinetic assays were found to be unstable (40% loss in activity over 5 h). Others have reported that certain OARs are stabilized by the presence of NADPH, but addition of 150 μM NADPH only had a marginal effect (30% loss). The addition of 150 μM NADP+ plus 1 mM EDTA and/or DTT improved stability with losses decreased to 20%. In contrast, addition of 1 mg·ml−1 BSA initially activated the enzyme by 14% with only 6% loss in activity over 5 h. Thus further enzymatic studies employed stock enzyme solutions that had been diluted into buffer containing 1 mg·ml−1 (15 μM) BSA, and the final concentration of BSA in the assay mixture was always less than 1 μM.
Kinetic mechanism
A full matrix of initial rates was determined with both substrates being varied in the absence of added products. This proved inconclusive as to whether the enzyme followed a Ping Pong or a ternary kinetic mechanism as the double-reciprocal plot could be construed to be either parallel or convergent. To distinguish between models, the products of the OAR reaction (NADP+ and β-hydroxybutyryl-CoA) were examined as inhibitors of the forward reaction (Figure 4). Analysis of the results in the form of double-reciprocal plots indicated that NADP+ was a linear competitive inhibitor with respect to NADPH (Figure 4A), but was non-competitive with respect to AcAcCoA (Figure 4C). In contrast, β-hydroxybutyryl-CoA was a non-competitive inhibitor of both NADPH (Figure 4B) and AcAcCoA (Figure 4D). These product inhibition patterns are indicative of a steady-state ordered mechanism with NADPH binding first (ordered Bi Bi mechanism in the nomenclature of Cleland [38]). The combined data from Figure 4 were then globally fitted to the rate equation for the steady-state ordered mechanism (eqn 2) with NADPH binding first, yielding absolute values for the kinetic constants: KiA=10.2±5.3 μM, KmA=179±37 μM (NADPH), KmB=2.6±0.5 mM (AcAcCoA) and kcat=265±35 s−1.
Figure 4. Product inhibition studies.

OAR was assayed in the presence of the products NADP+ and β-hydroxybutyryl-CoA, varying one substrate concentration and fixing the other. The rates for each product concentration were then fitted individually to the Michaelis–Menten equation (eqn 1) by non-linear regression and displayed as double-reciprocal plots. (A) NADPH varied in the presence of NADP+ (○, 0 μM; ●, 625 μM; □, 1250 μM; ■, 2500 μM) with AcAcCoA fixed at 250 μM. (B) NADPH varied in the presence of β-hydroxybutyryl-CoA (○, 0 μM; ●, 1250 μM; □, 2500 μM; ■, 5000 μM) with AcAcCoA fixed at 250 μM. (C) AcAcCoA varied in the presence of NADP+ (○, 0 μM; ●, 312.5 μM; □, 625 μM; ■, 1250 μM), with NADPH maintained at 50 μM. (D) AcAcCoA varied in the presence of β-hydroxybutyryl-CoA (○, 0 μM; ●, 1250 μM; □, 2500 μM) with NADPH fixed at 50 μM.
This conclusion was corroborated by showing that NADPH was able to bind to free OAR both by ultrafiltration and fluorescence enhancement experiments. Ultrafiltration of a 4:1 mixture of NADPH/OAR indicated that 6.7±1.4 μM NADPH was able to bind to 12.5 μM OAR, corresponding to a Kd of 38±11 μM. Fluorescence enhancement studies fitted to the equation for one-site binding (eqn 3) gave more precise estimates of the binding constant of 14.1±3.3 μM (results not shown), which is not significantly different from the KiA of 10.2±5.3 μM determined above. Owing to the high predicted Km for AcAcCoA, we were unable to establish whether AcAcCoA was also able to bind to the free enzyme.
Inhibition of P. falciparum OAR
A variety of potential inhibitors of OAR were screened at 20 μM for activity against the enzyme (Table 2). Triclosan, a potent inhibitor of ENR, was inactive against OAR (only 3% inhibition), whereas structural analogues such as hexachlorophene (an antiseptic), bithionol (an antiseptic and anthelminthic), di-resorcinol sulphide and bromochlorophen inhibited OAR by >75%. IC50 values for these latter compounds ranged from 2.05 μM for hexachlorophene to 15.4 μM for bromochlorophen (Table 2). EC50 values for growth inhibition of P. falciparum in vitro were somewhat higher, ranging from 6.2 μM for hexachlorophene to 88 μM for bromochlorophen (Table 2). EC50 values for chloroquine and triclosan are in agreement with published values for the 3D7 strain of P. falciparum [39,40].
Table 2. Inhibition of OAR and effects on growth of P. falciparum in vitro.
