

Abstract
Proteomic analysis of mouse kidney peroxisomes resulted in the identification of a novel nudix hydrolase designated RP2p, which is encoded by the D7RP2e gene. RP2p consists of 357 amino acids and contains two conserved domains: a nudix hydrolase domain and a CoA-binding domain. In addition, a PTS (peroxisomal targeting signal) type 1 (Ala-His-Leu) was found at the C-terminus. Analysis of the enzyme characteristics revealed that RP2p is a CoA diphosphatase with activity towards CoA, oxidized CoA and a wide range of CoA esters, including choloyl-CoA and branched-chain fatty-acyl-CoA esters. The enzymatic properties of RP2p indicate that at low substrate concentrations medium and long-chain fatty-acyl-CoA esters are the primary substrates. Enzyme activity was optimal at pH 9 or above, and required the presence of Mg2+ or Mn2+ ions. Subcellular fractionation studies revealed that all CoA diphosphatase activity in mouse kidney is restricted to peroxisomes.
Keywords: acyl-CoA ester, kidney, mouse, nudix hydrolase, peroxisome, proteomics
Abbreviations: BN, Blue native; DTT, dithiothreitol; IPTG, isopropyl β-D-thiogalactoside; MALDI–TOF, matrix-assisted laser-desorption ionization–time-of-flight; MBP, maltose-binding protein; PTS, peroxisomal targeting signal; Q-TOF, quadrupole time-of-flight
INTRODUCTION
Peroxisomes are small subcellular organelles that are present in virtually all eukaryotic cells. They are involved in a number of essential metabolic functions, including α- and β-oxidation of fatty acids, ether-phospholipid biosynthesis and L-pipecolic acid oxidation [1,2]. Peroxisomal proteins are encoded by nuclear genes, synthesized in the cytosol on free ribosomes and imported into the organelle. Newly synthesized proteins destined for peroxisomes contain a specific PTS (peroxisomal targeting signal) which directs them to the organelle [3,4].
Defects in one or more peroxisomal functions are associated with severe clinical manifestations. So far, 17 different diseases, including single and multiple enzyme deficiencies, belong to the group of peroxisomal disorders [5,6]. Thanks to the marked increase of new information with respect to the functions and biogenesis of peroxisomes in the past decade, the metabolic and molecular basis of most of the known peroxisomal disorders has been resolved. However, the total number of disorders linked to a dysfunction of peroxisomes may well increase as we gain more insight into the functions of this organelle. Therefore the elucidation of the complete protein composition of peroxisomes is of crucial importance for our understanding of their role in human metabolism and disease.
In the present paper, we describe the identification and characterization of a novel nudix hydrolase, named RP2p, from male mouse kidney peroxisomes. Mouse RP2p shows diphosphatase activity towards CoA and a wide range of CoA esters, including medium- and long-chain fatty-acyl-CoA esters. We also identified the human and rat homologue of RP2p in the GenBank® database. Our data indicate that this peroxisomal nudix hydrolase not only plays a role in the breakdown of free CoA in peroxisomes, but also converts fatty-acyl-CoA esters into acyl-4′-phosphopantetheine esters and 3′,5′-ADP.
EXPERIMENTAL
CoA and Complete, Mini™ protease inhibitor cocktail tablets were obtained from Roche Applied Science. CoA esters and 3′,5′-ADP were purchased from Sigma Chemical Co. All other chemicals used were of analytical grade.
Purification of peroxisomes from male mouse kidney
Kidneys were obtained from male Swiss mice, 4–6 months of age, and were homogenized in 5 mM Mops buffer, pH 7.4, containing 250 mM sucrose, 2 mM EDTA and 0.1% ethanol. A post-nuclear supernatant was produced by centrifugation of the homogenate at 600 g for 10 min at 4 °C. Peroxisomes were isolated by using equilibrium density-gradient centrifugation in a 20–35% (w/v) Nycodenz gradient with a 50% (w/v) cushion as described in [7]. Fractions of 2 ml were taken from the bottom of the gradient and were assayed for the marker enzymes glutamate dehydrogenase (mitochondria), catalase (peroxisomes), phosphoglucoisomerase (cytoplasm), β-hexosaminidase (lysosomes) and esterase (microsomes) as described previously [8,9]. Protein concentration was measured according to the method of Bradford [10], using BSA as standard.
Two dimensional gel-electrophoresis
BN (Blue native)-PAGE was performed at 4 °C according to the method of Schägger and von Jagow [11], using a 5–14% polyacrylamide gradient gel. Before electrophoresis, a fraction containing purified peroxisomes (approx. 0.5 mg of protein) was dissolved in loading buffer containing 1% (w/v) digitonin and was subsequently applied to the gel. Electrophoresis was continued until most of the dye was eluted from the gel. After electrophoresis, strips of approx. 0.5 cm width were sliced from the gel, and prepared for SDS/PAGE according to the method of Laemlli [12]. First the gel strips were covered with loading buffer and incubated for 15 min at 37 °C. After a brief wash with electrophoresis buffer, the strips were positioned on top of a standard SDS/PAGE gel (10% polyacrylamide separation gel with a 5% stacking gel). After electrophoresis, the proteins were stained with silver according to the method of Rabilloud et al. [13].
