

Abstract
Enzyme structure and dynamics may play a main role in substrate binding and the subsequent steps in the CYP (cytochrome P450) catalytic cycle. In the present study, changes in the structure of human CYP2D6 upon binding of the substrate are studied using steady-state and time-resolved fluorescence methods, focusing not only on the emission of the tryptophan residues, but also on emission of the substrate. As a substrate, MAMC [7-methoxy-4-(aminomethyl)-coumarin] was selected, a compound exhibiting native fluorescence. As well as the wild-type, the W128F (Trp128→Phe) mutant of CYP2D6 was studied. After binding, a variety of energy transfer possibilities exist, and molecular dynamics simulations were performed to calculate distances and relative orientations of donors and acceptors. Energy transfer from Trp128 to haem appeared to be important; its emission was related to the shortest of the three average tryptophan fluorescence lifetimes observed for CYP2D6. MAMC to haem energy transfer was very efficient as well: when bound in the active site, the emission of MAMC was fully quenched. Steady-state anisotropy revealed that besides the MAMC in the active site, another 2.4% of MAMC was bound outside of the active site to wild-type CYP2D6. The tryptophan residues in CYP2D6 appeared to be less accessible for the external quenchers iodide and acrylamide in presence of MAMC, indicating a tightening of the enzyme structure upon substrate binding. However, the changes in the overall enzyme structure were not very large, since the emission characteristics of the enzyme were not very different in the presence of MAMC.
Keywords: cytochrome P450, energy transfer, enzyme structure, substrate binding, time-resolved fluorescence
Abbreviations: CYP, cytochrome P450; DAS, decay-associated spectra; FRET, Förster resonance energy transfer; HAMC, 7-hydroxy-4-(aminomethyl)-coumarin; MAMC, 7-methoxy-4-(aminomethyl)-coumarin; MD, molecular dynamics; Ni-NTA, Ni2+-nitrilotriacetate; R.M.S.D., root-mean-square deviation; SASA, solvent-accessible surface area; TCSPC, time-correlated single-photon counting
INTRODUCTION
Among the large variety of CYPs (cytochrome P450s), one of the most important phase I drug-metabolizing enzymes in humans is CYP2D6. Although the hepatic expression levels are low (approx. 2% of the total amount of CYP) it is involved in the oxidative metabolism of approx. 30% of the currently marketed drugs, including β-blockers, neuroleptics, antidepressants and antiarrhythmics [1,2]. In addition, this enzyme is clinically relevant because of the large interindividual differences that exist in CYP2D6 activity, due to gene multiplicity and polymorphisms [3]. The scientific challenge is to rationalize why and how substrates are metabolized and to predict the metabolism of newly found or synthesized (drug-like) compounds. For this purpose, detailed structural information on CYP2D6 is essential, but so far no crystal structure of CYP2D6 has been resolved and any structural information on this enzyme still depends on homology modelling.
The present study focuses on some structural features of the enzyme, in particular the conformational changes induced by binding of the substrate. Since homology models as such cannot be used for this purpose, we made use of MD (molecular dynamics) and docking studies, as has been successfully employed for other CYPs [4]. Furthermore, both time-resolved and steady-state fluorescence spectroscopy were used, as has been applied before to CYP101 by Prasad et al. [5], and Prasad and Mitra [6]. In these studies on the dynamics of substrate binding to CYP101, the attention was focused on the emission of the five available tryptophan residues. Tryptophans are good endogenous probes, since both lifetimes and emission wavelengths may be influenced when the conformation of (parts of) the protein changes [7–9]. Analysis of the tryptophan fluorescence lifetime data for CYP101 indicated substrate-dependent dynamic behaviour of Trp42 in the wild-type and Y96A mutant enzymes. CYP2D6 contains seven tryptophan residues. Based on the homology model, the fluorescence of Trp128 in CYP2D6 is expected to be sensitive to substrate binding as well. Although Trp42 in CYP101 is located close to the substrate-access channel and Trp128 in CYP2D6 is not, both are close to the haem group. Moreover, Trp128 is thought to be bound to the propionate of pyrrole ring D of the porphyrin via hydrogen bonds, as are the analogous tryptophan residues in the crystal structures of mammalian CYPs 2C5, 2C9, 2B4 and 3A4 (PDB codes 1N6B, 1OG2, 1PO5 and 1TQN respectively) [10–13].
A useful feature of tryptophan fluorescence in haem proteins is resonance energy transfer to the haem prophyrin ring [14,15], which generally leads to a low tryptophan fluorescence quantum yield [16]. In the presence of substrates, other possible energy transfer pathways need consideration as well, i.e. from tryptophan to substrate and from substrate to haem. The CYP2D6 substrate MAMC [7-methoxy-4-(aminomethyl)-coumarin] is a fluorescing moderate affinity substrate [17,18]. Its long-wavelength absorption band overlaps with the tryptophan emission, so when bound to CYP2D6, energy transfer is expected. Furthermore, it can also be excited selectively, allowing for investigation of its properties even when bound to CYP2D6.
In the present study, the binding of MAMC to CYP2D6 was studied by steady-state and time-resolved fluorescence experiments on the tryptophan residues and MAMC, for both wild-type and W128F (Trp128→Phe) mutant CYP2D6. Quenching experiments with acrylamide and potassium iodide provided information regarding the accessibility of the tryptophan residues and the dynamics of the protein structure [19–21]. Experimental results were compared with MD simulations that were performed on a homology model [22]. The relative locations and orientations of the tryptophan residues, the haem group and MAMC during the simulations were related to the FRET (Förster resonance energy transfer) efficiency between the individual pairs of FRET partners and were used to support the interpretation of the intricate fluorescence signals.
MATERIALS AND METHODS
Materials
Professor Dr Ingelman-Sundberg (Division of Molecular Toxicology, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden) kindly provided the pSP19T7LT plasmid which contains the cDNA of human CYP2D6, His6-tagged at the C-terminus and linked (by a short linker region) to the cDNA of human NADPH–cytochrome P450 reductase. MAMC and HAMC [7-hydroxy-4-(aminomethyl)-coumarin] were synthesized as described in [17]. Emulgen 911 was purchased from KAO Chemicals (Tokyo, Japan). Ni-NTA (Ni2+-nitrilotriacetate)–agarose was from Qiagen. All other chemicals were of analytical grade and were obtained from standard suppliers.