The following compounds gave less than 20% inhibition when tested against OAR at 20 μM or were too insoluble to assay: bithionol sulphoxide, cerulenin, clofoctol, dichlorophene, diclazuril, fenticlor, fluazifop-butyl, haloxyfop-methyl, isoniazid, ponazuril, quizalofop-ethyl, 2,2′-thiobis-(4-t-octylphenol), triclocarban and toltrazuril. EC50 values are the weighted means±weighted S.D. for three separate experiments performed in triplicate. The standard drug chloroquine was included as a control (EC50 9.3±2.1 nM). IC50 values are the weighted means±weighted S.D. for two separate experiments. N.A., not active, 3% inhibition at 20 μM.
| Compound | Structure | IC50 OAR (μM) | EC50P. falciparum (μM) |
|---|---|---|---|
| Triclosan (antibacterial) | 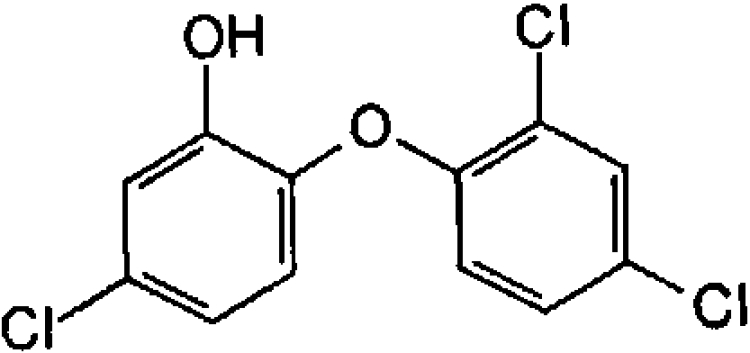 |
N.A. | 1.3±0.3 |
| Hexachlorophene (antimicrobial and anthelminthic) | 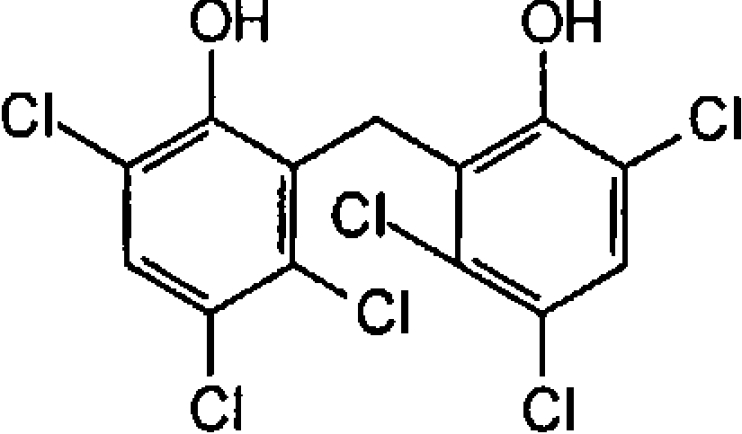 |
2.05±0.01 | 6.2±0.7 |
| Di-resorcinol sulphide | 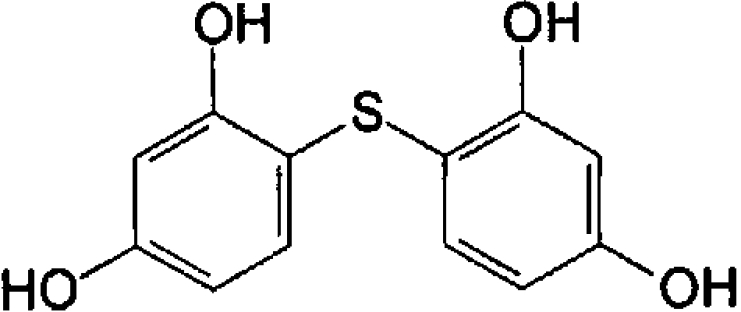 |
3.8±0.16 | 92±5 |
| Bithionol (antimicrobial and anthelminthic) | 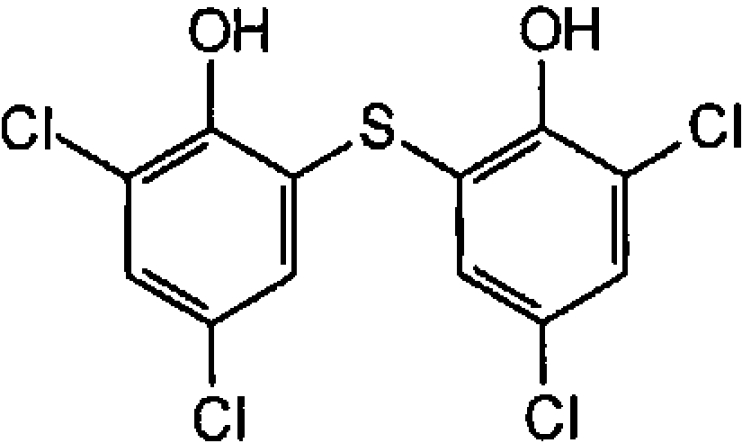 |
10.1±0.7 | 24±4 |
| Bromochlorophen (anthelminthic) | 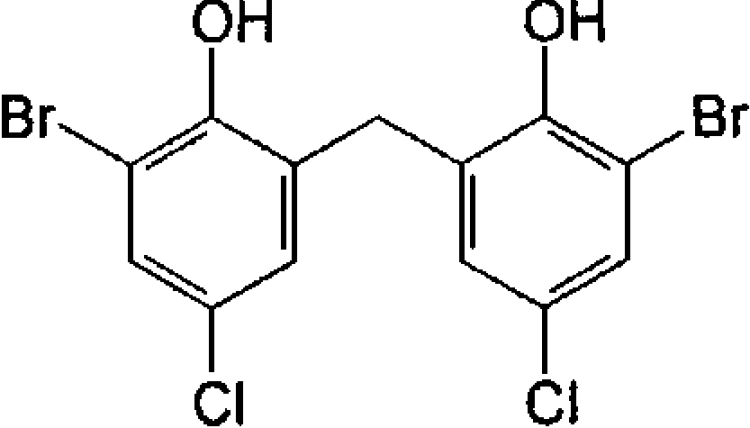 |
15.4±0.04 | 88±3 |
The mechanism of inhibition of OAR by hexachlorophene was investigated further by varying substrate concentrations in the presence of the inhibitor (Figure 5). When the NADPH concentration was varied, fitting the data for each inhibitor concentration independently to the Michaelis–Menten equation (eqn 1) led to a family of lines which intersected on the y-axis in double-reciprocal plots, indicating competitive inhibition (Figure 5A). With AcAcCoA as the variable substrate, the individual fits did not intersect at a single point, but appear to be consistent with a mixed mechanism (Figure 5B). Secondary plots (insets) of slope (Km/Vmax) against inhibitor concentration are upwardly curving, suggestive of parabolic inhibition in which more than one inhibitor molecule binds to same form of the enzyme. Unfortunately, all attempts to globally fit the data to equations describing various forms of parabolic inhibition proved unsuccessful.
