

Abstract
β-NAD+e (extracellular β-NAD+), present at nanomolar levels in human plasma, has been implicated in the regulation of [Ca2+]i (the intracellular calcium concentration) in various cell types, including blood cells, by means of different mechanisms. Here, we demonstrate that micromolar NAD+e (both the α and the β extracellular NAD+ forms) induces a sustained [Ca2+]i increase in human granulocytes by triggering the following cascade of causally related events: (i) activation of adenylate cyclase and overproduction of cAMP; (ii) activation of protein kinase A; (iii) stimulation of ADP-ribosyl cyclase activity and consequent overproduction of cADP-ribose, a universal Ca2+ mobilizer; and (iv) influx of extracellular Ca2+. The NAD+e-triggered [Ca2+]i elevation translates into granulocyte activation, i.e. superoxide and nitric oxide generation, and enhanced chemotaxis in response to 0.1–10 μM NAD+e. Thus extracellular β-NAD+e behaves as a novel pro-inflammatory cytokine, stimulating human granulocytes and potentially recruiting them at sites of inflammation.
Keywords: ADP-ribosyl cyclase, chemotaxis, cyclic ADP-ribose, granulocyte, intracellular calcium, NAD+
Abbreviations: ART, mono-ADP-ribosyltransferase; ATPe, extracellular ATP; (c)ADPR, (cyclic) ADP-ribose; [Ca2+]i, intracellular calcium concentration; [cADPR]i, intracellular cADPR concentration; [cAMP]i, intracellular cAMP concentration; CI, chemotaxis index; Cx43, connexin 43; 8-Br-cAMP, 8-bromo-cAMP; HBSS, Hanks balanced salt solution; IP3, inositol 1,4,5-trisphosphate; NAD+e, extracellular NAD+ (α and β forms); NT, nucleoside transporter; PCA, perchloric acid; PKA, protein kinase A; PLC, phospholipase C; RyR, ryanodine receptor; SOCC, store-operated Ca2+ channel
INTRODUCTION
Recent studies have demonstrated a role of NAD+e (extracellular NAD+) [1–4] as a signal metabolite, significantly expanding its physiological functions beyond the long-known intracellular ones. The common denominator of these extracellular activities is up-regulation of [Ca2+]i (the intracellular calcium concentration). Although earlier investigations suggested cell lysis as the possible source of NAD+e, recent findings indicate the involvement of specific release mechanisms that can be triggered by mechanical stress [3] or by electrical field stimulation [5]. NAD+ release can occur in several cell types by means of an equilibrative transport process across Cx43 (connexin 43) hemichannels: NAD+ efflux, potentially detrimental to cells, is down-regulated by high [Ca2+]i levels via protein kinase C-mediated Cx43 phosphorylation [1].
Incubation with NAD+e of several mammalian cell types (astrocytes, osteoblasts, CD38-transfected fibroblasts and HeLa cells [2,3,6]) expressing the ecto-ADP-ribosyl cyclase CD38, which converts NAD+ into the intracellular calcium mobilizer cADPR (cyclic ADP-ribose) [7,8], has been shown to up-regulate [Ca2+]i via a cADPR-induced intracellular Ca2+-release mechanism. Binding of cADPR to the intracellular RyRs (ryanodine receptors) [9–12] is ensured by several transport mechanisms for this cyclic nucleotide, which overcome the topological restrictions imposed by the ectocellular localization of the catalytic domain of the cyclase [1]. Thus: (i) CD38 itself also behaves as an active cADPR transporter across the cell membrane [6]; and (ii) extracellular cADPR can cross the plasma membrane through several NTs (nucleoside transporters), including concentrative NTs, widely expressed in most cell types [1]. NAD+e-induced internalization of CD38, which has been observed in carcinoma cell lines [13], may also act in concert to shift cADPR metabolism from the extracellular to the intracellular environment.
Ectocellular ARTs (mono-ADP-ribosyltransferases; [14]) can also affect cellular Ca2+-regulated functions in the presence of NAD+e. In murine T-lymphocytes, NAD+e is a substrate of ART-2, which ADP-ribosylates the purinoceptor P2X7 or a P2X7-associated protein, leading to pore formation, Ca2+ influx and eventually cell death [4,15,16]. In human monocytes too, NAD+e has been reported to increase the [Ca2+]i by eliciting influx of extracellular Ca2+: the underlying mechanism has not yet been elucidated, although an involvement of ectocellular CD38+ or P2X7-induced pore formation has been ruled out [17].
In granulocytes, respiratory burst and migration are known to be activated by a [Ca2+]i increase [18–20]. Involvement of cADPR in the regulation of granulocyte functions was unequivocally demonstrated by Partida-Sánchez et al. [21], who showed that deletion of CD38 renders mice susceptible to Streptococcus pneumoniae infection because of failure of their neutrophils to migrate to the infected lung tissue. This functional impairment of the CD38−/− neutrophils is caused by absence of the cADPR-induced [Ca2+]i increase, which in control mice is largely due to influx of extracellular Ca2+ across SOCCs (store-operated Ca2+ channels). In addition, cADPR proved to regulate chemotaxis through Ca2+ mobilization in human granulocytes and monocytes stimulated by engagement of selected chemoattractant receptors [22].
The emerging data on NAD+e as a signal molecule regulating cell functions and the key role of the CD38+/cADPR system in neutrophil chemotaxis prompted us to investigate whether NAD+e is involved in the activation of human polymorphonuclear granulocytes. Our results demonstrate that micromolar NAD+e triggers a signalling cascade that involves the overproduction of intracellular cADPR and results in the influx of extracellular Ca2+. The ensuing sustained [Ca2+]i elevation increases O2− and NO (nitric oxide) generation and activates chemotaxis towards NAD+e. The likely occurrence of increased (relative to basal plasma levels) NAD+e concentrations at the site of inflammation [16] supports the conclusion that NAD+e can exhibit functions of a pro-inflammatory cytokine targeting granulocytes via cADPR-mediated and Ca2+-dependent mechanisms.
EXPERIMENTAL
Materials
Fura/2AM (fura 2 acetoxymethyl ester), protein kinase inhibitors [staurosporine and PKA (protein kinase A) inhibitor peptide sequence 14–22; cell-permeant] and Ro-20-1724 [(3-butoxy-4-methoxybenzyl)imidazolidin-2-one] were obtained from Calbiochem (Milan, Italy). Sytox Green was from Molecular Probes (Eugene, OR, U.S.A.). The [3H]cAMP assay system and Ficoll-Paque™ Plus medium were from Amersham Bioscience AB (Uppsala, Sweden). All other chemicals were obtained from Sigma (Milan, Italy).