Mutagenesis and expression
The W128F mutation was introduced into pSP19T7LT using the QuikChange XL Site-Directed Mutagenesis kit (Stratagene). The sequences of the forward and reverse oligonucleotides respectively, with the mutated residue in italic, were as follows: 5′-CTATGGGCCCGCGTTTCGCGAGCAGAGGC-3′ and 5′-GATACCCGGGCGCAAAGCGCTCGTCTCCG-3′. After mutagenesis, the presence of the desired W128F mutation was confirmed by DNA sequencing. Both the W128F mutant and the wild-type pSP19T7LT plasmids were transformed into Escherichia coli, strain JM109. Expression and membrane isolation was carried out as described in [23].
Enzyme purification and sample preparation
Enzymes were purified as described previously [22]. In brief, membranes were resuspended in 50 mM potassium phosphate buffer, pH 7.4, with 10% (v/v) glycerol. Enzymes were solubilized by stirring in 50 mM potassium phosphate buffer, pH 7.4, with 10% (v/v) glycerol supplemented with 0.5% Emulgen 911 for 2 h at 4°C. Insoluble parts were removed by centrifugation (60 min, 120000 g at 4°C). The supernatant was incubated, rocking gently, with Ni-NTA agarose (Qiagen) for 60 min at 4°C. The column material was filtered out and washed with 10 vol. of 50 mM potassium phosphate buffer, pH 7.4, with 10% (v/v) glycerol, containing 2 mM histidine. CYP2D6 was eluted with 0.2 M histidine. After overnight dialysis in 50 mM potassium phosphate buffer, pH 7.4, with 10% (v/v) glycerol, the sample was concentrated on a Vivaspin 20 filtration tube [10 kDa MWCO (molecular weight cut-off) PES (polyethersulphone); Sartorius]. The amount of CYP was quantified by taking CO-difference absorbance spectra of the enzymes, using ϵ=91 cm−1·mM−1 at 450 nm [24]. Samples of 1 μM purified enzyme were used for fluorescence measurements. The MAMC concentration was 2 μM; at this concentration the emission intensities of CYP2D6 and MAMC were almost equal, and one signal did not overwhelm the other. For the quenching experiments, potassium iodide was added to obtain final concentrations in the range 0–0.5 M. To avoid formation of I2, 10 mM Na2S2O3 was added, and the ionic strength was kept constant by addition of KCl.
Enzyme activity
MAMC O-demethylation reactions were carried out in duplicate in a 96-well microtitre plate, in a total volume of 200 μl as described previously [23]. The reaction mixture consisted of 50 mM potassium phosphate buffer, pH 7.4, containing 5 mM MgCl2, 0–200 μM MAMC and E. coli membranes (40 nM wild-type or W128F mutant CYP2D6). The reactions were initiated by addition of an NADPH-regenerating system, resulting in final concentrations of 0.1 mM NADPH, 0.3 mM glucose 6-phosphate and 0.4 unit/ml glucose-6-phosphate dehydrogenase. The reaction was monitored for 30 min at 37°C on a Victor2 1420 multilabel counter (Wallac Oy, Turku, Finland) (maximum of excitation spectrum, λex=405 nm and maximum of emission spectrum, λem=460 nm). The metabolite of MAMC, i.e. HAMC, was quantified using the synthetic reference compound.
Fluorescence instrumentation
Time-resolved fluorescence experiments were performed using a laser system combined with time-resolved photon-counting detection. The laser system used was a Mira 900-P laser (Coherent, Santa Clara, CA, U.S.A.), emitting 3 ps pulses at a repetition rate of 76 MHz, pumped by a Verdi-8 diode laser (Coherent). The Mira 900-P is a mode-locked titanium–sapphire laser, tuneable from approx. 700 to 1000 nm. The laser emission was led through a pulse picker (Coherent), which reduced the repetition rate to 4.75 MHz in order to avoid double excitation of molecules. The output wavelength of 290 nm was generated by means of a frequency tripler (Coherent). This light was used to excite the protein sample in a cell with 1 cm pathlength (type 104F, Hellma GmbH & Co. KG, Müllheim, Germany). For the TCSPC (time-correlated single-photon counting), a SPC-630 (Becker & Hickl GmbH, Berlin, Germany) system with a time-resolution of approx. 15 ps was used. A laser pulse focused on a photodiode provided the synchronization signal. Fluorescence emission was collected at 90°, dispersed by a monochromator (TVC JarrellAsh Monospec 18, Grand Junction, CO, U.S.A.) and detected by a photomultiplier tube. Decays were recorded at wavelengths between 330 and 440 nm in 10 nm steps. Quenching experiments were performed using TCSPC detection at two wavelengths, 340 and 430 nm. Steady-state fluorescence emission and excitation spectra were recorded on an LS-50B instrument (PerkinElmer). Steady-state anisotropy measurements were conducted using a Fluorolog-tau2 system (Horiba; Jobin Yvon, Edison, NJ, U.S.A.).
Data analysis
Fluorescence decay curves were analysed using a fitting procedure based on the Levenberg–Marquardt algorithm. This procedure uses the instrument response signal (obtained by recording scattered laser light) for deconvolution of the recorded decays and determination of lifetimes and wavelength-dependent amplitudes. Each decay curve is fitted by mono- or multi-exponential curves, where I(t) is the intensity as a function of time t, Ai is the amplitude, and τi is the lifetime of fitted decays (eqn 1):
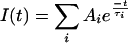 |
(1) |
The quality of the fit (‘goodness of fit’) was assessed on the basis of χ2, the covariance matrix and the distribution of residuals. The set of time constants was kept as low as possible; normally not more than three constants were needed to obtain a fit of sufficient quality. For measurements at a series of detection wavelengths, a global fit procedure was used, based on the method described above. Lifetimes are assumed to be constant for the decays included, while the amplitudes were varied. This assumption is correct for proteins in many cases [25].