Identification of proteins by MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight)-MS and Q-TOF (quadrupole time-of-flight) MS/MS analysis
Protein-containing gel slices were S-alkylated, digested with trypsin (Roche Molecular Biochemicals, sequencing grade), and extracted according to the method of Shevchenko et al. [14]. Extracted peptides were concentrated using a SpeedVac, and the pellet was taken up in 10 μl of 1% methanoic (formic) acid, 60% acetonitrile. When required, the extracted peptides were purified and concentrated using Zip-Tips (Millipore). Peptides were eluted from the Zip-Tips with 5–10 μl of 1% formic acid and 60% acetonitrile. The peptide solution was mixed with an equal volume of 10 mg/ml α-cyano-4-hydroxycinnamic acid (Sigma Chemical Co.) solution in acetonitrile/ethanol (1:1, v/v) with 1% trifluoroacetic acid and 1 mM ammonium acetate. A volume of 1 μl was spotted on the target and was allowed to dry at room temperature (20 °C). MALDI–TOF-MS spectra were acquired on a Micromass M@LDI-R equipped with a 2-GHz digitizer. The resulting peptide spectra were used to search a non-redundant protein sequence database (Swiss-Prot/TrEMBL) using the Proteinprobe program or the MASCOT search engine and database. For ESI-Q-TOF (electrospray ionization Q-TOF) MS, 2 μl of a peptide solution was introduced into a nanospray capillary, and positive mode spectra were recorded with a Q-TOF mass spectrometer (Micromass) equipped with a Z-spray source.
Cloning of the mouse D7RP2e cDNA
First-strand cDNA was prepared from mouse kidney as described in [15] and was used as a template to amplify the RP2p encoding cDNA by PCR using Taq DNA polymerase and the following primers: a forward primer, 5′-GGATCCATGAGCAGCTCGAGCAGC-3′, containing a BamHI endonuclease site (underlined) and a reverse primer, 5′-AAGCTTTTACAGGTGGGCAGTATATC-3′, with a HindIII endonuclease site (double underlined). The PCR protocol for amplification of the RP2p-encoding cDNA was 94 °C for 2 min followed by 30 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 2 min and a final extension step at 72 °C for 2 min. The PCR product was purified using the Prep-A-Gene Purification kit (Bio-Rad) and was cloned downstream of the IPTG (isopropyl β-D-thiogalactoside)-inducible PTAC promoter into the BamHI and HindIII sites of the bacterial pMAL-C2X expression vector (New England Biolabs) to express mouse RP2p as a fusion protein with MBP (maltose-binding protein). The open reading frame was sequenced to exclude sequence errors introduced during PCR amplification.
Expression and purification of mouse RP2p
Transformed Escherichia coli (BL21) were grown at 37 °C in 500 ml of LB (Luria–Bertani) medium supplemented with 0.1% (w/v) glucose and 100 μg/ml ampicillin to a D600 of approx. 0.5, and IPTG was added to a final concentration of 0.1 mM to induce protein expression. After 4 h at 37 °C, cells were collected by centrifugation for 5 min at 1000 g. The pellet was dissolved in PBS solution with 0.1% (v/v) Triton X-100 and protease inhibitor cocktail (one tablet of Complete, Mini™ per 10 ml of solution). Cell lysates were prepared by sonication for three periods of 20 s at 8–9 W in an ice water bath, followed by centrifugation at 12000 g for 15 min at 4 °C. The supernatant was used for the purification of the MBP-fusion protein using approx. 4 ml of amylose resin as described by the manufacturer (New England Biolabs).
Measurement of CoA diphosphatase activity
CoA diphosphatase activity was measured in a 100 mM Tris/HCl buffer, pH 9.0, containing 5 mM MgCl2, 10 mM DTT (dithiothreitol) and 2 mM CoA in a final volume of 125 μl. Reactions were started by adding the protein sample and were terminated after 15 min at 37 °C by the addition of 25 μl of 2 M perchloric acid. After neutralization with potassium carbonate, the samples were centrifuged for 10 min at 12000 g at 4 °C. After dilution of the supernatants with an equal volume of 100 mM ammonium phosphate (buffer A), 50 μl was used to analyse CoA and its metabolites on a HPLC system using a 250 mm×4.6 mm Supelcosil LC-18-S column equilibrated with buffer A at a flow rate of 1 ml/min. The metabolites were eluted from the column with a linear gradient of 20 min changing from 100% buffer A to 100% buffer B (mixture of buffer A/methanol, 50:50). Absorption was measured at 265 nm using a Gynkotek UVD 340S photodiode array detector with Chromeleon software, version 6.50. The amount of 3′,5′-ADP produced in the assay was calculated using a curve of 3′,5′-ADP standards analysed using the same method. When CoA esters were used as substrates, DTT was excluded from the incubation mixture as it stimulates the non-enzymatic hydrolysis of the ester-bond at high pH (results not shown), and the HPLC method was extended with a second linear gradient of 5 min changing from 100% buffer B to 100% buffer C (16.9 mM sodium phosphate, pH 6.9/acetonitrile, 20:80).
RESULTS
Identification of proteins from mouse kidney peroxisomes
Peroxisomes were isolated from male mice kidneys and subjected to a combination of BN-PAGE and SDS/PAGE followed by silver staining. A representative two-dimensional gel is shown in Figure 1. The two-dimensional gel shows several proteins migrating as larger complexes of proteins in the first, native, dimension as compared with their migration rate in the second, denaturing, dimension. This suggests that these proteins are part of a protein complex in their native state. To identify the individual proteins, spots were excised from the gel and prepared for MALDI–TOF MS as described in the Experimental section. This resulted in the identification of 13 distinct proteins as summarized in Table 1.