Figure 5. Hexachlorophene inhibition of OAR.

OAR was assayed in the presence of hexachlorophene while varying the concentration of one substrate, with the second substrate at a fixed concentration. Data sets for each inhibitor concentration were fitted by non-linear regression and are presented as Lineweaver–Burk transformations. The inset in both panels shows the secondary plot of Km/Vmax against hexachlorophene concentration. (A) NADPH varied in the presence of 0 (○), 10 (●), 15 (□) and 17.5 (■) μM hexachlorophene, with AcAcCoA maintained at 250 μM. (B) AcAcCoA varied in the presence of 0 (○), 10 (●), 15 (□) and 20 (▲) μM hexachlorophene, with NADPH maintained at 50 μM.
Structural studies
P. falciparum OAR was crystallized from 2-methyl-2,4-pentanediol solutions, synchrotron diffraction data were collected to 1.5 Å resolution, and the structure was solved by molecular replacement. Refinement produced a final model with an R-factor of 0.202 (Rfree=0.223) and good geometry [R.M.S.D. (root mean square deviation) from ideal bond lengths and angles is 0.01 Å and 1.5° respectively]. Residues 248–252 did not have well-defined density, and these correspond to the α6 helix present in the E. coli OAR and OAR–NADP+ structures [26,27]. The unit cell contains one monomer with a tetramer formed through crystallographic symmetry, in agreement with the analytical ultracentrifugation data and the previously solved OAR structures [25–27] (Figure 6A). Surprisingly, although the cofactor-binding site is empty, the structure appears to be in a conformation compatible with cofactor binding. Superposition on the E. coli apo-OAR structure gives an R.M.S.D. of 1.9 Å on 157 Cα atoms, whereas superposition on the OAR–NADP+ complex (PDB entry 1Q7B; [27]) gives an R.M.S.D. of 1.4 Å on 233 Cα atoms. Indeed, inspection of the active site shows that the catalytic triad, Ser199, Tyr212 and Lys216 point into the cofactor-binding site, with conformations similar to those observed in the E. coli OAR–NADP+ complex (Figure 6B). Interestingly, although the cofactor-binding site is unoccupied in the P. falciparum OAR structure, an ordered sulphate molecule from the crystallization solution occupies a position equivalent to the 2′-phosphate on the adenosine ribose (Figure 6B). This sulphate interacts with P. falciparum OAR in a similar way to that of the adenosine 2′-phosphate in the E. coli OAR–NADP+ structure. In P. falciparum OAR, the positively charged guanidino group of the conserved Arg72 stabilizes the negative charge of the sulphate, as observed for the equivalent Arg14 and the adenosine phosphate in E. coli OAR–NADP+. Conversely, in E. coli OAR without NADP+, the Arg14 side chain forms a salt bridge with the conserved α6 helix residue Asp187 (Asp248 in P. falciparum). In short-chain alcohol dehydrogenases, the α6 helix is usually disordered in the absence of cofactor and becomes ordered upon cofactor binding. Therefore it is tempting to speculate that the disordered α6 helix in P. falciparum OAR represents an intermediate state where it is not stabilized either by the formation of the salt bridge with Arg72 or by the presence of cofactor.
Figure 6. Quaternary structure and cofactor-binding site of P. falciparum OAR.

(A) The OAR homotetramer, obtained by crystallographic symmetry from the single monomer in the asymmetric unit, is shown in a surface representation. Each of the monomers is coloured separately, with the residues predicted to be involved in cofactor binding (by homology with the E. coli structure) shown in yellow. N marks the N-terminal region of OAR that forms significant hydrogen-bonding interactions across the dimer interface. (B) OAR is shown in a surface representation, in an orientation similar to the grey monomer in (A). The surface is coloured yellow for residues conserved with the E. coli structure. The structure of the NADP+ cofactor, obtained by superposition on the E. coli complex (PDB entry 1Q7B; [27]), is shown as a stick model, with green carbons. P. falciparum OAR residues lining the cofactor-binding site are shown as sticks with black carbons and are labelled. The ordered sulphate observed in the P. falciparum OAR structure is shown together with an unbiased |Fo|−|Fc|, Φcalc electron-density map contoured at 2.5 σ.