DAS (decay-associated spectra) provide information on the fluorescence emission per lifetime component. DAS are constructed by distributing the total intensity per decay curve over the lifetimes that make up the total intensity according to their ratio (amplitudes are obtained by global fit). The relative fluorescence intensity of lifetime component τj at wavelength λ, with amplitude A, can be expressed by the following equation, where Iτi(λ) is the intensity of the spectral component with lifetime τi at wavelength λ:
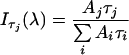 |
(2) |
FRET
FRET is the radiationless transfer of excitation energy from a donor to an acceptor chromophore, controlled by long-range dipole–dipole interactions [26,27]. It can be used for estimation of intermolecular distances, because the efficiency of energy transfer (ΦT) is dependent on the distance r between a donor with lifetime τD, and an acceptor, as expressed in equation 3:
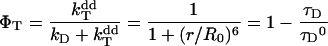 |
(3) |
where kTdd is the energy transfer rate, defined as,
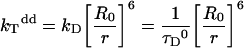 |
kD is the emission rate constant of the donor (the inverse of donor lifetime in the absence of energy transfer τD0), τD is the donor lifetime in the presence of energy transfer and r is the distance between donor and acceptor; R0 is the Förster radius, the distance at which transfer and spontaneous decay of the donor are equally probable. R0 can be determined experimentally and is given by eqn 4:
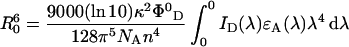 |
(4) |
where κ2 is the orientation factor of the donor and acceptor transition dipole moments, Φ0D is the fluorescence quantum yield of the donor in absence of energy transfer, NA is Avogadro's number, n is the average refractive index of the medium, and
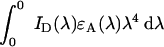 |
is the overlap integral of the (normalized) donor emission ID(λ) and acceptor absorption spectra ϵA(λ).
The distance dependence of energy transfer makes FRET useful to study structural changes. A complicating feature is the dependency of the FRET efficiency on the orientation of donor and acceptor transition dipoles via κ2. This orientation factor varies between 0 and 4, and can be calculated from atomic coordinates. Because of the dynamics of the protein, the exact orientations of the dipole transition moments are not constant, and values of κ2 (and also the distance r) will fluctuate in time.
Anisotropy
Steady-state anisotropy experiments were performed on a Fluorolog FL3-11-Tau3 system (Horiba Jobin-Yvon). Anisotropy (r) is defined as
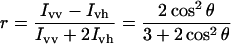 |
(5) |
in which Ivv and Ivh are the intensities of the vertically and horizontally polarized components of the fluorophore emission respectively, and θ is the angle between absorption and emission transition dipole moment. Eqn 5 can be adapted to an equation giving the ratio of concentrations of unbound, [S], to bound, [PS], substrate (see the Supplementary material at http://www.BiochemJ.org/bj/393/bj3930635add.htm for derivation), where θ1 is the angle between emission and absorption transition dipole moment for the substrate:
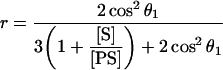 |
(6) |
with the (measured) anisotropy for the unbound substrate being 0. A solution of 50 μM MAMC in glycerol was used to determine the limiting anisotropy r0.
Computer simulations
The substrate MAMC was docked into the active site of the homology model with the programs Autodock and GOLD [28,29]. Five MD simulations were performed in explicit simple point charge water [30]: four with different initial orientations of the substrate and one with the wild-type CYP2D6 protein without substrate. Interatomic interactions were calculated from the GROMOS™ force field parameter set 43A2 [31–33]. All five simulations were performed for 10 ns (at picosecond frequency) using the GROMACS simulation package [34]. Simulation parameter settings were chosen as described previously [23].
The value of κ2/r6 for individual pairs of fluorescent groups is directly proportional to the FRET efficiency of the pair (eqns 3 and 4). In order to relate the structural dynamics to the FRET efficiency, dipole transition moments were chosen as follows. For tryptophan the transition moment 1La lies in the plane of the indole ring at an angle of −46° with respect to the indole molecular frame [35]. Only the 1La transition dipole moment of tryptophan was included, since it is the only emitting state (internal conversion from 1Lb is on a much faster timescale than fluorescence). For the haem moiety, two degenerate dipole transition moments in the plane of the haem were considered, one pointing from N1 to N3 and another perpendicular to this [15]. For the substrate MAMC, a transition dipole moment was estimated in analogy to previous studies on coumarin-type compounds point from atom 3 to atom 7 (Figure 1) [36]. κ2/r6 was calculated for every pair of tryptophan–haem, tryptophan–MAMC and MAMC–haem transition moments and averaged over the simulations.
Figure 1. Structures of MAMC (upper left), tryptophan (upper right) and the haem group (lower middle) in CYP2D6.

 indicates the transition dipole moment; for tryptophan and haem, two transitions are possible, as indicated.
indicates the transition dipole moment; for tryptophan and haem, two transitions are possible, as indicated.
Solvent-accessible surface areas of all tryptophan residues have been calculated from the MD simulation data according to the algorithm of Lee and Richards [37] for all tryptophan residues using a probe size of 0.14 nm.
RESULTS AND DISCUSSION
Expression, purification and activity of mutant W128F
The expression levels of the W128F mutant CYP2D6 were approx. 200 nM in an average culture, which is equal to those of the wild-type enzyme. P420 (inactive form of P450, determined as a peak at 420 nm in CO-difference spectrum) was present in the crude membranes, but was lost during the purification of the W128F mutant. The rate of MAMC O-demethylation by the W128F mutant is approx. 2-fold lower than that by wild-type CYP2D6, with a Vmax of 3.5±0.7 compared with 6.7±0.1 min−1 for wild-type, whereas the Km remained constant at 29.8±0.1 μM compared with 30±1 μM for mutant and wild-type respectively. From these similar kinetic parameters we see that Trp128 does not appear to be crucial for MAMC O-demethylating activity, nor for CYP2D6 stability. This is in agreement with MD simulations that show that the residue at position 128 is not the only residue forming hydrogen bonds to the propionate of pyrrole ring D of the porphyrin; the arginine residue at position 101 forms hydrogen bonds to the same atoms.
Fluorescence properties of CYP2D6 and MAMC
The fluorescence emission spectra of wild-type and W128F CYP2D6 in the presence and absence of MAMC, as well as for MAMC alone, are depicted in Figure 2. The corresponding excitation and emission maxima together with the fluorescence lifetimes are listed in Table 1.
Figure 2. Emission spectra of 1 μM wild-type CYP2D6, 1 μM W128F mutant, 2 μM MAMC (lighter traces) and mixtures of wild-type and W128F mutant CYP2D6 with MAMC (darker traces).