Figure 1. Two-dimensional electrophoresis of mouse kidney peroxisomes.

In the first dimension, proteins were separated using BN-PAGE and subsequently by SDS/PAGE in the second dimension. Subsequently, proteins were visualized by silver staining. See the Experimental section for details. Arrows indicate the direction of electrophoresis during the first dimension (A) and second dimension (B). Molecular mass markers (sizes in kDa) for the second dimension are on both sides of the gel.
Table 1. Identification of proteins from mouse kidney peroxisomes using MALDI–TOF-MS.
Protein spots derived from the two-dimensional gel were prepared for MALDI–TOF-MS analysis as described in the Experimental section. The resulting peptide spectra were used to search a non-redundant protein sequence database (Swiss-Prot/TrEMBL) using the Proteinprobe program or the MASCOT search engine and database. The molecular mass of the denatured proteins was calculated from their primary amino acid sequence as taken from the GenBank® database. DHAP, dihydroxyacetonephosphate.
| Molecular mass (kDa) | ||||
|---|---|---|---|---|
| No. | Identified protein | Native | Denatured | Remarks |
| 1 | Catalase | 240 | 59.8 | Homotetramer [17] |
| 2 | Acyl-CoA oxidase I | 140 | 74.6 | Homodimer [16] |
| 3 | L-Hydroxyacid oxidase 2 | 140–240 | 38.8 | Homomultimer |
| 4 | α-Methylacyl-CoA racemase | ∼80 | 41.7 | Homodimer |
| 5 | RP2p | <50 | 40.4 | Accession number P11930 |
| 6 | D-Amino acid oxidase | <50 | 38.7 | Monomer |
| 7 | trans-2-enoyl-CoA reductase | ∼30/∼60 | 32.5 | Monomer and homodimer |
| 8 | Epoxide hydrolase | ∼70 | 62.5 | Accession number P34914 |
| 9 | Peroxisomal bifunctional enzyme | ∼80/∼160 | 78.2 | Monomer and oligomer |
| 10 | Acyl-CoA oxidase 3 | >100 | 78.4 | Homodimer |
| 11 | Δ3,Δ2-Enoyl-CoA isomerase | <50 | 35.2 | Riken 1810022C23 gene |
| 12 | Alkyl-DHAP synthase | ∼70 | 71.7 (67.0) | Precursor (mature form) |
| 13 | Carnitine O-octanoyltransferase | ∼80 | 70.3 | Accession number P11466 |
Most of the proteins identified have already been reported as peroxisomal. This includes acyl-CoA oxidase I (Figure 1, spots numbered 2). The calculated molecular mass of the full-length protein, based on the amino acid sequence obtained from the GenBank® database (accession number Q9R0H0, mouse peroxisomal palmitoyl-CoA oxidase), is 74.6 kDa. However, the protein was observed mainly as two fragments with apparent molecular masses of approx. 50 and 20 kDa. Using peptide mass finger-printing analysis, it could be deduced that the 50 kDa protein is the N-terminal part, while the 20 kDa protein contains the C-terminal part of acyl-CoA oxidase I. Integration of the known molecular masses and the relative position of the identified proteins on the two-dimensional gel leads us to the conclusion that acyl-CoA oxidase I forms a homodimer and mainly consists of two cleaved polypeptides. These results are in line with the work of Miyazawa et al. [16] on acyl-CoA oxidase from rat liver. Several of the other proteins were also observed as oligomers in the two-dimensional gel. This includes catalase (Figure 1, spot number 1), L-hydroxyacid oxidase 2 (Figure 1, spot number 3) and α-methylacyl-CoA racemase (Figure 1, spot number 4).
One protein was identified that had not been linked to peroxisomes previously. This protein is the mouse testosterone-regulated RP2 protein encoded by the D7Rp2e gene (accession number P11930, androgen-regulated protein rp2, SW:RP2_MOUSE, Chr 7 (7B1) Roswell Park 2 complex, LocusID: 110959 UniGene Cluster Mm.27194). Six peptides could reliably be matched, resulting in 25.8% coverage (results not shown). Mouse RP2p consists of 357 amino acids with a calculated molecular mass of 40.3 kDa. This is in line with the apparent molecular mass of the protein observed in the two-dimensional gel (Figure 1, spot number 5). Analysis of the amino acid sequence of RP2p revealed two conserved domains. The first domain was found at amino acids 30–50 and contains a putative binding motif for CoA and its derivatives with the signature LLTXR(SA)X3RX3GX3FPGG designated UPF0035 in the PROSITE database. This motif is also present in mouse Nudt7 and yeast Pcd1, two nudix hydrolases with activity towards CoA [19,20]. The second domain was found at amino acids 92–119 and contains the nudix box sequence signature motif (nudix or NUDT motif) GX5EX5(UA)XRE(UA)XEEXGU (where U indicates a hydrophobic amino acid). Furthermore, a peroxisomal targeting signal type 1 was found at the C-terminus (Ala-His-Leu). A human (GenBank® accession number XM_372723) as well as a rat (GenBank® accession number XP_218512) homologue of RP2p were also identified in the GenBank® database. Both the human and rat proteins have a typical PTS type 1 at their C-terminus, SHL and ARL respectively. Figure 2 shows the alignment of the two conserved domains of RP2p from mouse, man and rat with other nudix hydrolases with CoA diphosphatase activity; mouse Nudt7 [19], Saccharomyces cerevisiae Pcd1 [20] and Caenorhabditis elegans Ndx8 [21]. Comparison of the organization of the two motifs within mouse RP2p and Nudt7 shows that, in Nudt7, both motifs are positioned close to each other, while, in RP2p, the motifs are separated by a stretch of 42 amino acids (Figure 3A). Further analysis of the amino acid sequence of mouse RP2p was carried out using the RADAR (Rapid Automatic Detection and Alignment of Repeats software at EMBL-EBI) program, and the result is shown in Figure 3(B).