HPLC analyses of nucleotides and purification of α- and β-NAD+
HPLC analyses of nucleotides, to evaluate their grade of purity, were performed as described in [23]. Either α- or β-NAD+ (1 μmol; Sigma) was purified by preparative phosphate HPLC analysis, performed on a C18 column (300×7.8 mm, 15 μm-diameter; Waters); buffers and gradient were prepared as described in [23], and the flow rate was 2 ml/min. The α- or β-NAD+ peaks were collected, dried, resuspended in 250 μl water and submitted to preparative formate analysis on the same column type; buffer A was 10 mM formic acid containing 5 mM tetraethylammonium, pH 4.0, and buffer B was buffer A also containing 50% (v/v) methanol. The solvent programme was: 100% A for 5 min, linearly increasing to 100% B for 15 min; 100% B for 10 min; flow rate 2 ml/min. The α- or β-NAD+ peaks were collected, dried and resuspended in water: the purity of the nucleotides was confirmed by the analytical phosphate analysis. Nucleotides were identified and quantified by comparison of their absorption spectra and peak areas with those of computer-stored standards.
Isolation of human granulocytes
Buffy coats, prepared from freshly drawn blood of healthy volunteers, were provided by Galliera Hospital, Genova, Italy: granulocytes were isolated by density centrifugation through Ficoll–Paque™ Plus medium and hypotonic lysis of erythrocytes, and resuspended at 60×106/ml in RPMI 1640 medium. The preparation contained >95% granulocytes, as verified by May–Grunwald–Giemsa staining, and >98% viable cells, as measured by the Trypan Blue dye exclusion method.
Fluorimetric measurements of [Ca2+]i
Granulocytes (30×106/ml) were loaded with 10 μM Fura/2AM for 45 min at 25 °C in RPMI medium, washed with HBSS (Hanks balanced salt solution; cat. no. H8264; Sigma) and resuspended in the same solution or in a Ca2+-free HBSS (cat. no. H6648; Sigma) at 5×106 cells/ml. Following cell sedimentation on a 20 mm coverslip mounted on a 200 μl volume chamber, Ca2+ measurements were performed as described in [24].
Determination of [cADPR]i (intracellular cADPR concentration) levels
Granulocytes (30×106/ml) were incubated at 25 °C with 0, 1, 10 or 100 μM α-NAD+. At 0, 1, 2.5, 5, 15 and 60 min, a 500 μl aliquot of the suspension was withdrawn and processed to measure the cADPR content [25].
Determination of [cAMP]i (intracellular cAMP concentration) levels
Cells were suspended in HBSS (30×106/ml) and pre-incubated for 5 min at 25 °C in the presence of the cAMP phosphodiesterase inhibitor Ro-20-1724 (10 μM final concentration). After incubation with 0, 1, 10 or 100 μM α- or β-NAD+, a 300 μl aliquot of the suspension was withdrawn at different times and the reaction was stopped by adding 20 μl of 9 M PCA (perchloric acid). PCA was removed as described in [25]. [cAMP]i was determined using the [3H]cAMP assay system.
Assays of ADP-ribosyl cyclase activity
Intact cells were resuspended in HBSS (60×106/ml) and incubated at 25 °C with or without 100 μM α-NAD+ for 10 min, or 500 μM 8-Br-cAMP (8-bromo-cAMP) for 30 min. After addition of protease inhibitors (10 μM pepstatin, 0.1 mM aprotinin and leupeptin) and a 1:100 dilution of phosphatase inhibitor cocktail (Sigma cat. no. P2850), control and either α-NAD+- or 8-Br-cAMP-stimulated cells were lysed by sonication on ice for 1 min at 3 W using a W380 ultrasonic processor (Heat-System Ultrasonics, New York, NY, U.S.A.). ADP-ribosyl cyclase activity and protein content were measured on cell lysates as described in [25].
Oxidative burst of granulocytes
Superoxide generation was quantified using the cytochrome c reduction assay [26]. Cells were resuspended in HBSS at 5×106 cells/ml, and 225 μl of the suspension was incubated with 50 μl cytochrome c (0.2 mM) and stimulated (or not) with 1, 10 or 100 μM α- or β-NAD+ for 30 min at 37 °C. Samples were centrifuged at 5000 g for 15 s and the absorbance of the supernatants was recorded at 550 nm (reduced cytochrome c peak absorbance) and 540 nm (reference). Results are expressed as the difference in absorbance between the two wavelengths.
Determination of NO
Cells (60×106/ml in RPMI medium) were incubated for 60 min at 37 °C with or without α- or β-NAD+. Samples were centrifuged at 5000 g for 15 s, supernatants were recovered and diluted with an equal volume of Griess reagent (0.1% naphthyl ethylenediamine dihydrochloride in distilled water/1% sulphanylamide in 5% concentrated H3PO4), and the absorbance at 545 nm was estimated for 100 μl aliquots after 10 min. A standard curve was generated in parallel with known amounts of NaNO2.
Chemotaxis
Granulocytes were resuspended at 10×106/ml in chemotaxis buffer (HBSS, PBS and 5% albumin; 39:16:1 by vol.). Chemotaxis assays were performed using 96-well ChemoTx system microplates (Neuro Probe, Inc., Gaithersburg, MD, U.S.A.) with a 3-μm pore size polycarbonate filter. α- or β-NAD+ (0.1–100 μM) was diluted in chemotaxis buffer and added in the bottom wells. Cell suspensions (25 μl) were placed on top of the filter. Granulocytes were incubated for 60 min at 37 °C. The transmigrated cells were transferred into a 96-well plate and quantified by adding 60 μl of a solution composed of 0.2% (v/v) Nonidet P40 and 1 μM Sitox Green. After 20 min incubation at 37 °C, fluorescence was recorded (excitation wavelength, 485 nm; emission wavelength, 520 nm). A standard curve was obtained by placing a serial dilution of the cell suspension in the bottom wells. The results were expressed as the chemotaxis index (CI):
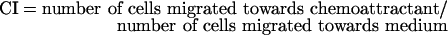 |
A CI>1 indicates chemoattraction of cells.
RESULTS
cADPR-mediated Ca2+ response of human granulocytes to NAD+e
Intact, freshly isolated granulocytes were incubated with 1, 10 or 100 μM β-NAD+e: a sustained and concentration-dependent Ca2+ increase was consistently observed within 3 min of addition of β-NAD+e. As shown in Figure 1(A), 1 μM β-NAD+e was sufficient to induce a significant [Ca2+]i elevation.
Figure 1. β-NAD+e, α-NAD+e and ATPe increase [Ca2+]i in human granulocytes.

(A) Fura2/AM-loaded granulocytes were treated with increasing concentrations of β-NAD+ and the [Ca2+]i measured at 10 min is shown (n=6 for 1 and 10 μM β-NAD+, P<0.005; n=9 for 100 μM β-NAD+, P<0.0001). (B, C and E) Traces shown are individual experiments representative of results obtained upon addition (arrow) of 100 μM β-NAD+ (B) or α-NAD+ (C) or ATP (E) to either untreated (traces #1) or 8-Br-cADPR-pre-treated (100 μM for 90 min; traces #2) cells in HBSS, or to untreated cells in Ca2+-free HBSS containing 1 mM EGTA (traces #3) (n≥3 for each condition). (D) Mean [Ca2+]i values±S.D. obtained upon addition of 100 μM β-NAD+ (white circles) or α-NAD+ (black circles). Each point is the mean of the [Ca2+]i values recorded every 20 s in nine traces for β-NAD+ and five traces for α-NAD+.