Intensity was measured in arbitrary units (a.u.).
Table 1. Fluorescence maxima (nm), relative intensities (peak area, arbitrary units) and emission lifetimes (ns) for wild-type (WT) and W128F mutant CYP2D6 and MAMC.
For the lifetimes, the relative contributions to the total signal are indicated in parentheses (%). Iem is the emission intensity. Results are means±S.D.
| λex (nm) | λem (nm) | Iem (area) | τ1 (ns) | τ2 (ns) | τ3 (ns) | |
|---|---|---|---|---|---|---|
| WT | 278 | 336 | 1.38±0.003 | 0.13±0.05 (80%) | 1.06±0.03 (17%) | 3.81±0.08 (3%) |
| WT+MAMC | 278 | 335 | 1.28±0.006 | 0.12±0.06 (83%) | 1.30±0.27 (14%) | 4.00±0.17 (3%) |
| W128F | 280 | 338 | 1.07±0.002 | 0.07±0.02 (84%) | 1.09±0.06 (13%) | 4.17±0.06 (3%) |
| W128F+MAMC | 280 | 333 | 0.99±0.004 | 0.14±0.04 (81%) | 1.40±0.24 (16%) | 3.96±0.30 (3%) |
| MAMC | 328 | 406 | 0.56±0.018 | 0.75±0.12 (52%) | 2.53±0.06 (48%) | |
| MAMC+WT | 328 | 400 | 0.36±0.006 | 0.68±0.07 (56%) | 2.62±0.11 (44%) | |
| MAMC+W128F | 327 | 400 | 0.36±0.006 | 0.71±0.16 (50%) | 2.77±0.17 (50%) |
The emission maximum at 335–340 nm indicates moderately buried tryptophan residues [38]. The emission spectrum of the W128F mutant is 2 nm red-shifted compared with wild-type CYP2D6 and is approx. 22% lower in intensity. In the presence of MAMC, excitation and emission maxima of wild-type and W128F mutant CYP2D6 do not shift, indicating that no large conformational change of the enzyme is needed to accommodate the substrate, but their intensities decrease by approx. 6%. A notable decrease in the MAMC emission intensity is seen in the presence of the enzyme, and this is the same for the mutant W128F (35%) as for the wild-type CYP2D6 (35%), indicative of some form of energy transfer or quenching. Also, in the presence of both CYP2D6 enzymes, a blue shift in MAMC emission of 6 nm is observed. This shift is most likely to be the result of a smaller Stokes shift (the excitation spectrum does not change) and to indicate a less polar environment for MAMC in the presence of CYP2D6 than for pure MAMC in aqueous solution, which is, of course, expected.
Fitting of the fluorescence intensity decay curves yielded three tryptophan lifetimes for wild-type and W128F mutant CYP2D6 and two for MAMC, as shown in Table 1. The origin of this double exponential decay is not trivial; it may be caused by a charge transfer state, similar to that described for coumarin-type molecules [26,39]. The fluorescence lifetimes of both wild-type and W128F mutant CYP2D6 do not change dramatically upon addition of MAMC.
The tryptophan DAS of the wild-type and W128F mutant CYP2D6 are shown in Figure 3. For the wild-type enzyme, all three components have similar relative intensities, and their emission maxima are close as well. Interestingly, the spectrum associated with τ1 of the W128F mutant has a 3-fold lower intensity than that of τ2 and τ3. This suggests that Trp128 contributes predominantly to the shortest, 130 ps, lifetime component of the overall tryptophan emission. It was expected that this tryptophan residue would have such a short fluorescence lifetime, since its location at position 128 is close to the haem and its emission will therefore be quenched (by a FRET mechanism) more strongly than the other residues. On the other hand, the remaining signal indicates that the shortest lifetime is not solely due to Trp128 and in fact is even shorter than in wild-type CYP2D6. The shorter τ1 for W128F compared with wild-type CYP2D6 was unexpected: the tryptophan closest to haem is removed, so, at first sight, the residual lifetime should be longer. However, in view of the large number of possible relaxation times of the six tryptophan residues in W128F, this seemingly shortened lifetime may be the result of a shift in the distribution of all tryptophan lifetimes over the three lifetimes obtained from the fit of the data. In addition, lifetimes shorter than 100 ps are not uncommon in proteins, even in cases when no haem or other energy-transfer mechanism is present [40,41], and may be caused by close-lying quenching amino acids such as tyrosine, cysteine or histidine [42].
Figure 3. Intensity was measured in arbitrary units (a.u.).

Decay-associated spectra of wild-type CYP2D6 (A) and W128F (B) and the corresponding lifetimes (△, total spectrum; ◇, τ1; □, τ2; ×, τ3).
The decay times of MAMC do not change significantly when either wild-type CYP2D6 or W128F mutant is present. Although a change would be expected due to interactions with the protein, it seems that these interactions do not influence the lifetimes or that the changes are too small to be observed in the overwhelming emission of the unbound MAMC. A more feasible explanation may be that the fluorescence quantum yield of bound MAMC is negligible due to quenching by the close-lying haem. To assess the latter explanation, the FRET ‘network’ in the CYP2D6–MAMC system was evaluated and is discussed below.
Quenching experiments
The accessibility of the tryptophan residues was studied using acrylamide and iodide as dynamic quenchers. In both cases, quenching only takes place if the quencher and the fluorescing tryptophan residue are in physical contact; long-range quenching does not occur. Acrylamide is a small uncharged molecule that can diffuse into the protein; its quenching behaviour provides information on the overall accessibility of the tryptophan residues in the protein [19]. Iodide is a charged ion, which cannot enter the protein interior and will quench only exposed tryptophan residues on the periphery of the protein [21]. The fluorescence intensity quenching experiments using acrylamide yielded linear Stern–Volmer curves for both wild-type and W128F CYP2D6. Apparently, static quenching in ground-state tryptophan–acrylamide complexes is negligible. Using iodide as a quencher, slightly downwardly curved Stern–Volmer curves were obtained, indicating that some tryptophan residues are inaccessible to iodide [19]. For MAMC emission, upward curving Stern–Volmer graphs are observed in the case of iodide quenching, indicating a static component. Analysis of the data using a modified version of the Stern-Volmer equation allows for the separation of the static and dynamic parts of the observed quenching. This showed that quenching in this case is not caused by ground-state complexes, but rather by close proximity of quencher and fluorophore. This results in almost 100% quenching probability [20], presumably caused by iodide being attracted to the positive charge of the substrate.