P. falciparum OAR assumes the active conformation in the absence of cofactor
A previous study on E. coli OAR suggests that it is an allosteric enzyme which exhibits negative co-operativity upon cofactor binding [26]. In the proposed mechanism, the binding of NADPH to one of the monomers increases the affinity for ACP towards that monomer, while reducing ACP affinity in the other sites of the tetramer.
For negative co-operativity to take effect, interactions across the dimer interface must occur. A comparison of the E. coli OAR and OAR–NADP+ structures has shown that significant conformational changes occur within the monomer upon cofactor binding [27]. These include rearrangement of active site residues and the associated β5–α5 loop to a catalytically active conformation and a shift of the β4–α4 loop [27] (Figures 7A and 7B). These changes allow the acyl-ACP substrate to access the active site and interact with the nicotinamide moiety of the bound cofactor (Figure 7B). In these regions, the structure of the P. falciparum OAR (Figure 7C) without cofactor shows higher similarity to the E. coli OAR–NADP+ structure (Figure 7B) than to that of OAR without cofactor (Figure 7A).
Figure 7. Conformational differences in P. falciparum OAR and E. coli OAR in the absence of NADP+ cofactor.

(A) Monomer of E. coli OAR without NADP+ (PDB entry 1I01; [26]). (B) Monomer of E. coli OAR with bound NADP+ (PDB entry 1Q7B; [27]). (C) Crystallographic dimer of P. falciparum OAR with bound sulphate. Protein backbones are shown as grey ribbons covered by a surface representation. The second monomer in the P. falciparum OAR structure is coloured by electrostatic potential as calculated with the PyMOL APBS tool (red, −5 kT; blue, +5 kT). The two main areas of conformational change are highlighted in green (the β4–α4 loop) and magenta (the β5–α5 loop). Cofactors/ligands are shown as sticks. The nucleotide sequence data reported for Plasmodium falciparum 3-oxoacyl-acyl-carrier-protein reductase are available in the DDBJ, EMBL, GenBank® and GSDB Nucleotide Sequence Databases under accession number AF527538.
In the E. coli OAR–NADP+ structure, the adjustment at the monomer–monomer interface occurs when Glu168 at the C-terminus of the α5 helix forms new hydrogen bonds with the backbone nitrogens of Leu95 (Phe156 in P. falciparum) and Gly147 (Gly208 in P. falciparum). This results in an upward shift of the β5′–α5′ loop towards the α4′ helix (Figure 7). In P. falciparum OAR, this glutamate residue is conserved (Glu229) and is hydrogen-bonded to backbone amides of Phe156 and Gly208 that are consistent with the upward shift of the α4′ helix. Furthermore, when the NADP+ of the OAR–NADP+ structure is superimposed on P. falciparum OAR, no structural clashes are observed between the cofactor and protein, and thus the malaria enzyme appears to be in a conformation already compatible with cofactor binding.
Interestingly, the N-terminal region of P. falciparum OAR also contributes to the dimer interface where Asn56 and Tyr57 form hydrogen bonds across the interface to Asn56′ and Tyr57′ of an adjacent subunit (Figure 6A). We note that these residues are absent in the recombinant protein construct of Pillai et al. [13] (X in Figure 1) and in our insoluble expression constructs C1 and C2 (Figures 1 and 2). Although the precise start of the mature processed P. falciparum OAR in the apicoplast remains unknown, these interactions suggest that these residues may be critical for stability (Lys54 is the first residue for which good density is observed).
ACP recognition
Previous studies have identified a conserved positively charged patch on the surface of E. coli fatty acid biosynthesis enzymes [41,42]. This positively charged patch is positioned at the entrance of the active site and is thought to be involved in recognition of the highly conserved and negatively charged α2 helix of ACP. The α2 helix of P. falciparum ACP shows high sequence identity with that of E. coli: 11 of the 15 residues are identical, and a further two are conservative substitutions. Therefore it might be expected that P. falciparum OAR would also contain a positively charged region at the active site entrance that is capable of interacting with the negatively charged region on ACP. Mutagenesis studies of E. coli OAR showed that two arginine residues (Arg129 and Arg172) present in this patch are central to the binding of ACP [42]. These residues are conserved in P. falciparum OAR (Arg190 and Arg233; Figure 1). The electrostatic surface potential of P. falciparum OAR was calculated (Figure 7), and these arginine residues form part of a highly positively charged region on the opposing dimer, consistent with reports that ACP binds across the dimer interface [27].