Figure 2. Partial sequence alignment of RP2p to related nudix hydrolases.

Sections of the RP2p amino acid sequence from mouse (MmRP2p), rat (RnRP2p) and human (HsRP2p) containing the CoA-binding motif (A) with the PROSITE UPF0035 signature and the nudix domain (B) were aligned to mouse Nudt7α (MmNudt7; GenBank® accession number NP_077757), S. cerevisiae Pcd1 (ScPcd1; GenBank® accession number NP_013252) and C. elegans Ndx-8 (CeNdx8; GenBank® accession number NP_493372). The start and end amino acid positions are indicated in parentheses before and after each sequence.
Figure 3. Analysis of the amino acid sequence containing the conserved domains of mouse RP2p.

(A) Amino acid sequence alignment containing the putative CoA-binding domain with the signature LLTXR(SA)X3RX3GX3FPGG designated UPF0035 in the PROSITE database (underlined) and the nudix box sequence signature motif (nudix or NUDT motif) GX5EX5(UA)XRE(UA)XEEXGU (double underlined) of mouse RP2p and Nudt7. (B) Alignment of the two amino acid sequences of the observed repeat in mouse RP2p. Start and end amino acid positions are indicated in parentheses before and after each sequence.
Characterization of RP2p
Mouse RP2p was expressed in E. coli as a fusion protein with MBP and was purified using amylose resin. The purity of the isolated fusion protein was established by SDS/PAGE (Figure 4). The enzymatic properties were determined using a broad range of substrates, including ATP, ADP, NAD+, NADH, FAD, NADP+, NADPH and CoA. The initial assay conditions used were as described by Gasmi and McLennan [19] for mouse Nudt7. Only in the case of CoA as substrate did we observe the formation of a product which was identified as 3′,5′-ADP using the commercially available compound for comparison in the HPLC analysis (results not shown). Subsequently, the optimal conditions for enzyme activity were determined using CoA as substrate. As shown in Figure 5(A), activity was highest at alkaline pH. To keep non-enzymatic breakdown of CoA to a minimum, however, all subsequent incubations were performed at pH 9.0.
Figure 4. SDS/PAGE of purified recombinant mouse RP2p.
Mouse RP2p was expressed in E. coli as an MBP-fusion protein and was subsequently purified using amylose resin. The indicated amounts of the purified protein and molecular mass markers (M) were applied to an SDS/polyacrylamide gel. The gel was stained with Brilliant Blue G according to the method described by Neuhoff et al. [18]. Molecular-mass sizes are given in kDa.
Figure 5. Enzymatic characterization of mouse RP2p.
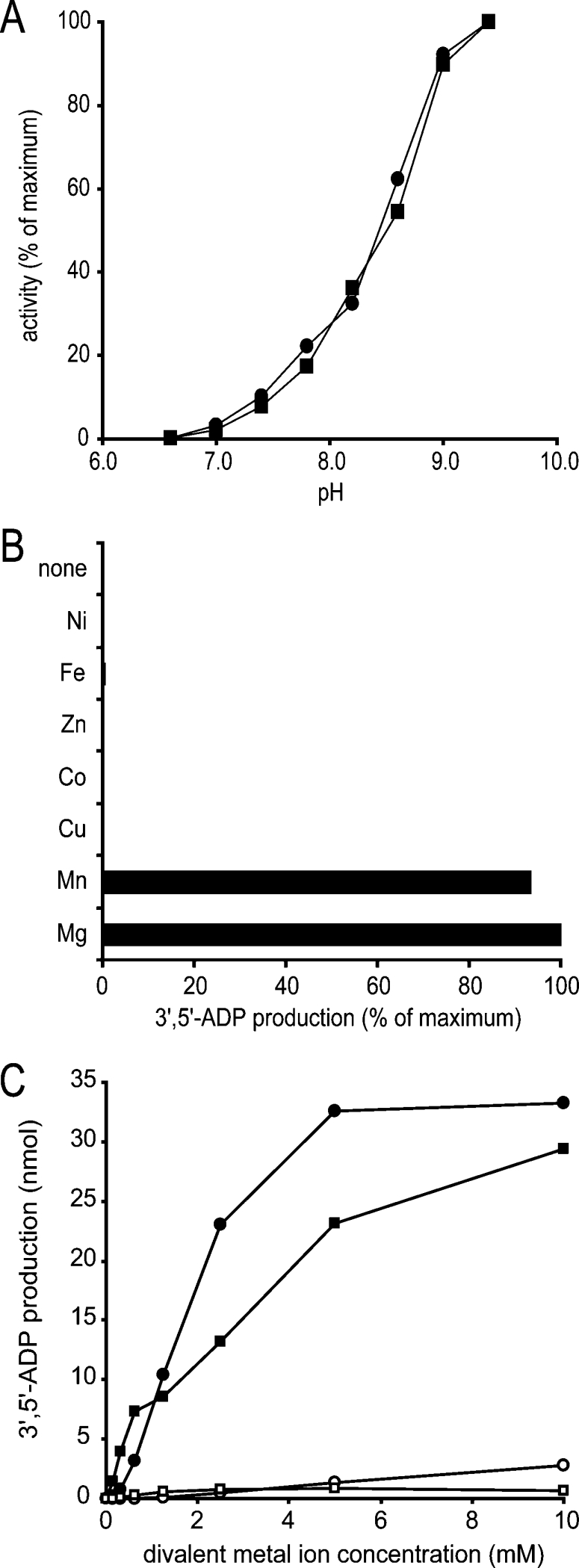
(A) The pH-dependency of CoA diphosphatase activity of purified mouse kidney peroxisomes (■, 10 μg protein) and recombinant mouse RP2p (●, 2.8 μg of protein) was determined under the assay conditions as described in the Experimental section, except that Tris/HCl buffer was replaced with glycine/Hepes buffer (100 mM each) at the indicated pH. (B) Requirement of bivalent metal ions for CoA diphosphatase activity. Purified recombinant mouse RP2p (2.8 μg of protein) was incubated under the standard assay conditions as described in the Experimental section in the presence or absence of several bivalent metal ions as indicated on the left at a final concentration of 5 mM. (C) Mg2+ and Mn2+ ion dependency of CoA diphosphatase activity. CoA diphosphatase activity was measured under the conditions described above in the presence of Mg2+ (●,○) or Mn2+ (■,□) at the indicated concentrations using a Tris/HCl buffer at pH 9.0 (■,●) or 7.4 (□,○).