Supplementation of 100 μM β-NAD+e in the presence of 1 mM extracellular EGTA in Ca2+-free HBSS resulted in the complete inhibition of Ca2+ elevation, demonstrating that influx of extracellular Ca2+ is involved in the generation of the sustained Ca2+ increase (Figure 1B).
Replacement of β-NAD+e with α-NAD+e did not modify the pattern of Ca2+ response in granulocytes and its inhibition by EGTA (Figure 1C). On the contrary, no Ca2+ responses were recorded upon incubating granulocytes with 100 μM NADH or NADP+. Figure 1(D) shows the average traces of the [Ca2+]i increases obtained upon addition of 100 μM α-NAD+e (n=5) or β-NAD+e (n=9): the kinetics of the [Ca2+]i increases proved to be very similar with both dinucleotide forms.
We then evaluated the presence of contaminating nucleotides in commercial batches of β- and α-NAD+e: HPLC analysis demonstrated <5% and <1% ADPR respectively, whereas ATP was not detected. The addition of the amount of ADPR expected to be present in 100 μM β-NAD+e (i.e. 5 μM) did not result in any Ca2+ increase, thus ruling out the possibility that the effects obtained with 100 μM β- and α-NAD+e were due to contaminating ADPR. Nevertheless, we also purified both β- and α-NAD+ by means of a HPLC preparative method (see the Experimental section). The addition of 100 μM HPLC-purified β-NAD+ or α-NAD+ showed a superimposable long-lasting Ca2+ increase as obtained with commercial preparations, thus excluding the possibility that other contaminating compounds could be responsible for the effects on [Ca2+]i.
Supplementation of granulocytes with 100 μM ATPe (extracellular ATP) induced a rapid and transient [Ca2+]i rise, completely different from the sustained NAD+-induced [Ca2+]i increase (Figure 1E). The ATPe-induced [Ca2+]i peak was not affected by EGTA (results not shown), confirming intracellular Ca2+ mobilization as the underlying mechanism [27]. Indeed, pre-treatment of granulocytes with U73122, a membrane-permeant PLC (phospholipase C) inhibitor, completely abolished the ATP-induced [Ca2+]i increase, thus implicating IP3 (inositol 1,4,5-trisphosphate) as the signal metabolite responsible for this transient Ca2+ event.
The [Ca2+]i rise elicited by β-NAD+e or by α-NAD+e was markedly inhibited by pre-incubation with either 8-Br-cADPR at 100 μM (Figures 1B and 1C, and Table 1), a membrane-permeant cADPR antagonist [28], or ryanodine at 50 μM (Table 1), a concentration known to inhibit Ca2+ release from cADPR-responsive stores [11]. The extent of inhibition of the Ca2+ increase was approx. 80% with both 8-Br-cADPR and ryanodine, with no significant differences between the effects afforded on granulocytes by β-NAD+e and α-NAD+e respectively. The Ca2+ elevation evoked by ATP was only slightly inhibited (25%) by 8-Br-cADPR (Figure 1E), in agreement with previous observations on CD38+ and CD38− 3T3 cells, demonstrating the co-operation between IP3 and cADPR in the generation, and especially in the shaping, of the ATP-induced calcium response [29].
Table 1. Effect of different compounds on the α- and β-NAD+e-induced Ca2+ response of human granulocytes.
Before exposure to 100 μM α- or β-NAD+, granulocytes were pre-incubated at 25 °C in HBSS under the following conditions: with 100 μM 8-Br-cADPR or 50 μM ryanodine for 90 min; with 10 μM SKF96365 for 1 min; with 5 μM U73122 for 10 min; with 2 μM staurosporine for 60 min; or with 1 μM PKA inhibitor for 15 min. The [Ca2+]i and the [cADPR]i were measured 10 and 15 min respectively after exposure to NAD+. Results are expressed as percentages of the inhibition of α- or β-NAD+-induced responses in control cells, not pre-incubated with the inhibitors. Each value is the mean±S.D. for at least three different experiments. n.d., not determined.
| [Ca2+]i (%) | [cADPR]i (%) | ||
|---|---|---|---|
| Inhibitor | α-NAD+ | β-NAD+ | α-NAD+ |
| 8-Br-cADPR | 80±6 | 75±5 | n.d. |
| Ryanodine | 81±6 | 77±5 | n.d. |
| SKF96365 | 75±4 | 70±8 | n.d. |
| U73122 | 0 | 0 | n.d. |
| Staurosporine | 75±6 | 78±7 | 90±5 |
| PKA inhibitor | 55±7 | 52±8 | 65±6 |
These results implicate cADPR as being responsible for the NAD+-induced Ca2+ increase, which was abrogated in the presence of extracellular EGTA (Figure 1). Previous reports have suggested, in several cell types, either that cADPR might directly trigger Ca2+ influx across still-undefined plasma-membrane Ca2+ channels or, alternatively, that the cADPR-induced Ca2+ release from ryanodine-gated stores might activate plasma-membrane SOCCs [30–34]. Through either mechanism, cADPR would induce extracellular Ca2+ influx into granulocytes. SKF96365 (10 μM), an inhibitor of SOCCs [33,35], caused a remarkable reduction in the Ca2+ response to both β- and α-NAD+e (Table 1), suggesting that SOCCs may indeed be involved in the generation of the cADPR-mediated sustained [Ca2+]i increase.
The [Ca2+]i event elicited by α- and β-NAD+e was not modified by pre-treatment of the granulocytes with U73122 (Table 1), which rules out any involvement of IP3 in this process.
The close qualitative and quantitative similarities between the Ca2+ events triggered by β-NAD+e (a substrate of ADP-ribosyl cyclases, which are able to convert it into nicotinamide, cADPR and ADPR) and α-NAD+e, which is neither a substrate nor an inhibitor of the ADP-ribosyl cyclase activity [8], strongly suggest that the effects on [Ca2+]i in human granulocytes were due directly to NAD+ itself, and not to the extracellular generation of its metabolites cADPR and ADPR. Nevertheless, we performed most subsequent experiments using both the natural β-NAD+ and the α-NAD+ form, except for the evaluation of [cADPR]i, which was assayed only after addition of α-NAD+ (see below).