DISCUSSION
A preliminary characterization of recombinant OAR from P. falciparum has been reported previously by Pillai et al. [13]. However, our findings differ considerably from this report in a number of respects. Their protein is only 12 amino acid residues shorter at the N-terminus (X in Figure 1) than our construct (C4 in Figure 1), yet the specific activity (0.03 unit·mg−1) for their protein is more than 3 orders of magnitude lower than ours (62.5 units·mg−1; Table 1). Even allowing for suboptimal assay conditions of low ionic strength and high pH (50 mM Tris/HCl, pH 7.5) used by these authors, this cannot account for more than a 2-fold difference with our data (cf. Figure 3). Moreover, the specific activity of our recombinant enzyme is of the same order as those for E. coli and B. napus (Table 1). The shorter two of our expression constructs (C1 and C2) that bracket the start site of their construct were found to be expressed mainly as insoluble inclusion bodies. This is compatible with the observation that the N-terminus has a defined structural role in the context of the tetramer (Figure 6), possibly accounting for the extremely low specific activity reported by Pillai et al. [13] and the pronounced discrepancies in other kinetic parameters. For example, the apparent Km for NADPH is ≫200 μM (with 250 μM AcAcCoA) compared with 47.5 μM (with 500 μM AcAcCoA) in our study. This might account for the even larger discrepancy between apparent kcat values (0.014 s−1 compared with 265 s−1; Table 1). The robust nature of our kinetic data is supported by the reasonable agreement of the KiA for NAPDH of 10.2 μM with the binding constant of 14.1 μM determined by fluorescence enhancement. Furthermore, the apparent Km values for the model substrates AcAcCoA and AcAcNAC are broadly similar to those of the bacterial and plant enzymes (Table 1). However, the physiological substrate is acetoacetyl-ACP, which has a Km (app) 100-fold lower than AcAcCoA for the E. coli enzyme [43]. Further studies are planned to determine the kinetic behaviour with this bulky carrier protein substrate.
The physicochemical properties of our enzyme are broadly similar to those reported for the B. napus and E. coli enzymes (Table 1). However, the dimeric or trimeric structure predicted by gel filtration is not entirely consistent either with other OARs (Table 1) or with our own AUC or structural studies, which indicate a tetramer. Interestingly, MabA (FabG1) from Mycobacterium tuberculosis has been reported to undergo a dimer-to-tetramer self-association with a dissociation constant of 22 μM [44], and crystallographic studies noted that the interface between dimers in the tetramer were stabilized by caesium ions [24]. Thus the physiologically relevant form of the malaria enzyme may depend on the OAR concentration and ionic strength within the apicoplast. However, the discrepancy between the AUC and gel-filtration results could also be due to non-specific interactions between the gel matrix and OAR retarding elution, hence displaying an apparent smaller molecular size.
There are no studies in the literature describing a full kinetic mechanism for any OAR, no doubt due to the widely reported instability of these enzymes at high dilution and the high cost of substrates. Our kinetic analyses, including product inhibition and binding studies, are consistent with a steady-state ordered bi-reaction system, with NADPH binding first before formation of a ternary complex. This concurs with structural and modelling studies of Price et al. [26] for the E. coli enzyme. These authors compared their structure with the structure of B. napus OAR complexed with NADP+ [25] and predicted that large conformational changes must occur for NADPH to bind, which subsequently affect the binding of the ACP to the surface of OAR and, as a result, the delivery of the acyl chain to the active site. However, the P. falciparum OAR structure reported in the present study shows a conformation compatible with cofactor binding, although it was crystallized in the absence of NADP+. The ordered sulphate, which binds in a position equivalent to one of the phosphates in the cofactor, could contribute to ordering of the active site, as observed previously for other phospho-ligand-binding proteins (see, for example, [45,46]). However, the mechanism responsible for the rearrangement of the active-site residues and movements of the β4–α4, β5–α5 loops and the dimer interface upon sulphate binding is not clear at present. To date, attempts to determine the structure of the enzyme in the absence of sulphate or in the presence of inhibitors have proved unsuccessful.
The antiseptic triclosan, a known inhibitor of ENR, is inactive against malaria OAR. This compound is a slow tight-binding inhibitor that forms an ENR–NAD+–triclosan ternary complex [15,47–50]. Hexachlorophene, another antiseptic with anthelminthic activity, is a structural analogue of triclosan. However, we found no evidence for time-dependent inhibition of malaria OAR by hexachlorophene in the presence of NADP+. This is consistent with the parabolic competitive inhibition pattern with respect to NADPH, where the non-linear upward-curving nature of the re-plots of slope versus inhibitor concentration suggests a mechanism in which more than one hexachlorophene binds to the same form of the enzyme. Cooperative inhibition by hexachlorophene has been reported for a wide range of NAD(P)+-linked dehydrogenases [51], arginase [52], acetylcholinesterase [53] and succinate dehydrogenase [54]. BSA, an avid binder of hexachlorophene [55], can reverse arginase inhibition [52], possibly complicating the kinetic analysis of the present study due to the inclusion of BSA as a stabilizer in the enzyme diluent. However, binding to BSA seems unlikely given the fact that hexachlorophene was in 10–20-fold excess over BSA in these experiments. Complexes of OAR with hexachlorophene would aid in defining the location and stoichiometry of binding.
A variety of structural analogues of hexachlorophene were examined as inhibitors of OAR. Of these, di-resorcinol sulphide, bithionol and bromochlorophen were found to be active against OAR with IC50 values in the low micromolar range. Significantly, hexachlorophene and other bis-(2-hydroxyphenyl)methane/sulphide analogues were also active in vitro against the intact parasite. However, the correlation between enzyme potency and antimalarial activity is poor, and further studies are required to determine whether any of these compounds act specifically as inhibitors of fatty acid biosynthesis.
In conclusion, OAR represents an attractive additional drug target in the malaria fatty acid biosynthetic pathway. Further kinetic and structural studies are underway to identify more potent and selective inhibitors.