So far, all known nudix hydrolases strictly require bivalent metal ions in order to be active. Therefore we tested the activity of RP2p in the presence of a series of bivalent metal ions, including Mg2+. This revealed that the enzyme was active only in the presence of Mg2+ and Mn2+ with no activity with any of the other bivalent cations tested (Figure 5B). Finally, the activity of RP2p was measured both at pH 9.0 and at pH 7.4, a more physiological pH, using different concentrations of Mg2+ or Mn2+ ions. The results (Figure 5C) show that the highest activity was obtained with Mg2+ at a concentration of 5 mM. At bivalent metal ion concentrations below 1 mM, however, RP2p activity was higher with Mn2+ than with Mg2+. Fluoride, a common inhibitor of nudix hydrolases, also inhibited the purified recombinant enzyme with an IC50 value of approx. 50 μM (results not shown).
To determine the substrate specificity of mouse RP2p, the purified recombinant protein was tested with a wide range of CoA esters (Figure 6). Diphosphatase activity was observed with all substrates used, but the highest activity was obtained with medium-chain acyl-CoA esters. Higher rates of enzyme activity were found with the unsaturated acyl-CoA esters compared with the saturated esters. Also with choloyl-CoA and dimethylnonanoyl-CoA, a branched-chain fatty-acyl-CoA ester, diphosphatase activity was readily detectable, whereas with pristanoyl-CoA, only minor activity was observed.
Figure 6. Substrate specificity of mouse RP2p.

Purified recombinant mouse RP2p (2.8 μg of protein) was incubated for 15 min under the standard assay conditions as described in the Experimental section, except that DTT was omitted from the incubation mixture. All substrates, indicated on the left, were added to a final concentration of 200 μM. Diphosphatase activity is given as the mean for two independent experiments and do not vary by more than 5%. DMN-CoA, dimethylnonanoyl-CoA.
To analyse further the substrate specificity of mouse RP2p, the enzyme activity was measured using several substrates, including free CoA, 3′-dephospho-CoA, oxidized CoA, lauroyl-CoA (C12:0-CoA) and palmitoyl-CoA (C16:0-CoA), at different substrate concentrations. Lineweaver–Burk plots were used to calculate both Km and Vmax values, and results are summarized in Table 2. The highest rate of enzyme activity was obtained when free CoA was used as a substrate. When 3′-dephospho-CoA was used as a substrate, no product (5′-AMP) was observed. In addition, the hydrolysis of free CoA was not inhibited when 3′-dephospho-CoA was added in excess amounts (results not shown). The Vmax values as determined for lauroyl-CoA and palmitoyl-CoA were much lower when compared with that of CoA; however, the Km values calculated for these substrates were also much lower. As a consequence, the Vmax/Km ratios for these substrates were even higher when compared with that of free CoA.
Table 2. Analysis of the enzymatic properties of mouse RP2p.