[cADPR]i in NAD+e-stimulated granulocytes
Prevention of the sustained NAD+e-induced Ca2+ response by the cADPR antagonist 8-Br-cADPR and by ryanodine prompted us to investigate whether an increase in [cADPR]i occurs following NAD+e stimulation. In order to avoid ectocellular generation of cADPR from β-NAD+e, which would interfere with the determination of possible changes in the [cADPR]i, we used α-NAD+ as an agonist. Exposure of granulocytes to α-NAD+e resulted in a concentration-dependent increase in [cADPR]i, which was found to be 30.53±6.63, 39.69±6.23, 41.52±8.30 and 44.26±9.07 pmol/109 cells after 15 min of incubation with 0, 1, 10 and 100 μM α-NAD+e respectively (n=5; P<0.05) (Figure 2A). The [cADPR]i values recorded after 60 min incubation with α-NAD+e were only 10% higher than those measured at 15 min (results not shown). Figure 2(B) shows the time course of the [cADPR]i elevation during incubation of granulocytes with 100 μM α-NAD+e. The [cADPR]i was consistently increased at 1 min after addition of α-NAD+e (109±3% of control values; n=5).
Figure 2. [cAMP]i, [cADPR]i and [Ca2+]i levels in α-NAD+e-stimulated granulocytes.

(A) After addition of α-NAD+ at the concentrations indicated in the abscissa, [cAMP]i (white bars) and [cADPR]i (grey bars) levels were determined at 1 min and at 15 min, and expressed as percentages of the values measured in unstimulated cells (control). [Ca2+]i values (black bars) are expressed as percentages of the basal [Ca2+]i, before addition of α-NAD+ (50±4 nM). (B) After addition of 100 μM α-NAD+, [cAMP]i (●) and [cADPR]i (■) levels were determined at the times indicated and expressed as percentages of control values, recorded on cells at zero time. Each point is the mean for five experiments. [Ca2+]i levels (▲) are from a representative trace, and each point is the mean of values recorded every minute, expressed as the percentage of the values in unstimulated cells. Values of S.D. are omitted for the sake of clarity.
These data indicate that α-NAD+e triggers production of intracellular cADPR from intracellular β-NAD+. In a number of cell types, different concentrations of functionally active (i.e. [Ca2+]i-increasing) intracellular cADPR have been shown to correlate with different quantitative levels of ADP-ribosyl cyclase activity [24,25,36]. Thus, in addition to analysing the [cADPR]i, we also assayed the levels of ADP-ribosyl cyclase activity in α-NAD+e-stimulated granulocytes. Cells were incubated for 10 min in the presence or absence of 100 μM α-NAD+e, and levels of ADP-ribosyl cyclase activity were then measured in the cell lysates using β-NAD+ as substrate. Cyclase activity increased from 2.85±0.25 (control cells) to 3.68±0.33 pmol of cADPR/min per mg of protein in α-NAD+-treated cells (n=3; P<0.05). Therefore rapid up-regulation of ADP-ribosyl cyclase activity follows stimulation of human granulocytes with α-NAD+, resulting in a significant increase in the [cADPR]i.
Role of PKA in the NAD+e-induced increases in [cADPR]i and [Ca2+]i
Stimulation of ADP-ribosyl cyclase activity by hormones or extracellular agonists in a variety of cell types has been reported to occur via PKA-mediated phosphorylation of the cyclase [37–40]. To investigate a possible involvement of protein kinases in the NAD+e-induced activation of ADP-ribosyl cyclase, human granulocytes were pre-incubated with either staurosporine, a non-specific inhibitor of protein kinases, or a cell-permeant, PKA-specific myristoylated peptide, before incubation with α-NAD+e. The increase in the [cADPR]i induced by 100 μM α-NAD+e was almost completely abrogated (90%) by staurosporine (n=4; P<0.05; Table 1). The PKA inhibitor was consistently, though not completely, effective (65% inhibition of the [cADPR]i increase induced by 100 μM α-NAD+e; n=4; P<0.05; Table 1), suggesting that other protein kinases may also contribute to activation of ADP-ribosyl cyclase under these conditions. The inhibitory effects afforded by staurosporine and by the PKA inhibitor on the α-NAD+e-induced [cADPR]i increase were paralleled by similar effects on the [Ca2+]i elevation (Table 1). Comparable results were obtained when staurosporine- and PKA inhibitor-pretreated granulocytes were challenged with β-NAD+e instead of α-NAD+e (Table 1).
In order to confirm a role for PKA in the elevation of both [cADPR]i and [Ca2+]i levels, we investigated whether NAD+e treatment induced an increase in the [cAMP]i. The [cAMP]i of human granulocytes (basal value of 15.9±2.9 pmol/107 cells; n=10) increased immediately after stimulation with micromolar β-NAD+e in a concentration-dependent way, with 1, 10 and 100 μM β-NAD+e inducing a 32%, 42% and 61% increase respectively of the [cAMP]i at 1 min (n=5; P<0.05 for 1 and 10 μM; P<0.005 for 100 μM). A similar increase in the [cAMP]i was recorded in cells stimulated with 100 μM α-NAD+e (Figure 2A). In addition, as summarized in Figure 2(A), α-NAD+e also induced a concentration-dependent increase in the [cADPR]i. As shown in Figure 2(B), comparison of the time course of NAD+e-triggered cAMP and cADPR production clearly shows that the latter lags behind the former: this finding localizes cAMP upstream of cADPR in the signalling pathway triggered by NAD+e (α or β) that results in the sustained [Ca2+]i increase.
To obtain direct evidence for the involvement of PKA in the NAD+e-induced activation of ADP-ribosyl cyclase, granulocytes were incubated with 500 μM 8-Br-cAMP (a cell-permeant PKA activator) and the [cADPR]i was measured: at 15 and 30 min after addition of 8-Br-cAMP, the [cADPR]i (basal value of 29.36±5.54 pmol/107 cells) was increased to 54.95±10.99 and 79.38±15.38 pmol/107 cells (n=3; P<0.05) respectively. The increase in the [cADPR]i was paralleled by stimulation of the ADP-ribosyl cyclase activity, which was 1.2-fold after 30 min pre-incubation of intact granulocytes with 500 μM 8-Br-cAMP (from 2.79±0.23 pmol of cADPR/min per mg of protein in control cells to 3.35±0.26 pmol of cADPR/min per mg of protein in 8-Br-cAMP-treated cells; n=3; P<0.05). The 8-Br-cAMP-induced increase in the [cADPR]i was also paralleled by a sustained [Ca2+]i increase (Figure 3). Pre-incubation of cells with 8-Br-cADPR prevented this 8-Br-cAMP-induced [Ca2+]i elevation (Figure 3) almost completely, unequivocally proving that cAMP by itself can trigger a cADPR-mediated Ca2+ response in human granulocytes.
Figure 3. The 8-Br-cAMP-induced [Ca2+]i increase in human granulocytes is inhibited by 8-Br-cADPR.

Traces shown, representative of three different experiments, were obtained upon addition (arrow) of 500 μM 8-Br-cAMP to untreated (trace 1) or to 8-Br-cADPR-pre-treated (100 μM for 90 min; trace 2) cells in HBSS.