Acknowledgments
A.H.F. is a Wellcome Principal Research Fellow. S.R.W. and K.A.I. were funded by studentships from the Wellcome Trust and the BBSRC, respectively. D.v.A. is supported by a Wellcome Trust Senior Research Fellowship and the EMBO Young Investigator Programme. We would like to thank Sandra Oza and John Richardson for useful advice, and Irene Hallyburton and Dougie Lamont of the Postgenomics and Molecular Interactions Centre, University of Dundee for assistance with the ultracentrifugation and mass spectrometric studies respectively. We also thank the European Synchrotron Radiation Facility, Grenoble, for the time at beamline BM14.
References
- 1.Snow R. W., Guerra C. A., Noor A. M., Myint H. Y., Hay S. I. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature (London) 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Geneva: World Health Organization; 2002. The World Health Report 2002: Reducing Risks, Promoting Healthy Life. [Google Scholar]
- 3.Kohler S., Delwiche C. F., Denny P. W., Tilney L. G., Webster P., Wilson R. J. M., Palmer J. D., Roos D. S. A plastid of probable green algal origin in apicomplexan parasites. Science. 1997;275:1485–1489. doi: 10.1126/science.275.5305.1485. [DOI] [PubMed] [Google Scholar]
- 4.Wilson R. J. M., Williamson D. H., Preiser P. Malaria and other apicomplexans – the plant connection. Infect. Agents Dis. 1994;3:29–37. [PubMed] [Google Scholar]
- 5.Waller R. F., Keeling P. J., Donald R. G. K., Striepen B., Handman E., Lang-Unnasch N., Cowman A. F., Besra G. S., Roos D. S., McFadden G. I. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12352–12357. doi: 10.1073/pnas.95.21.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schweizer E., Hofmann J. Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol. Mol. Biol. Rev. 2004;68:501–517. doi: 10.1128/MMBR.68.3.501-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith S., Witkowski A., Joshi A. K. Structural and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 2003;42:289–317. doi: 10.1016/s0163-7827(02)00067-x. [DOI] [PubMed] [Google Scholar]
- 8.Campbell J. W., Cronan J. E., Jr Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu. Rev. Microbiol. 2001;55:305–332. doi: 10.1146/annurev.micro.55.1.305. [DOI] [PubMed] [Google Scholar]
- 9.Surolia N., Surolia A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat. Med. 2001;7:167–173. doi: 10.1038/84612. [DOI] [PubMed] [Google Scholar]
- 10.Ralph S. A., van Dooren G. G., Waller R. F., Crawford M. J., Fraunholz M. J., Foth B. J., Tonkin C. J., Roos D. S., McFadden G. I. Metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat. Rev. Microbiol. 2004;2:203–216. doi: 10.1038/nrmicro843. [DOI] [PubMed] [Google Scholar]
- 11.Waters N. C., Kopydlowski K. M., Guszczynski T., Wei L., Sellers P., Ferlan J. T., Lee P. J., Li Z. Y., Woodard C. L., Shallom S., et al. Functional characterization of the acyl carrier protein (PfACP) and β-ketoacyl ACP synthase III (PfKASIII) from Plasmodium falciparum. Mol. Biochem. Parasitol. 2002;123:85–94. doi: 10.1016/s0166-6851(02)00140-8. [DOI] [PubMed] [Google Scholar]
- 12.Prigge S. T., He X., Gerena L., Waters N. C., Reynolds K. A. The initiating steps of a type II fatty acid synthase in Plasmodium falciparum are catalyzed by pfACP, pfMCAT, and pfKASIII. Biochemistry. 2003;42:1160–1169. doi: 10.1021/bi026847k. [DOI] [PubMed] [Google Scholar]
- 13.Pillai S., Rajagopal C., Kapoor M., Kumar G., Gupta A., Surolia N. Functional characterization of β-ketoacyl-ACP reductase (FabG) from Plasmodium falciparum. Biochem. Biophys. Res. Commun. 2003;303:387–392. doi: 10.1016/s0006-291x(03)00321-8. [DOI] [PubMed] [Google Scholar]
- 14.Sharma S. K., Kapoor M., Ramya T. N. C., Kumar S., Kumar G., Modak R., Sharma S., Surolia N., Surolia A. Identification, characterization, and inhibition of Plasmodium falciparum β-hydroxyacyl-acyl carrier protein dehydratase (FabZ) J. Biol. Chem. 2003;278:45661–45671. doi: 10.1074/jbc.M304283200. [DOI] [PubMed] [Google Scholar]
- 15.Perozzo R., Kuo M., Sidhu A. S., Valiyaveettil J. T., Bittman R., Jacobs W. R., Jr, Fidock D. A., Sacchettini J. C. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J. Biol. Chem. 2002;277:13106–13114. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 16.Heath R. J., Yu Y. T., Shapiro M. A., Olson E., Rock C. O. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J. Biol. Chem. 1998;273:30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 17.Heath R. J., Rubin J. R., Holland D. R., Zhang E. L., Snow M. E., Rock C. O. Mechanism of triclosan inhibition of bacterial fatty acid synthesis. J. Biol. Chem. 1999;274:11110–11114. doi: 10.1074/jbc.274.16.11110. [DOI] [PubMed] [Google Scholar]