CoA diphosphatase activity was measured as described in the Experimental section. Km and Vmax values were calculated from Lineweaver–Burk plots using at least five data points measured in duplicate. N.D., not detectable.
| Substrate | Km (mM) | Vmax (μmol/mg per min) | Vmax/Km |
|---|---|---|---|
| CoA | 0.60 | 0.56 | 0.93 |
| 3′-Dephospho-CoA | N.D. | N.D. | – |
| Oxidized CoA | 0.58 | 0.012 | 0.02 |
| Lauroyl-CoA | 0.08 | 0.2 | 2.5 |
| Palmitoyl-CoA | 0.01 | 0.013 | 13 |
| Choloyl-CoA | 0.1 | 0.17 | 1.7 |
| Pristanoyl-CoA | 0.1 | 0.009 | 0.09 |
To determine the subcellular distribution of CoA diphosphatase activity in mouse kidney, an isopycnic density-gradient experiment was performed (Figure 7). Analysis of the density gradient showed that CoA diphosphatase activity co-localized with catalase, the marker enzyme for peroxisomes. The bulk of CoA diphosphatase activity was found in the high-density fractions with two minor peaks at lower densities in which catalase activity was also observed.
Figure 7. Subcellular distribution of CoA diphosphatase activity in mouse kidney.

A post-nuclear supernatant of kidneys from nine male mice was prepared and subjected to equilibrium density-gradient centrifugation using a linear 20–35% (w/v) Nycodenz gradient as described by Wanders et al. [7]. After centrifugation, the gradient was fractionated, and marker enzymes and CoA diphosphatase activities were measured in all fractions: (A) catalase (peroxisomes); (B) β-hexosaminidase (lysosomes); (C) CoA diphosphatase; (D) glutamate dehydrogenase (mitochondria); (E) esterase (microsomes); and (F) total protein concentration. Results are percentages of total activity observed (A–E) or mg/ml protein (F).
Previously, Gasmi and McLennan [19] reported the presence of a nudix hydrolase, Nudt7, with activity towards CoA in peroxisomes. These investigators found that Nudt7 activity was optimal at pH 8 [19]. For RP2p, however, we observed the highest enzyme activity at pH 9.0 and above. Therefore, to discriminate which enzyme is most active in mouse kidney peroxisomes, we compared the pH-dependency of CoA diphosphatase activity in our purified mouse kidney peroxisomes with that of the purified recombinant enzyme. The result, shown in Figure 5(A), revealed that the pH-dependency of CoA diphosphatase activity is identical in both samples, implying that RP2p is responsible for most, if not all, of the CoA diphosphatase activity in mouse kidney peroxisomes.
DISCUSSION
Proteomic analysis of mouse kidney peroxisomes revealed a novel peroxisomal nudix hydrolase, designated RP2p. It is encoded by the D7RP2e gene and is also known as androgen-regulated protein rp2. The primary structure of RP2p consists of 357 amino acids. The protein contains a putative CoA-binding site, a nudix box sequence signature motif and a PTS type 1 (Ala-His-Leu) at the C-terminus. Based on the results from the two-dimensional gel-electrophoresis experiments, RP2p appears to be monomeric under native conditions. Alignment of the two conserved domains of mouse RP2p with nudix hydrolases with CoA diphosphatase activity from other species showed a high degree of similarity. However, it should be noted that the CoA-binding domain and the nudix hydrolase domain in MmNudt7, CeNdx-8 and ScPcd1 are in close proximity to each other, while, in RP2p, these domains are separated by a stretch of approx. 40 amino acids. Analysis of the primary structure of mouse RP2p suggests that the stretch of amino acids which separates the two domains could originate from a duplication of the nucleotide sequence encoding amino acids 22–56 containing the complete CoA-binding domain. This is supported by the fact that comparison of the coding nucleotide sequences reveals a high degree of similarity.
Characterization of the enzymatic properties showed that RP2p can hydrolyse CoA into 3,5-ADP and 4′-phosphopantetheine. Like other nudix hydrolases, RP2p requires the presence of Mg2+ or Mn2+ ions for its enzyme activity and is inhibited by fluoride. Besides CoA, RP2p can also use medium- and long-chain fatty-acyl-CoA esters, as well as pristanoyl-CoA and choloyl-CoA, as substrates. Unsaturated fatty-acyl-CoA esters were hydrolysed at an even higher rate compared with saturated CoA esters. The highest rate of diphosphatase activity was obtained with free CoA as substrate, although the Km value for CoA is also relatively high. The affinity of RP2p for medium- and long-chain fatty-acyl-CoA esters, however, is much higher, which implies that, at physiological substrate concentrations, CoA esters could be the preferred substrates. When the ‘inactive’ forms of CoA, oxidized CoA and dephospho-CoA, were used as substrate, little or no activity was observed. In addition, the Km value for oxidized CoA was comparable with that of CoA. From this we can conclude that it is unlikely that RP2p plays a role in the elimination of these substances from the peroxisomal lumen.
Analysis of the subcellular distribution of CoA diphosphatase activity in mouse kidney showed that most, if not all, of the activity is indeed located in peroxisomes. Although most of the activity was found in the high-density fractions, two minor peaks of activity were observed at intermediate and low densities. As all of these fractions also contain catalase activity, we believe that the peak observed at intermediate density represents a peroxisomal subpopulation, while the peak at low density is due to enzymes leaked from the peroxisomal lumen.