Taken together, these data support the conclusion that stimulation of ADP-ribosyl cyclase by NAD+e in human granulocytes occurs mainly via PKA.
Superoxide and NO generation by β-NAD+e-stimulated granulocytes
The respiratory burst and the production of nitrite in activated neutrophils are known to be regulated by the [Ca2+]i [18–20]. Thus we investigated whether the [Ca2+]i increase induced by NAD+e could stimulate superoxide (O2−) and nitrite generation. As shown in Figure 4(A), incubation of neutrophils with β-NAD+e resulted in a concentration-dependent increase in O2− and nitrite production. As low as 1 μM extracellular dinucleotide was effective, similarly to what was observed in the case of the [Ca2+]i (Figure 1), inducing a 30% increase in both O2− and nitrite generation.
Figure 4. β-NAD+e-induced superoxide and nitrite generation in human granulocytes.

(A) Granulocytes were treated for 30 min at 37 °C with β-NAD+e at the concentrations indicated, and O2− generation (white bars) was measured. For nitrite measurements, granulocytes were incubated for 60 min at 37 °C with β-NAD+e at the concentrations indicated (grey bars), or pre-incubated for 90 min with 100 μM 8-Br-cADPR and then stimulated for 60 min at 37 °C with 100 μM β-NAD+e (black bar). For both of these parameters, results are expressed as percentages of control values measured in unstimulated cells, and are the means±S.D. for four different experiments. The basal value of O2− production was 10.67±5.65 ΔmAU (n=15); the basal concentration of nitrite was 0.76±0.43 μM (n=14). (B) Before stimulation with β-NAD+e, granulocytes were pre-incubated at 25 °C as follows: with 100 μM 8-Br-cADPR or with 50 μM ryanodine for 90 min; with 50 μM Cibacron Blue for 20 min; with 5 μM U73122 for 10 min; or with 20 mM nicotinamide for 1 min. Pre-treated and untreated (control) granulocytes were then incubated for 30 min at 37 °C with or without 100 μM β-NAD+e, and the O2− production was measured. Results are expressed as the percentage of inhibition observed with each compound, and are the means±S.D. for at least three experiments.
We then tested the effect on O2− generation of a number of compounds known to interfere at various sites in the signalling cascade triggered by β-NAD+e and resulting in the sustained [Ca2+]i increase. As shown in Figure 4(B), O2− production triggered by 100 μM NAD+e was reduced by 60% upon pre-incubation of granulocytes with 100 μM 8-Br-cADPR, with 50 μM ryanodine, or with the ADP-ribosyl cyclase inhibitors nicotinamide (20 mM) and Cibacron Blue 3G-A (50 μM) [11]. Thus cADPR appears to have a causal role in the NAD+e-induced stimulation of O2− release. The PLC inhibitor U73122 (5 μM) had no effect on NAD+e-stimulated O2− generation, in agreement with what was observed for the [Ca2+]i increase (Table 1).
Pre-incubation of granulocytes with 100 μM 8-Br-cADPR (Figure 4A) or with 50 μM ryanodine also reduced nitrite production by 100 μM β-NAD+e-stimulated cells (by 65% and 61% respectively).
α-NAD+e was as effective as β-NAD+e in increasing nitrite production (results not shown), but it proved to be less effective than β-NAD+e in stimulating O2− release from human neutrophils.
NAD+e stimulates chemotaxis of human granulocytes
Migration of granulocytes in response to chemoattractants is critical to ensure the fast deployment of these cells and of their defensive functions (phagocytosis, O2− and NO production) at the site of inflammation [21,22]. Therefore we investigated whether β-NAD+e, the naturally occurring NAD+ form, can attract granulocytes along a concentration gradient, as would occur at sites of inflammation, where [NAD+]e is expected to increase well above the nanomolar levels present in blood plasma [16]. When different concentrations of β-NAD+e were placed in the bottom well of a chemotaxis chamber, a bell-shaped response was observed, with maximal migration being recorded in response to 10 μM β-NAD+ (Figure 5). α-NAD+e was as effective as β-NAD+e in stimulating cell migration (Figure 5), whereas NADH and NADP+ did not evoke any chemotactic response at the same concentration, in agreement with their failure to evoke a Ca2+ response. The CI calculated for 10 μM β-NAD+ was similar to that observed with the same concentration of ATP, a nucleotide known to behave as a chemoattractant [27].
Figure 5. Chemotaxis of human granulocytes towards different concentrations of α- and β-NAD+.

Migration of granulocytes through 3 μm pore membranes towards a solution containing β-NAD+ (white bars) or α-NAD+ (grey bars), at the concentrations indicated, was measured in ChemoTx chambers and expressed as the CI. Results shown are the means±S.D. from five experiments (P<0.01 for 1 μM NAD+; P<0.0001 for 10 μM NAD+). Granulocytes pre-incubated with 100 μM 8-Br-cADPR for 90 min were also challenged with 10 μM β-NAD+ (black bar) or α-NAD+ (hatched bar). Results shown are the means±S.D. for three experiments (P<0.001 compared with CI values of granulocytes not pre-treated with 8-Br-cADPR).
Pre-incubation of cells with 100 μM 8-Br-cADPR completely abrogated the migration induced by 10 μM α- or β-NAD+ (Figure 5), confirming a major role of the cADPR/[Ca2+]i signalling cascade in the chemotactic response of granulocytes to NAD+e.
DISCUSSION
In a number of mammalian tissues, NAD+e, released through Cx43 hemichannels from ADP-ribosyl cyclase-positive and cADPR-generating stromal cells, has a hormone-like role, regulating specific parenchymal cell functions (smooth myocyte contraction, glutamate release from astrocytes and expansion of haemopoietic progenitors) [1]. In these cell systems, cADPR behaves as a second messenger, and the cADPR-induced [Ca2+]i increase is the ultimate intracellular signal modulating cell functions [1].
In other cell systems, e.g. in blood cells, NAD+e is an agonist of important functional responses by means of mechanisms other than direct, ADP-ribosyl cyclase-catalysed conversion of NAD+e to cADPR. Gerth et al. [17] demonstrated that NAD+e triggers the influx of extracellular Ca2+ in human monocytes. In murine T-lymphocytes, ART-2 mediates ADP-ribosylation of surface proteins, which results in the activation of the P2X7 receptor followed by extensive Ca2+ influx, pore formation and cell death [4,15,16].
The present findings demonstrate for the first time that NAD+e triggers a signalling event in human neutrophils, eventually leading to their activation. Addition of micromolar (1–100 μM) concentrations of NAD+e results in a long-lasting [Ca2+]i increase, which is determined by influx of extracellular Ca2+ (Figure 1). The experimental data are consistent with the following NAD+e-induced cascade of events: (i) activation of adenylate cyclase and a rapid increase in [cAMP]i; (ii) PKA-mediated stimulation of ADP-ribosyl cyclase activity and elevation of the [cADPR]i; and (iii) a sustained [Ca2+]i rise (Figure 6).