- 18.Kapoor M., Gopalakrishnapai J., Surolia N., Surolia A. Mutational analysis of the triclosan-binding region of enoyl-ACP (acyl-carrier protein) reductase from Plasmodium falciparum. Biochem. J. 2004;381:735–741. doi: 10.1042/BJ20040302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biagini G. A., O'Neill P. M., Nzila A., Ward S. A., Bray P. G. Antimalarial chemotherapy: young guns or back to the future? Trends Parasitol. 2003;19:479–487. doi: 10.1016/j.pt.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y. M., Lu Y. J., Rock C. O. The reductase steps of the type II fatty acid synthase as antimicrobial targets. Lipids. 2004;39:1055–1060. doi: 10.1007/s11745-004-1330-3. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y. M., Rock C. O. Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty-acid synthase. J. Biol. Chem. 2004;279:30994–31001. doi: 10.1074/jbc.M403697200. [DOI] [PubMed] [Google Scholar]
- 22.Lambert J. D., Yang C. S. Mechanisms of cancer prevention by tea constituents. J. Nutr. 2003;133:3262S–3267S. doi: 10.1093/jn/133.10.3262S. [DOI] [PubMed] [Google Scholar]
- 23.Navarro-Peran E., Cabezas-Herrera J., Garcia-Canovas F., Durrant M. C., Thorneley R. N., Rodriguez-Lopez J. N. The antifolate activity of tea catechins. Cancer Res. 2005;65:2059–2064. doi: 10.1158/0008-5472.CAN-04-3469. [DOI] [PubMed] [Google Scholar]
- 24.Cohen-Gonsaud M., Ducasse S., Hoh F., Zerbib D., Labesse G., Quemard A. Crystal structure of MabA from Mycobacterium tuberculosis, a reductase involved in long-chain fatty acid biosynthesis. J. Mol. Biol. 2002;320:249–261. doi: 10.1016/S0022-2836(02)00463-1. [DOI] [PubMed] [Google Scholar]
- 25.Fisher M., Kroon J. T. M., Martindale W., Stuitje A. R., Slabas A. R., Rafferty J. B. The X-ray structure of Brassica napus β-keto acyl carrier protein reductase and its implications for substrate binding and catalysis. Structure. 2000;8:339–347. doi: 10.1016/s0969-2126(00)00115-5. [DOI] [PubMed] [Google Scholar]
- 26.Price A. C., Zhang Y. M., Rock C. O., White S. W. Structure of β-ketoacyl-[acyl carrier protein] reductase from Escherichia coli: negative cooperativity and its structural basis. Biochemistry. 2001;40:12772–12781. doi: 10.1021/bi010737g. [DOI] [PubMed] [Google Scholar]
- 27.Price A. C., Zhang Y. M., Rock C. O., White S. W. Cofactor-induced conformational rearrangements establish a catalytically competent active site and a proton relay conduit in FabG. Structure. 2004;12:417–428. doi: 10.1016/j.str.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Clos J., Brandau S. pJC20 and pJC40 – 2 high-copy-number vectors for T7 RNA polymerase-dependent expression of recombinant genes in Escherichia coli. Protein Expression Purif. 1994;5:133–137. doi: 10.1006/prep.1994.1020. [DOI] [PubMed] [Google Scholar]
- 29.Trager W., Jensen J. B. Human malaria parasites in continuous culture. Nature (London) 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 30.Desjardins R. E., Canfield C. J., Haynes J. D., Chulay J. D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979;16:710–718. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr. Sect. A Found. Crystallogr. 1994;A50:157–163. [Google Scholar]
- 33.Perrakis A., Morris R., Lamzin V. S. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 34.Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr. Sect. A Found. Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 36.Bendtsen J. D., Nielsen H., von Heijne G., Brunak S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 37.Zuegge J., Ralph S., Schmuker M., McFadden G. I., Schneider G. Deciphering apicoplast targeting signals – feature extraction from nuclear-encoded precursors of Plasmodium falciparum apicoplast proteins. Gene. 2001;280:19–26. doi: 10.1016/s0378-1119(01)00776-4. [DOI] [PubMed] [Google Scholar]
- 38.Cleland W. W. Steady state kinetics. In: Boyer P. D., editor. The Enzymes. New York and London: Academic Press; 1970. pp. 1–65. [Google Scholar]
- 39.Su X. Z., Kirkman L. A., Fujioka H., Wellems T. E. Complex polymorphisms in an 330 kDa protein are linked to chloroquine resistant P. falciparum in Southeast Asia and Africa. Cell. 1998;91:593–603. doi: 10.1016/s0092-8674(00)80447-x. [DOI] [PubMed] [Google Scholar]