The expression of the RP2 gene in mouse kidney has been investigated extensively in the past, although the function of the gene product remained unknown [22–27]. Depending on the mouse strain, RP2 mRNA levels (also known as MAK mRNA and pMK908) can undergo 2–12-fold induction in response to testosterone. The expression of RP2 is driven by at least two promoters, only one of which is androgen-inducible [25]. It is intriguing that the expression of this gene is induced by androgens, including testosterone, at least in mouse kidney and liver, but not in heart, brain, muscle, testes and submaxillary gland [23]. Androgens are not known to affect the expression of other genes that code for peroxisomal enzymes, with the exception of D-amino acid oxidase, which is known to catalyse the oxidative deamination of D-amino acids [28]. There is no obvious metabolic link between RP2p and D-amino acid oxidase, which makes it difficult to see what role RP2p and/or D-amino acid oxidase play in androgen-regulated metabolism.
The highest levels of RP2 mRNA have been found in mouse kidney, but RP2 mRNA is also observed in heart, brain, muscle, testes, submaxillary gland and, in lower amounts, in liver [19]. Even under testosterone-induced conditions, the RP2 mRNA levels in liver are still lower compared with those in uninduced kidney. Nudt7, however, another peroxisomal nudix hydrolase with CoA diphosphatase activity, is primarily expressed in liver and, to a lesser extent, in kidney [19]. So far, the substrate specificity of this enzyme has been tested only for a small number of CoA esters, including acetyl-CoA and succinyl-CoA, and it is unclear whether Nudt7 is capable of using long-chain CoA esters as substrates. Therefore it cannot be concluded that RP2p and Nudt7 share the same function in peroxisomal metabolism.
The results of the present study show that the products of peroxisomal β-oxidation are the primary substrates for RP2p. This suggests that RP2p is involved in the catabolism of the peroxisomal β-oxidation products. To date, two peroxisomal nudix hydrolases with activity towards CoA and CoA esters, Nudt7 and RP2p, have been identified. This could indicate that the formation of acyl-phosphopantetheine esters from acyl-CoA esters is not a minor pathway. Recently, Antonenkov et al. [29] postulated that peroxisomal nudix hydrolases could play a role in the export of metabolites from the peroxisomal matrix. The authors demonstrated that the peroxisomal membrane is permeable for small molecules and cleavage of large bulky molecules like CoA into smaller molecules could make them more suitable for export [29]. Our results suggest that this export pathway is accessible not only to free CoA, but also possibly to all products of the peroxisomal β-oxidation. In addition, free CoA plays an important role in the peroxisomal β-oxidation, as it is essential for the activity of 3-oxo-acyl-CoA thiolase. Depletion of this substrate from the peroxisomal lumen would render the enzyme inactive and subsequently halt peroxisomal β-oxidation. The relatively low affinity of RP2p for CoA supports this. We believe that RP2p may well regulate free CoA levels by preventing CoA accumulation in peroxisomes.
Until now, the presence and possible functions of acyl-phosphopantetheine esters have never been studied and therefore it is still unclear whether these esters are synthesized only to accommodate their export from the peroxisomal matrix to the cytosol or whether they also serve other, as yet unknown, functions. It will therefore be important to elucidate the role of these esters in human metabolism.
Acknowledgments
We gratefully acknowledge Dr Hans Waterham and Dr Stephan Kemp for critically reading the manuscript. We thank Lida Zoetekouw and Simone Denis for expert technical assistance and H. L. Dekker for expert Q-TOF MS/MS analysis. This work was supported by the FP6 European Union Project ‘Peroxisomes’ (LSHG-CT-2004-512018).
References
- 1.Wanders R. J., Tager J. M. Lipid metabolism in peroxisomes in relation to human disease. Mol. Aspects Med. 1998;19:69–154. doi: 10.1016/s0098-2997(98)00003-x. [DOI] [PubMed] [Google Scholar]
- 2.Mannaerts G. P., Van Veldhoven P. P. Metabolic pathways in mammalian peroxisomes. Biochimie. 1993;75:147–158. doi: 10.1016/0300-9084(93)90072-z. [DOI] [PubMed] [Google Scholar]
- 3.Gould S. J., Keller G. A., Hosken N., Wilkinson J., Subramani S. A conserved tripeptide sorts proteins to peroxisomes. J. Cell Biol. 1989;108:1657–1664. doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swinkels B. W., Gould S. J., Bodnar A. G., Rachubinski R. A., Subramani S. A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J. 1991;10:3255–3262. doi: 10.1002/j.1460-2075.1991.tb04889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wanders R. J. Metabolic and molecular basis of peroxisomal disorders: a review. Am. J. Med. Genet. 2004;126:355–375. doi: 10.1002/ajmg.a.20661. [DOI] [PubMed] [Google Scholar]
- 6.Weller S., Gould S. J., Valle D. Peroxisome biogenesis disorders. Annu. Rev. Genomics Hum. Genet. 2003;4:165–211. doi: 10.1146/annurev.genom.4.070802.110424. [DOI] [PubMed] [Google Scholar]