The quenching constants for all compounds are collected in Table 2. The acrylamide quenching constants for tryptophan are similar for the samples considered, so no large differences in permeability exist between wild-type and W128F mutant CYP2D6. In both cases, the quenching rates are somewhat smaller in the presence of MAMC (12.5 and 13.0 compared with 9.7 and 10.5 M−1·s−1), indicating a slightly more compact structure. The structure of CYP2D6 is less compact than CYP101, which has a 3-fold lower quenching constant (4.5 and 1.45 M−1 respectively) [6]. The accessible fraction of tryptophan emission for quenching by acrylamide is, as expected, around 100%. The smaller accessible fraction of wild-type CYP2D6 in the presence of MAMC, some 20% lower, might be caused by MAMC blocking the pathway for acrylamide in the substrate-access channel, in line with the somewhat decreased quenching constant found in this case. A similar effect was not seen for the W128F mutant, which indicates that indeed the access channel to the active site is blocked: apparently it is Trp128, which lies in the active site, that cannot be reached any more by acrylamide when MAMC is present.
Table 2. Fluorescence quenching parameters of wild-type (WT) CYP2D6, W128F mutant CYP2D6, MAMC alone and MAMC in the presence of the enzymes, obtained from (linear) Stern–Volmer plots.
Ksv, quenching constant; kq, quenching rate; τav, average fluorescence lifetime; fa, accessible fraction.
| Acrylamide | Iodide | |||||||
|---|---|---|---|---|---|---|---|---|
| KSV (M−1) | kq (109×M−1·s−1) | τav (ns) | fa | kq (M−1) | (109×M−1·s−1) | τav (ns) | fa | |
| WT | 4.5 | 12.5 | 0.36 | 1.1 | 0.7 | 1.8 | 0.40 | 0.3 |
| WT+MAMC | 3.8 | 9.7 | 0.39 | 0.8 | 0.6 | 1.5 | 0.40 | 0.3 |
| W128F | 3.9 | 13.0 | 0.30 | 1.1 | 0.9 | 3.0 | 0.30 | 0.4 |
| W128F+MAMC | 4.1 | 10.5 | 0.39 | 1.0 | 0.8 | 2.1 | 0.39 | 0.3 |
| MAMC | 2.7 | 1.5 | 1.76 | 0.9 | 14.1 | 17.2 | 0.82 | 1.1 |
| MAMC+WT | 3.1 | 1.9 | 1.55 | 0.8 | 15.2 | 10.3 | 1.47 | 1.1 |
| MAMC+W128F | 2.9 | 1.8 | 1.59 | 0.9 | 18.4 | 11.6 | 1.59 | 1.1 |
As expected, the tryptophan residues in both W128F and wild-type CYP2D6 are less easily accessible to iodide than to acrylamide, and the fraction of the total emission accessible to quenching is accordingly smaller (30–40%). This is somewhat higher than for, for example, the 16% accessible for substrate-free CYP101 [6]. In addition the quenching constants for CYP2D6 are approx. 5-fold larger, indicating a higher degree of exposure of the tryptophan residues in CYP2D6 than in CYP101. The quenching constant of W128F is slightly higher than that of wild-type CYP2D6 (0.9 compared with 0.7 M−1). Its structure may be different, and/or the remaining tryptophans may be more exposed, resulting in a somewhat higher accessible fraction (0.4 instead of 0.3). In the presence of MAMC, the quenching rate is slightly lower, as was the case with acrylamide. This supports the explanation that the substrate-bound enzyme has a less accessible structure.
Calculation of the SASAs (solvent-accessible surface areas) revealed that Trp152, Trp262 and especially Trp75 were accessible to bulk solvent, while Trp128, Trp316 and Trp409 were not found to interact strongly with water. Consequently, we expect the iodide quenching to be mostly acting on Trp75. Therefore it is not surprising that the accessible fraction of emission of W128F found with the quenching experiments is not much different from the wild-type. In the presence of the substrate MAMC, the SASAs do not change much, confirming the fluorescence quenching results. Also in that case, Trp75 is the most accessible residue, and the accessibilities of the other residues are maximally 10%. As acrylamide is much smaller and may penetrate the protein, it is likely to interact with all tryptophans and calculations of the SASAs will not yield useful results for this probe.
FRET efficiencies
In CYP2D6, three chromophoric groups are present: the porphyrin, the tryptophan residues and the coumarin, which are all absorbing, and the coumarin and tryptophan residues which are also emitting, in generally the same spectral range. The spectral overlap may give rise to energy transfer pathways, in particular of the Förster type. The location and orientation of MAMC in the active site of CYP2D6, corresponding to one of the initial structures of the simulations, is illustrated in Figure 4. Three FRET pathways are possible when MAMC is in close proximity to CYP2D6: (i) from the tryptophan residues to haem, a well-known and efficient energy transfer route in haem proteins [14], (ii) from the tryptophan residues to MAMC, and (iii) from MAMC to the haem moiety.
Figure 4. Homology model of CYP2D6 based on rabbit CYP2C5, indicating the positions of the six tryptophan residues (red; purple for Trp128), the haem group (green) and the substrate MAMC (yellow).

Trp28 is not present in the homology model.
Over the course of the 10 ns MD simulations, the overall structure of the protein was well maintained. Atom-positional R.M.S.D.s (root-mean-square deviations) from the initial homology model levelled off to approx. 0.3–0.4 nm; important secondary-structure elements and hydrogen bonds were maintained throughout the simulations. The molecular dynamics simulations to determine the κ2/r6 factor of FRET yielded slightly different numbers for the starting configurations from the two docking algorithms used (Autodock and GOLD), but the trends were identical.