- 40.Kuo M. R., Morbidoni H. R., Alland D., Sneddon S. F., Gourlie B. B., Staveski M. M., Leonard M., Gregory J. S., Janjigian A. D., Yee C., et al. Targeting tuberculosis and malaria through inhibition of enoyl reductase. J. Biol. Chem. 2003;278:20851–20859. doi: 10.1074/jbc.M211968200. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y. M., Rao M. S., Heath R. J., Price A. C., Olson A. J., Rock C. O., White S. W. Identification and analysis of the acyl carrier protein (ACP) docking site on β-ketoacyl-ACP synthase III. J. Biol. Chem. 2001;276:8231–8238. doi: 10.1074/jbc.M008042200. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y. M., Wu B. N., Zheng J., Rock C. O. Key residues responsible for acyl carrier protein and β-ketoacyl-acyl carrier protein reductase (FabG) interaction. J. Biol. Chem. 2003;278:52935–52943. doi: 10.1074/jbc.M309874200. [DOI] [PubMed] [Google Scholar]
- 43.Schulz H., Wakil S. J. Studies on the mechanism of fatty acid synthesis. XXV. On the mechanism of β-ketoacylacyl carrier protein reductase from Escherichia coli. J. Biol. Chem. 1971;246:1895–1901. [PubMed] [Google Scholar]
- 44.Marrakchi H., Ducasse S., Labesse G., Montrozier H., Margeat E., Emorine L., Charpentier X., Daffe M., Quemard A. MabA (FabG1), a Mycobacterium tuberculosis protein involved in the long-chain fatty acid elongation system FAS-II. Microbiology. 2002;148:951–960. doi: 10.1099/00221287-148-4-951. [DOI] [PubMed] [Google Scholar]
- 45.Villa F., Deak M., Bloomberg G. B., Alessi D. R., van Aalten D. M. F. Crystal structure of the PTPL1/FAP-1 human tyrosine phosphatase mutated in colorectal cancer: evidence for a second phosphotyrosine substrate recognition pocket. J. Biol. Chem. 2005;280:8180–8187. doi: 10.1074/jbc.M412211200. [DOI] [PubMed] [Google Scholar]
- 46.Lietzke S. E., Bose S., Cronin T., Klarlund J., Chawla A., Czech M. P., Lambright D. G. Structural basis of 3-phosphoinositide recognition by pleckstrin homology domains. Mol. Cell. 2000;6:385–394. doi: 10.1016/s1097-2765(00)00038-1. [DOI] [PubMed] [Google Scholar]
- 47.Kapoor M., Jamal D. M., Surolia A., Surolia N. Kinetic determinants of the interaction of enoyl-ACP reductase from Plasmodium falciparum with its substrates and inhibitors. Biochem. Biophys. Res. Commun. 2001;289:832–837. doi: 10.1006/bbrc.2001.6061. [DOI] [PubMed] [Google Scholar]
- 48.Levy C. W., Roujeinikova A., Sedelnikova S., Baker P. J., Stuitje A. R., Slabas A. R., Rice D. W., Rafferty J. B. Molecular basis of triclosan activity. Nature (London) 1999;398:383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- 49.Qiu X. Y., Janson C. A., Court R., Smyth M. G., Payne D. J., Abdel-Meguid S. S. Molecular basis for triclosan activity involves a flipping loop in the active site. Protein Sci. 1999;8:2529–2532. doi: 10.1110/ps.8.11.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ward W. H. J., Holdgate G. A., Rowsell S., McLean E. G., Pauptit R. A., Clayton E., Nichols W. W., Colls J. G., Minshull C. A., Jude D. A., et al. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry. 1999;38:12514–12525. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- 51.Wang J. L., Buhler D. R. Inhibition of dehydrogenase enzymes by hexachlorophene. Biochem. Pharmacol. 1978;27:2947–2953. doi: 10.1016/0006-2952(78)90213-7. [DOI] [PubMed] [Google Scholar]
- 52.Prasad G. V., Rajendra W., Indira K. In vitro kinetics of the mouse hepatic arginase inhibition by hexachlorophene. Pestic. Biochem. Physiol. 1987;28:308–317. [Google Scholar]
- 53.Matsumura H., Matsuoka M., Igisu H., Ikeda M. Cooperative inhibition of acetylcholinesterase activities by hexachlorophene in human erythrocytes. Arch. Toxicol. 1997;71:151–156. doi: 10.1007/s002040050369. [DOI] [PubMed] [Google Scholar]
- 54.Lokanatha V., Sailaja P., Rajendra W. In vitro kinetics of the rat brain succinate dehydrogenase inhibition by hexachlorophene. J. Biochem. Mol. Toxicol. 1999;13:303–306. doi: 10.1002/(sici)1099-0461(1999)13:6<303::aid-jbt3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 55.Gulden M., Morchel S., Seibert H. Serum albumin binding at cytotoxic concentrations of chemicals as determined with a cell proliferation assay. Toxicol. Lett. 2003;137:159–168. doi: 10.1016/s0378-4274(02)00399-5. [DOI] [PubMed] [Google Scholar]
- 56.Sheldon P. S., Kekwick R. G., Smith C. G., Sidebottom C., Slabas A. R. 3-Oxoacyl-[ACP] reductase from oilseed rape (Brassica napus) Biochim. Biophys. Acta. 1992;1120:151–159. doi: 10.1016/0167-4838(92)90263-d. [DOI] [PubMed] [Google Scholar]
- 57.Toomey R. E., Wakil S. J. Studies on the mechanism of fatty acid synthesis. XV. Preparation and general properties of β-ketoacyl acyl carrier protein reductase from Escherichia coli. Biochim. Biophys. Acta. 1966;116:189–197. doi: 10.1016/0005-2760(66)90001-4. [DOI] [PubMed] [Google Scholar]