- 7.Wanders R. J., van Roermund C. W., Schor D. S., ten Brink H. J., Jakobs C. 2-Hydroxyphytanic acid oxidase activity in rat and human liver and its deficiency in the Zellweger syndrome. Biochim. Biophys. Acta. 1994;1227:177–182. doi: 10.1016/0925-4439(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 8.Wanders R. J., Kos M., Roest B., Meijer A. J., Schrakamp G., Heymans H. S., Tegelaers W. H., van den Bosch H., Schutgens R. B., Tager J. M. Activity of peroxisomal enzymes and intracellular distribution of catalase in Zellweger syndrome. Biochem. Biophys. Res. Commun. 1984;123:1054–1061. doi: 10.1016/s0006-291x(84)80240-5. [DOI] [PubMed] [Google Scholar]
- 9.Wanders R. J., van Roermund C. W., de Vries C. T., van den Bosch H., Schrakamp G., Tager J. M., Schram A. W., Schutgens R. B. Peroxisomal β-oxidation of palmitoyl-CoA in human liver homogenates and its deficiency in the cerebro-hepato-renal (Zellweger) syndrome. Clin. Chim. Acta. 1986;159:1–10. doi: 10.1016/0009-8981(86)90160-9. [DOI] [PubMed] [Google Scholar]
- 10.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 11.Schägger H., von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 12.Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 13.Rabilloud T., Carpentier G., Tarroux P. Improvement and simplification of low-background silver staining of proteins by using sodium dithionite. Electrophoresis. 1988;9:288–291. doi: 10.1002/elps.1150090608. [DOI] [PubMed] [Google Scholar]
- 14.Shevchenko A., Wilm M., Vorm O., Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 15.IJlst L., Wanders R. J., Ushikubo S., Kamijo T., Hashimoto T. Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of the major disease-causing mutation in the α-subunit of the mitochondrial trifunctional protein. Biochim. Biophys. Acta. 1994;1215:347–350. doi: 10.1016/0005-2760(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 16.Miyazawa S., Hayashi H., Hijikata M., Ishii N., Furuta S., Kagamiyama H., Osumi T., Hashimoto T. Complete nucleotide sequence of cDNA and predicted amino acid sequence of rat acyl-CoA oxidase. J. Biol. Chem. 1987;262:8131–8137. [PubMed] [Google Scholar]
- 17.Deisseroth A., Dounce A. L. Catalase: physical and chemical properties, mechanism of catalysis, and physiological role. Physiol. Rev. 1970;50:319–375. doi: 10.1152/physrev.1970.50.3.319. [DOI] [PubMed] [Google Scholar]
- 18.Neuhoff V., Arold N., Taube D., Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988;9:255–262. doi: 10.1002/elps.1150090603. [DOI] [PubMed] [Google Scholar]
- 19.Gasmi L., McLennan A. G. The mouse Nudt7 gene encodes a peroxisomal nudix hydrolase specific for coenzyme A and its derivatives. Biochem. J. 2001;357:33–38. doi: 10.1042/0264-6021:3570033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cartwright J. L., Gasmi L., Spiller D. G., McLennan A. G. The Saccharomyces cerevisiae PCD1 gene encodes a peroxisomal nudix hydrolase active toward coenzyme A and its derivatives. J. Biol. Chem. 2000;275:32925–32930. doi: 10.1074/jbc.M005015200. [DOI] [PubMed] [Google Scholar]
- 21.AbdelRaheim S. R., McLennan A. G. The Caenorhabditis elegans Y87G2A.14 Nudix hydrolase is a peroxisomal coenzyme A diphosphatase. MBC Biochem. 2002;3:5. doi: 10.1186/1471-2091-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger F. G., Gross K. W., Watson G. Isolation and characterization of a DNA sequence complementary to an androgen-inducible messenger RNA from mouse kidney. J. Biol. Chem. 1981;256:7006–7013. [PubMed] [Google Scholar]
- 23.Snider L. D., King D., Lingrel J. B. Androgen regulation of MAK mRNAs in mouse kidney. J. Biol. Chem. 1985;260:9884–9893. [PubMed] [Google Scholar]
- 24.King D., Sun Y. H., Lingrel J. B. Amino acid sequence of the testosterone-regulated mouse kidney RP2 protein deduced from its complementary DNA sequence. Nucleic Acids Res. 1986;14:5159–5170. doi: 10.1093/nar/14.13.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rheaume C., Barbour K. W., Tseng-Crank J., Berger F. G. Molecular genetics of androgen-inducible RP2 gene transcription in the mouse kidney. Mol. Cell. Biol. 1989;9:477–483. doi: 10.1128/mcb.9.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaudhuri A., Barbour K. W., Berger F. G. Evolution of messenger RNA structure and regulation in the genus Mus: the androgen-inducible RP2 mRNAs. Mol. Biol. Evol. 1991;8:641–653. doi: 10.1093/oxfordjournals.molbev.a040675. [DOI] [PubMed] [Google Scholar]
- 27.Singh N., Barbour K. W., Berger F. G. Evolution of transcriptional regulatory elements within the promoter of a mammalian gene. Mol. Biol. Evol. 1998;15:312–325. doi: 10.1093/oxfordjournals.molbev.a025928. [DOI] [PubMed] [Google Scholar]
- 28.Endahl B. R., Kochakian C. D. Role of castration and androgens in D-amino acid oxidase activity of tissues. Am. J. Physiol. 1956;185:250–256. doi: 10.1152/ajplegacy.1956.185.2.250. [DOI] [PubMed] [Google Scholar]
- 29.Antonenkov V. D., Sormunen R. T., Hiltunen J. K. The rat liver peroxisomal membrane forms a permeability barrier for cofactors but not for small metabolites in vitro. J. Cell Sci. 2004;117:5633–5642. doi: 10.1242/jcs.01485. [DOI] [PubMed] [Google Scholar]