Tryptophan to haem
Apart from spectral overlap, the FRET efficiencies for the different residues will depend not only on their distance from the haem group but also on the relative orientation of the donor and acceptor transition dipoles, expressed in κ2. The value of κ2/r6 was calculated from MD simulations for six of the tryptophans to the porphyrin ring in CYP2D6. The results for Trp29 could not be calculated, as its position is in the membrane-anchoring α-helix, which is not included in the homology model. The κ2/r6 factors thus obtained differ by several orders of magnitude for the tryptophan residues concerned (Table 3). The tryptophan-to-haem κ2/r6 factor was found to be much higher for Trp128 than for the other tryptophans. This supports the suggestion that Trp128 mainly contributes to the shortest fluorescence lifetime, although it should be realized, however, that energy transfer to the haem may not be the only cause of lifetime-shortening, as mentioned above. The energy transfer from the other tryptophan residues to the haem does not seem to be important; the quantum yield of CYP2D6 as a whole is not extremely low (6% for wild-type CYP2D6, 4% for W128F), and the lifetimes are mostly in the nanosecond range.
Table 3. Orientation factors (κ2/r6 values in nm−6) of the tryptophans to the haem and to MAMC, and of MAMC to the haem, in the CYP2D6 homology model obtained from MD simulations.
*Both dipole directions added.
| κ2/r6 (nm−6) | ||||||
|---|---|---|---|---|---|---|
| Haem* | Haem* | |||||
| (MAMC present) | (no MAMC present) | MAMC | ||||
| Average | R.M.S.D. | Average | R.M.S.D. | Average | R.M.S.D. | |
| W75 | 0.007 | 0.004 | 0.022 | 0.011 | 0.005 | 0.005 |
| W128 | 0.330 | 0.192 | 0.362 | 0.145 | 0.079 | 0.048 |
| W152 | 0.048 | 0.025 | 0.036 | 0.016 | 0.002 | 0.002 |
| W262 | 0.002 | 0.002 | 0.003 | 0.003 | 0.001 | 0.000 |
| W316 | 0.069 | 0.022 | 0.083 | 0.019 | 0.009 | 0.006 |
| W409 | 0.017 | 0.013 | 0.029 | 0.011 | 0.003 | 0.003 |
| MAMC | 7.289 | 6.365 | ||||
Tryptophan to MAMC
The spectral overlap of the tryptophan emission and the MAMC absorption is very large. However, as can be seen from Table 1, the fluorescence lifetimes of CYP2D6 are not shorter in the presence of MAMC, and, furthermore, a negative exponent (indicating an increase of intensity) is not needed to make a good fit of the MAMC intensity decay. Despite this, not only does the tryptophan emission spectrum in the presence of MAMC decrease by approx. 6%, but also the intensity of the MAMC spectrum itself decreases (Figure 2). Calculations of the κ2/r6 value between the various tryptophan residues and MAMC from the MD simulations show no significant changes for most tryptophans, and indicate only a possibility for Trp128 to transfer its energy to MAMC. However, in that case, no decrease in emission intensity would be expected for the W128F mutant, in contrast with the results. On the other hand, because the haem group is positioned between MAMC and Trp128, the tryptophan emission will instead transfer its energy to haem instead of to MAMC. An explanation for the reduced tryptophan emission intensity upon the addition of MAMC may be a conformational change in the protein, repositioning the tryptophan residues closer to quenching amino acids or the haem moiety.
The R.M.S.D. values of κ2/r6 give the width of the distribution around the average value and are an indication of flexibility of the protein. As is seen in Table 3, the relative R.M.S.D. values are not much different for the six tryptophan residues with and without MAMC present. Monitoring of the positions of the tryptophan transition dipole moments yielded the largest fluctuations for Trp262 for the substrate-free CYP2D6, and for Trp75 in the presence of MAMC. However, the fluctuations are smaller than 0.5 nm, which is small on the scale of the total protein (approx. 7 nm diameter).
MAMC to haem
When considering whether MAMC acts as the donor and the haem group as the acceptor, it is important to remember that the spectral overlap is extensive. The 35% reduction of the MAMC emission intensity in the presence of both the wild-type and W128F mutant CYP2D6 indicates that, indeed, such an energy transfer takes place. For the MAMC–haem pair the predicted κ2/r6 value is very high. The values for tryptophan to MAMC are, in general, relatively small, and here the highest value is found for Trp128, which is the closest to the active site. The MD simulations of r6 and κ2 revealed a FRET efficiency of almost 100%, meaning that the excitation energy is completely transferred from MAMC to haem and that no emission of MAMC when bound in the active site can be seen at all. This would explain why the lifetimes of MAMC in presence of CYP2D6 are not shortened and why the quenching properties are not changed: emission is only observed from unbound substrate. Thus, from the intensity reduction in MAMC emission (Table 1), it can be concluded that approx. 35% of the total amount of MAMC added to the sample is bound to CYP2D6, both for wild-type and W128F mutant. This is also reflected in the similar enzymatic activities. Unfortunately, this explanation also implies that the emission spectra do not reveal any information on the properties of bound MAMC.
On the basis of these results, MAMC to enzyme dissociation constants can be estimated using Eqn 7, where [P] is the protein concentration and Kd is the dissociation constant:
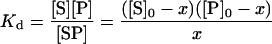 |
(7) |
[S]0 and [P]0 are the initial MAMC and enzyme concentrations (2 μM and 1 μM respectively) and x is the concentration of MAMC bound in the active site. It is calculated that Kd=0.56±0.10 μM for both wild-type and W128F CYP2D6.
Anisotropy
The results of steady-state anisotropy experiments are shown in Figure 5. The anisotropy of the tryptophan residues in the protein is approx. 0.2, as is expected for tryptophans rotating slowly on the fluorescence lifetime scale [26]. The slow rotation is caused by overall tumbling of the protein; side-chain rotations are too fast to be seen. Free MAMC in solution also rotates much faster than its fluorescence decay, and no residual anisotropy is observed.
Figure 5. Steady-state anisotropy traces of 1 μM wild-type CYP2D6 (dotted line), 2 μM MAMC (dashed line) and a mixture of both (black line).

Although the analysis of FRET suggests that MAMC bound to CYP2D6 will not contribute to the emission spectrum at all, a residual anisotropy of 0.015 was seen in the steady-state anisotropy traces of both wild-type and W128F mutant CYP2D6. This anisotropy can most probably be explained by invoking a third type of MAMC present in the sample: MAMC which is also bound in some way to CYP2D6 (to be denoted as ‘associated MAMC’, Table 4). Its emission will be partly polarized, as it rotates with the enzyme, but it is not located close enough to the haem to be quenched by FRET. It may be that the substrate first binds to the access channel of the enzyme, which undergoes a conformational change before allowing it to enter [43,44], or it is located in the access channel. Using eqns 5 and 6 and the initial anisotropy r0=0.39, the ratio of free MAMC/associated MAMC is calculated to be 41±6, implying that 2.4±0.5% of the amount of MAMC that is not bound in the active site is associated in another way to the protein. As these MAMC molecules will experience a slightly more apolar environment, their emission is blue-shifted, as can be seen in Figure 2. However, the observed shift of 6 nm is relatively large, if only 2.4% of MAMC is associated to the enzyme. Presumably the associated MAMC molecules still have a large degree of rotational freedom, causing the amount to be underestimated when calculated from anisotropy experiments.