Figure 6. Schematic representation of the mechanism of the NAD+e-induced [Ca2+]i increase leading to functional activation of human granulocytes.

Interaction of NAD+e with an unidentified plasma membrane receptor induces activation of adenylate cyclase, overproduction of cAMP, PKA-mediated stimulation of ADP-ribosyl cyclase and an increase in [cADPR]i. Downstream of cADPR, two mechanisms might co-operate to induce the observed increase in the [Ca2+]i: extracellular Ca2+ influx through SOCCs (a) or direct gating of a plasma-membrane Ca2+ channel by cADPR (b) (see the Discussion). The increased [Ca2+]i level stimulates functional responses (overproduction of reactive oxygen species, release of NO and chemotaxis towards NAD+e).
The cADPR-induced [Ca2+]i increase, inhibitable to a large extent (80%) by 8-Br-cADPR and ryanodine, was completely abolished by EGTA (Figure 1B), indicating extracellular Ca2+ influx. A Ca2+ influx triggered by cADPR generation has been documented already in Jurkat T-lymphocytes [31], human monocytes and neutrophils [22], and also in lymphokine-activated killer cells [34]. Specifically, in human neutrophils the engagement of a discrete subset of chemoattractant receptors elicited a significant, 8-Br-cADPR-inhibitable influx of extracellular Ca2+ [22]. How cADPR, an intracellular Ca2+ mobilizer, stimulates Ca2+ influx is still unknown, but two possible mechanisms have been postulated [22,34]: (i) direct opening by intracellular cADPR of a Ca2+ channel on the plasma membrane, and (ii) activation of SOCCs induced by Ca2+ released from RyRs. In the present experimental system, no evidence of an intracellular Ca2+ release could be detected in the presence of millimolar concentrations of extracellular EGTA (Figure 1). On the other hand, the negative effect of the SOCC inhibitor SFK96365 on the [Ca2+]i increase in NAD+-stimulated neutrophils (Table 1) suggests involvement of SOCCs in the cADPR-induced Ca2+ influx.
Kolisek et al. [41] have demonstrated that the transient receptor potential channel of the melastatin-related family TRPM2, a Ca2+ channel known to be activated by ADPR [42,43] and to be expressed in human granulocytes [44], can be gated by cADPR itself, and that cADPR and ADPR synergize in activating this channel. Activation of CD38 in NAD+e-stimulated granulocytes is indeed expected to enhance the intracellular conversion of cADPR into ADPR via its cADPR hydrolase activity. Thus TRPM2 may also be involved in the NAD+e-induced Ca2+ influx in human granulocytes under conditions where cADPR metabolism is up-regulated. The fact that the cADPR antagonists 8-Br-cADPR and ryanodine, although significantly inhibiting the Ca2+ rise (80%), do not completely abrogate it, in contrast with what was observed with EGTA, suggests the contribution of other cADPR-independent mechanisms of extracellular Ca2+ influx to the global [Ca2+]i rise triggered by NAD+e in granulocytes. A role for the P2X7 purinoceptor, also expressed on human granulocytes, in mediating influx of extracellular Ca2+ has been proposed recently in NAD+e-stimulated T-lymphocytes through the use of purinoceptor antagonists [4,15,16].
The [Ca2+]i elevation induced by micromolar NAD+e concentrations results in functional activation of granulocytes. Thus as low as 1 μM NAD+e stimulates superoxide and NO generation, and behaves as a chemoattractant. Interestingly, we observed a significant chemotactic effect in response to low (0.1–10 μM) NAD+e concentrations, and no cell migration in response to 100 μM NAD+e. A comparable analysis of different responses to ATPe was recently demonstrated by Idzko et al. [45] in immature dendritic cells: thus ATPe triggered a concentration-dependent Ca2+ signal, in contrast with a bell-shaped chemotactic response. Since no substantial differences were observed between α-NAD+e and β-NAD+e in eliciting the chemotactic response, the weak effect recorded at the highest concentration cannot be related to the extracellular generation of the β-NAD+e metabolites cADPR and ADPR. The biphasic pattern of chemotaxis seems rather to represent an intrinsic feature of this granulocytic response to various agonists [45].
The high sensitivity of granulocytes to the chemotactic attraction of NAD+ (with 10 μM eliciting the maximal response; Figure 5) supports the view of a role of the dinucleotide in recruiting granulocytes at sites of infection/inflammation, where NAD+e levels are expected to be enhanced [16].
The fact that α-NAD+ induced the same responses (in terms of [Ca2+]i and [cAMP]i increases, NO and reactive oxygen species generation, and chemotaxis) as β-NAD+, whereas β-NADH was completely inactive, may suggest that an oxidized nicotinamide moiety is important to induce these functional effects in human granulocytes. In fact, α-NAD+ has been observed to behave similarly to β-NAD+ in other experimental systems, either binding to an oxidized NAD+-specific site on the renal brush-border membrane from rat kidney [46] or functioning as a substrate or as a competitive inhibitor of NAD+-dependent dehydrogenases from mammalian, yeast and bacterial sources [47,48]. Thus apparently α-NAD+ can specifically interact with NAD+-binding sites on some receptors and dehydrogenases. The physiological significance of these observations remains to be elucidated.
The present results indicate that NAD+e can play an as-yet-unrecognized role as a pro-inflammatory cytokine, potentially activating the natural defences of the host against pathogens. Therefore this multifaceted dinucleotide is able to have a significant involvement in innate immunity, triggering activation of granulocytes via cADPR overproduction and sustained [Ca2+]i elevation.
Acknowledgments
This work was supported in part by grants from the AIRC (Associazione Italiana per la Ricerca sul Cancro), the Italian Ministry of Education, University and Scientific Research (MIUR-PRIN 2003, MIUR FIRB RBAUO19A3C and MIUR FIRB RBNE01ERXR), the University of Genova and Fondazione Cassa di Risparmio di Genova e Imperia.
References
- 1.De Flora A., Zocchi E., Guida L., Franco L., Bruzzone S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N. Y. Acad. Sci. 2004;1028:176–191. doi: 10.1196/annals.1322.021. [DOI] [PubMed] [Google Scholar]
- 2.Verderio C., Bruzzone S., Zocchi E., Fedele E., Schenk U., De Flora A., Matteoli M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001;78:1–13. doi: 10.1046/j.1471-4159.2001.00455.x. [DOI] [PubMed] [Google Scholar]
- 3.Romanello M., Bicego M., Pirulli D., Crovella S., Moro L., D'Andrea P. Extracellular NAD+: a novel autocrine/paracrine signal in osteoblast physiology. Biochem. Biophys. Res. Commun. 2002;299:424–431. doi: 10.1016/s0006-291x(02)02665-7. [DOI] [PubMed] [Google Scholar]
- 4.Seman M., Adriouch S., Scheuplein F., Krebs C., Freese D., Glowacki G., Deterre P., Haag F., Koch-Nolte F. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity. 2003;19:571–582. doi: 10.1016/s1074-7613(03)00266-8. [DOI] [PubMed] [Google Scholar]
- 5.Smyth L. M., Bobalova J., Mendoza M. G., Lew C., Mutafova-Yambolieva V. N. Release of beta-nicotinamide adenine dinucleotide upon stimulation of postganglionic nerve terminals in blood vessels and urinary bladder. J. Biol. Chem. 2004;279:48893–48903. doi: 10.1074/jbc.M407266200. [DOI] [PubMed] [Google Scholar]
- 6.Franco L., Guida L., Bruzzone S., Zocchi E., Usai C., De Flora A. The transmembrane glycoprotein CD38 is a catalytically active transporter responsible for generation and influx of the second messenger cyclic ADP-ribose across membranes. FASEB J. 1998;12:1507–1520. doi: 10.1096/fasebj.12.14.1507. [DOI] [PubMed] [Google Scholar]
- 7.Lee H. C., Walseth T. F., Bratt G. T., Hayes R. N., Clapper D. L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989;264:1608–1615. [PubMed] [Google Scholar]
- 8.Lee H. C., Aarhus R. ADP-ribosyl cyclase: an enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991;2:203–209. doi: 10.1091/mbc.2.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galione A., Lee H. C., Busa W. B. Ca2+-induced Ca2+ release in sea urchin egg homogenates: modulation by cyclic ADP-ribose. Science. 1991;253:1143–1146. doi: 10.1126/science.1909457. [DOI] [PubMed] [Google Scholar]
- 10.Li P. L., Tang W. X., Valdivia H. H., Zou A. P., Campbell W. B. cADP-ribose activates reconstituted ryanodine receptors from coronary arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H208–H215. doi: 10.1152/ajpheart.2001.280.1.H208. [DOI] [PubMed] [Google Scholar]
- 11.Lee H. C. Norwell, MS: Kluwer Academic Publishers; 2002. Cyclic ADP-ribose and NAADP: structures, metabolism and functions. [Google Scholar]
- 12.Galione A., Churchill G. C. Interactions between calcium release pathways: multiple messengers and multiple stores. Cell Calcium. 2002;32:343–354. doi: 10.1016/s0143416002001902. [DOI] [PubMed] [Google Scholar]
- 13.Zocchi E., Usai C., Guida L., Franco L., Bruzzone S., Passalacqua M., De Flora A. Ligand-induced internalization of CD38 results in intracellular Ca2+ mobilization: role of NAD+ transport across cell membranes. FASEB J. 1999;13:273–283. doi: 10.1096/fasebj.13.2.273. [DOI] [PubMed] [Google Scholar]
- 14.Glowacki G., Braren R., Firner K., Nissen M., Kuhl M., Reche P., Bazan F., Cetkovic-Cvrlje M., Leiter E., Haag F., Koch-Nolte F. The family of toxin-related ecto-ADP-ribosyltransferases in humans and the mouse. Protein Sci. 2002;11:1657–1670. doi: 10.1110/ps.0200602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawamura H., Aswad F., Minagawa M., Malone K., Kaslow H., Koch-Nolte F., Schott W. H., Leiter E. H., Dennert G. P2X7 receptor-dependent and -independent T cell death is induced by nicotinamide adenine dinucleotide. J. Immunol. 2005;174:1971–1979. doi: 10.4049/jimmunol.174.4.1971. [DOI] [PubMed] [Google Scholar]
- 16.Krebs C., Adriouch S., Braasch F., Koestner W., Leiter E. H., Seman M., Lund F. E., Oppenheimer N., Haag F., Koch-Nolte F. CD38 controls ADP-ribosyltransferase-2-catalyzed ADP-ribosylation of T cell surface proteins. J. Immunol. 2005;174:3298–3305. doi: 10.4049/jimmunol.174.6.3298. [DOI] [PubMed] [Google Scholar]
- 17.Gerth A., Nieber K., Oppenheimer N. J., Hauschildt S. Extracellular NAD+ regulates intracellular free calcium concentration in human monocytes. Biochem. J. 2004;382:849–856. doi: 10.1042/BJ20040979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hallett M. B., Davies E. V., Campbell A. K. Oxidase activation in individual neutrophils is dependent on the onset and magnitude of the Ca2+ signal. Cell Calcium. 1990;11:655–663. doi: 10.1016/0143-4160(90)90020-u. [DOI] [PubMed] [Google Scholar]
- 19.Park N. H., Han E. S., Lee C. S. The inhibitory effect of ambroxol on respiratory burst, degranulation and cytosolic Ca2+ change in degraded immunoglobulin G-activated neutrophils. Pharmacol. Toxicol. 1999;84:81–87. doi: 10.1111/j.1600-0773.1999.tb00878.x. [DOI] [PubMed] [Google Scholar]
- 20.Lucas R., Alves M., del Olmo E., San Feliciano A., Paya M. LAAE-14, a new in vitro inhibitor of intracellular calcium mobilization, modulates acute and chronic inflammation. Biochem. Pharmacol. 2003;65:1539–1549. doi: 10.1016/s0006-2952(03)00120-5. [DOI] [PubMed] [Google Scholar]
- 21.Partida-Sánchez S., Cockayne D. A., Monard S., Jacobson E. L., Oppenheimer N., Garvy B., Kusser K., Goodrich S., Howard M., Harmsen A., Randall T. D., Lund F. E. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 2001;7:1209–1216. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 22.Partida-Sánchez S., Iribarren P., Moreno-Garcia M. E., Gao J. L., Murphy P. M., Oppenheimer N., Wang J. M., Lund F. E. Chemotaxis and calcium responses of phagocytes to formyl peptide receptor ligands is differentially regulated by cyclic ADP ribose. J. Immunol. 2004;172:1896–1906. doi: 10.4049/jimmunol.172.3.1896. [DOI] [PubMed] [Google Scholar]
- 23.Guida L., Franco L., Zocchi E., De Flora A. Structural role of disulfide bridges in the cyclic ADP-ribose related bifunctional ectoenzyme CD38. FEBS Lett. 1995;368:481–484. doi: 10.1016/0014-5793(95)00715-l. [DOI] [PubMed] [Google Scholar]
- 24.Zocchi E., Daga A., Usai C., Franco L., Guida L., Bruzzone S., Costa A., Marchetti C., De Flora A. Expression of CD38 increases intracellular calcium concentration and reduces doubling time in HeLa and 3T3 cells. J. Biol. Chem. 1998;273:8017–8024. doi: 10.1074/jbc.273.14.8017. [DOI] [PubMed] [Google Scholar]