Table 4. Summary of the three types of MAMC in the presence of CYP2D6.
WT, wild-type
| MAMC type | WT | W128F |
|---|---|---|
| I Unbound | 62.6±2.0% | 62.4±2.1% |
| II Bound in active site | 35.0±1.5% | 35.2±1.6% |
| III Associated to enzyme | 2.4±0.5% | 2.4±0.5% |
Conclusions
Wild-type and W128F mutant CYP2D6 were investigated by steady-state and time-resolved fluorescence experiments to determine the extent of conformational changes in the protein, and the properties of MAMC upon binding. Because both the enzyme and the substrate contain fluorophores, it was possible to study emission properties of both compounds.
The experiments do not show indications of large-scale conformational changes in CYP2D6 in the presence of the substrate MAMC: there are only small changes in emission maxima and lifetimes when MAMC is present. However, the tryptophan residues in substrate-bound enzyme are quenched less efficiently than in substrate-free CYP2D6, which may be caused either by shielding of the tryptophans by MAMC or by a ‘contraction’ of the structure, induced by binding of the substrate. MD simulations indicated that the tryptophan residues are more flexible in the presence of MAMC, but these changes are small on the scale of the total protein; no large fluctuations in protein structure are found. Mutagenesis of more tryptophan residues would be helpful in the assignment of lifetimes to individual residues and may provide more information on possible local conformational changes in the enzyme upon substrate binding.
Several FRET pathways were evaluated from which we concluded that emission of MAMC when bound to CYP2D6 is transferred completely to the haem, and is therefore not observed. From anisotropy experiments, it could be inferred that not all observable MAMC is free in solution, but that at least 2.4% is associated to the protein. No differences were found in the dissociation constants.
Online Data
Acknowledgments
We thank Mr Ed Groot (LACDR, Division of Molecular Toxicology, Department of Pharmacochemistry, Vrije Universiteit, Amsterdam, The Netherlands) for his help in expressing and purifying the enzymes.
References
- 1.Zanger U. M., Raimundo S., Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch. Pharmacol. 2004;369:23–37. doi: 10.1007/s00210-003-0832-2. [DOI] [PubMed] [Google Scholar]
- 2.Shimada T., Yamazaki H., Mimura M., Inui Y., Guengerich F. P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 3.Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol. Sci. 2004;25:193–200. doi: 10.1016/j.tips.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 4.De Graaf C., Vermeulen N. P. E., Feenstra K. A. Cytochrome P450 in silico: An integrative modeling approach. J. Med. Chem. 2005;48:2725–2755. doi: 10.1021/jm040180d. [DOI] [PubMed] [Google Scholar]
- 5.Prasad S., Mazumdar S., Mitra S. Binding of camphor to Pseudomonas putida cytochrome P450cam: steady-state and picosecond time-resolved fluorescence studies. FEBS Lett. 2000;477:157–160. doi: 10.1016/s0014-5793(00)01745-2. [DOI] [PubMed] [Google Scholar]
- 6.Prasad S., Mitra S. Role of protein and substrate dynamics in catalysis by Pseudomonas putida cytochrome P450cam. Biochemistry. 2002;41:14499–14508. doi: 10.1021/bi026379e. [DOI] [PubMed] [Google Scholar]
- 7.Beechem J. M., Brand L. Time-resolved fluorescence of proteins. Annu. Rev. Biochem. 1985;54:43–71. doi: 10.1146/annurev.bi.54.070185.000355. [DOI] [PubMed] [Google Scholar]
- 8.Eftink M. R. The use of fluorescence methods to monitor unfolding transitions in proteins. Biochemistry (Moscow) 1998;63:276–284. [PubMed] [Google Scholar]
- 9.Engelborghs Y. Correlating protein structure and protein fluorescence. J. Fluoresc. 2003;13:9–16. [Google Scholar]
- 10.Wester M. R., Johnson E. F., Marques-Soares C., Dansette P. M., Mansuy D., Stout C. D. Structure of a substrate complex of mammalian cytochrome P450 2C5 at 2.3 Å resolution: evidence for multiple substrate binding modes. Biochemistry. 2003;42:6370–6379. doi: 10.1021/bi0273922. [DOI] [PubMed] [Google Scholar]
- 11.Williams P. A., Cosme J., Ward A., Angove H. C., Matak Vinkovic D., Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature (London) 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 12.Scott E. E., He Y. A., Wester M. R., White M. A., Chin C. C., Halpert J. R., Johnson E. F., Stout C. D. An open conformation of mammalian cytochrome P4502B4 at 1.6 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13196–13201. doi: 10.1073/pnas.2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yano J. K., Wester M. R., Schoch G. A., Griffin K. J., Stout C. D., Johnson E. F. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05 Å resolution. J. Biol. Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 14.Weber G., Teale F. J. W. Electronic energy transfer in haem proteins. Disc. Faraday Soc. 1959;27:134–141. [Google Scholar]
- 15.Gryczynski Z., Lubkowski J., Bucci E. Intrinsic fluorescence of hemoglobins and myoglobins. Methods Enzymol. 1997;278:538–569. doi: 10.1016/s0076-6879(97)78030-3. [DOI] [PubMed] [Google Scholar]
- 16.Hochstrasser R. M., Negus D. K. Picosecond fluorescence decay of tryptophans in myoglobin. Proc. Natl. Acad. Sci. U.S.A. 1984;81:4399–4403. doi: 10.1073/pnas.81.14.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onderwater R. C., Venhorst J., Commandeur J. N., Vermeulen N. P. E. Design, synthesis, and characterization of 7-methoxy-4-(aminomethyl)coumarin as a novel and selective cytochrome P450 2D6 substrate suitable for high-throughput screening. Chem. Res. Toxicol. 1999;12:555–559. doi: 10.1021/tx980248q. [DOI] [PubMed] [Google Scholar]