- 25.Bruzzone S., De Flora A., Usai C., Graeff R., Lee H. C. Cyclic ADP-ribose is a second messenger in the lipopolysaccharide-stimulated proliferation of human peripheral blood mononuclear cells. Biochem. J. 2003;375:395–403. doi: 10.1042/BJ20030556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen H. J., Chovaniec M. E. Superoxide generation by digitonin-stimulated guinea pig granulocytes. A basis for a continuous assay for monitoring superoxide production and for the study of the activation of the generating system. J. Clin. Invest. 1978;61:1081–1087. doi: 10.1172/JCI109007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Virgilio F., Chiozzi P., Ferrari D., Falzoni S., Sanz J. M., Morelli A., Torboli M., Bolognesi G., Baricordi O. R. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood. 2001;97:587–600. doi: 10.1182/blood.v97.3.587. [DOI] [PubMed] [Google Scholar]
- 28.Walseth T. F., Lee H. C. Synthesis and characterization of antagonists of cyclic-ADP-ribose-induced Ca2+ release. Biochim. Biophys. Acta. 1993;1178:235–242. doi: 10.1016/0167-4889(93)90199-y. [DOI] [PubMed] [Google Scholar]
- 29.Bruzzone S., Kunerth S., Zocchi E., De Flora A., Guse A. H. Spatio-temporal propagation of Ca2+ signals by cyclic ADP-ribose in 3T3 cells stimulated via purinergic P2Y receptors. J. Cell Biol. 2003;163:837–845. doi: 10.1083/jcb.200307016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parekh A., Penner R. Store depletion and calcium influx. Physiol. Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 31.Guse A. H., da Silva C. P., Berg I., Skapenko A. L., Weber K., Heyer P., Hohenegger M., Ashamu G. A., Schulze-Koops H., Potter B. V., Mayr G. W. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature (London) 1999;398:70–73. doi: 10.1038/18024. [DOI] [PubMed] [Google Scholar]
- 32.Hashii M., Minabe Y., Higashida H. cADP-ribose potentiates cytosolic Ca2+ elevation and Ca2+ entry via L-type voltage-activated Ca2+ channels in NG108-15 neuronal cells. Biochem. J. 2000;345:207–215. [PMC free article] [PubMed] [Google Scholar]
- 33.Kiselyov K., Shin D. M., Shcheynikov N., Kurosaki T., Muallem S. Regulation of Ca2+-release-activated Ca2+ current (Icrac) by ryanodine receptors in inositol 1,4,5-trisphosphate-receptor-deficient DT40 cells. Biochem. J. 2001;360:17–22. doi: 10.1042/0264-6021:3600017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rah S. Y., Park K. H., Han M. K., Im M. J., Kim U. H. Activation of CD38 by interleukin-8 signaling regulates intracellular Ca2+ level and motility of lymphokine-activated killer cells. J. Biol. Chem. 2005;280:2888–2895. doi: 10.1074/jbc.M409592200. [DOI] [PubMed] [Google Scholar]
- 35.Merritt J. E., Armstrong W. P., Benham C. D., Hallam T. J., Jacob R., Jaxa-Chamiec A., Leigh B. K., McCarthy S. A., Moores K. E., Rink T. J. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 1990;271:515–522. doi: 10.1042/bj2710515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munshi C. B., Graeff R., Lee H. C. Evidence for a causal role of CD38 expression in granulocytic differentiation of human HL-60 cells. J. Biol. Chem. 2002;277:49453–49458. doi: 10.1074/jbc.M209313200. [DOI] [PubMed] [Google Scholar]
- 37.Morita K., Kitayama S., Dohi T. Stimulation of cyclic ADP-ribose synthesis by acetylcholine and its role in catecholamine release in bovine adrenal chromaffin cells. J. Biol. Chem. 1997;272:21002–21009. doi: 10.1074/jbc.272.34.21002. [DOI] [PubMed] [Google Scholar]
- 38.Zocchi E., Carpaneto A., Cerrano C., Bavestrello G., Giovine M., Bruzzone S., Guida L., Franco L., Usai C. The temperature-signaling cascade in sponges involves a heat-gated cation channel, abscisic acid, and cyclic ADP-ribose. Proc. Natl. Acad. Sci. U.S.A. 2001;98:14859–14864. doi: 10.1073/pnas.261448698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boittin F. X., Dipp M., Kinnear N. P., Galione A., Evans A. M. Vasodilation by the calcium-mobilizing messenger cyclic ADP-ribose. J. Biol. Chem. 2003;278:9602–9608. doi: 10.1074/jbc.M204891200. [DOI] [PubMed] [Google Scholar]
- 40.Xie G. H., Rah S. Y., Kim S. J., Nam T. S., Ha K. C., Chae S. W., Im M. J., Kim U. H. ADP-ribosyl cyclase couples to cyclic AMP signaling in the cardiomyocytes. Biochem. Biophys. Res. Commun. 2005;330:1290–1298. doi: 10.1016/j.bbrc.2005.03.114. [DOI] [PubMed] [Google Scholar]
- 41.Kolisek M., Beck A., Fleig A., Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 42.Perraud A. L., Fleig A., Dunn C. A., Bagley L. A., Launay P., Schmitz C., Stokes A. J., Zhu Q., Bessman M. J., Penner R., et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature (London) 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 43.Sano Y., Inamura K., Miyake A., Mochizuki S., Yokoi H., Matsushime H., Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- 44.Heiner I., Eisfeld J., Lückhoff A. Role and regulation of TRP channels in neutrophil granulocytes. Cell Calcium. 2003;33:533–540. doi: 10.1016/s0143-4160(03)00058-7. [DOI] [PubMed] [Google Scholar]
- 45.Idzko M., Dichmann S., Ferrari D., Di Virgilio F., la Sala A., Girolomoni G., Panther E., Norgauer J. Nucleotides induce chemotaxis and actin polymerization in immature but not mature human dendritic cells via activation of pertussis toxin-sensitive P2Y receptors. Blood. 2002;100:925–932. doi: 10.1182/blood.v100.3.925. [DOI] [PubMed] [Google Scholar]
- 46.Braun-Werness J. L., Jackson B. A., Werness P. G., Dousa T. P. Binding of nicotinamide adenine dinucleotide by the renal brush border membrane from rat kidney cortex. Biochim. Biophys. Acta. 1983;732:553–561. doi: 10.1016/0005-2736(83)90231-6. [DOI] [PubMed] [Google Scholar]
- 47.Oppenheimer N. J. The stereospecificity of oxidation of alpha-[4R-2H]NADH by dehydrogenases. J. Biol. Chem. 1986;261:12209–12212. [PubMed] [Google Scholar]
- 48.Uotila L., Mannervik B. A steady-state-kinetic model for formaldehyde dehydrogenase from human liver. A mechanism involving NAD+ and the hemimercaptal adduct of glutathione and formaldehyde as substrates and free glutathione as an allosteric activator of the enzyme. Biochem. J. 1979;177:869–878. doi: 10.1042/bj1770869. [DOI] [PMC free article] [PubMed] [Google Scholar]