- 18.Venhorst J., Onderwater R. C., Meerman J. H., Vermeulen N. P. E., Commandeur J. N. Evaluation of a novel high-throughput assay for cytochrome P450 2D6 using 7-methoxy-4-(aminomethyl)-coumarin. Eur. J. Pharm. Sci. 2000;12:151–158. doi: 10.1016/s0928-0987(00)00150-0. [DOI] [PubMed] [Google Scholar]
- 19.Eftink M. R., Ghiron C. A. Exposure of tryptophanyl residues in proteins: quantitative determination by fluorescence quenching studies. Biochemistry. 1976;15:672–680. doi: 10.1021/bi00648a035. [DOI] [PubMed] [Google Scholar]
- 20.Lakowicz J. R. Quenching of fluorescence. In: Lakowicz J. R., editor. Principles of Fluorescence Spectroscopy. New York: Plenum Press; 1983. pp. 257–295. [Google Scholar]
- 21.Lehrer S. S. Solute perturbation of protein fluorescence: the quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry. 1971;10:3254–3263. doi: 10.1021/bi00793a015. [DOI] [PubMed] [Google Scholar]
- 22.Keizers P. H. J., de Graaf C., de Kanter F. J. J., Oostenbrink C., Feenstra K. A., Commandeur J. N. M., Vermeulen N. P. E. Metabolic regio- and stereoselectivity of cytochrome P450 2D6 towards 3,4-methylenedioxy-N-alkylamphetamines: in silico predictions and experimental validation. J. Med. Chem. 2005;48:6117–6127. doi: 10.1021/jm050338+. [DOI] [PubMed] [Google Scholar]
- 23.Keizers P. H. J., Lussenburg B. M. A., De Graaf C., Mentink L., Vermeulen N. P. E., Commandeur J. N. M. Influence of phenylalanine 120 on cytochrome P450 2D6 catalytic selectivity and regiospecificity: Crucial role in 7-methoxy-4-(aminomethyl)-coumarin metabolism. Biochem. Pharmacol. 2004;68:2263–2271. doi: 10.1016/j.bcp.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Omura T., Sato R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 25.Beechem J. M. Global analysis of biochemical and biophysical data. Methods Enzymol. 1992;210:37–55. doi: 10.1016/0076-6879(92)10004-w. [DOI] [PubMed] [Google Scholar]
- 26.Valeur B. Weinheim: Wiley-VCH; 2002. Molecular Fluorescence: Principles and Applications. [Google Scholar]
- 27.Cheung H. C. Resonance energy transfer. In: Lakowicz J. R., editor. Topics in Fluorescence Spectroscopy 2: Principles. New York: Plenum Press; 1991. pp. 127–173. [Google Scholar]
- 28.Morris G. M., Goodsell D. S., Halliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 29.Jones G., Willett P., Glen R. C., Leach A. R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 30.Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces. Dordrecht: Reidel; 1981. pp. 331–342. [Google Scholar]
- 31.van Gunsteren W. F., Billeter S. R., Eising A. A., Hünenberger P. H., Krüger P., Mark A. E., Scott W. R. P., Tironi I. G. Zürich: Vdf Hochschulverlag AG an der ETH Zürich; 1996. Biomolecular Simulation: the GROMOS 96 Manual and User Guide. [Google Scholar]
- 32.Davra X., Mark A. E., van Gunsteren W. F. Parametrization of aliphatic CHn united atoms of GROMOS96 force field. J. Comput. Chem. 1998;19:535–547. [Google Scholar]
- 33.Schuler L. D., van Gunsteren W. F. On the choice of dihedral angle potential energy functions for n-alkanes. Mol. Simul. 2000;25:301–319. [Google Scholar]
- 34.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 35.Callis P. 1La and 1Lb transitions of tryptophan: applications of theory and experimental observations to fluorescence of proteins. Methods Enzymol. 1997;278:113–150. doi: 10.1016/s0076-6879(97)78009-1. [DOI] [PubMed] [Google Scholar]
- 36.Maroncelli M., Fleming G. R. Picosecond solvation dynamics of coummarin 153: the importance of molecular aspects of solvation. J. Chem. Phys. 1987;86:6221–6239. [Google Scholar]
- 37.Lee B., Richards F. M. Interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 38.Lakowicz J. R. Protein fluorescence. In: Lakowicz J. R., editor. Principles of Fluorescence Spectroscopy. New York: Plenum Press; 1983. pp. 341–379. [Google Scholar]
- 39.Sharma V. K., Saharo P. D., Sharma N., Rastogi R. C., Ghoshal S. K., Mohan D. Influence of solvent and substituent on excited state characteristics of laser grade coumarin dyes. Spectrochim. Acta Part A. 2003;59:1161–1170. doi: 10.1016/s1386-1425(02)00296-2. [DOI] [PubMed] [Google Scholar]
- 40.Stortelder A., Buijs J. B., Bulthuis J., Gooijer C., Van Der Zwan G. Time-resolved fluorescence of the bacteriophage T4 capsid protein gp23. J. Photochem. Photobiol. B. 2005;78:53–60. doi: 10.1016/j.jphotobiol.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 41.Larsen O. F. A., Van Stokkum I. H. M., Pandit A., Van Grondelle R., Van Amerongen H. Ultrafast polarized fluorescence measurements on tryptophan and a tryptophan-containing peptide. J. Phys. Chem. B. 2003;107:3080–3085. [Google Scholar]
- 42.Chen Y., Barkley M. D. Toward understanding tryptophan fluorescence in proteins. Biophys. J. 1998;81:1765–1775. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 43.Lüdemann S. K., Lounnas V., Wade R. C. How do substrates enter and products exit the buried active site of cytochrome P450cam? 1. Random expulsion molecular dynamics investigation of ligand access channels and mechanisms. J. Mol. Biol. 2000;303:797–811. doi: 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]
- 44.Lüdemann S. K., Lounnas V., Wade R. C. How do substrates enter and products exit the buried active site of cytochrome P450cam? 2. Steered molecular dynamics and adiabatic mapping of substrate pathways. J. Mol. Biol. 2000;303:813–830. doi: 10.1006/jmbi.2000.4155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.