

Abstract
The minimal signalling unit for tyrosine kinase receptors is two protomers dimerized by one or more ligands. However, it is clear that maximal signalling requires the formation of larger complexes of many receptors at discrete foci on the cell surface. The biological interactions that lead to this are likely to be diverse and have system specific components. In the present study, we demonstrate that, in the FGF (fibroblast growth factor)–FGFR (FGF receptor) system, multimers of the minimal complex composed of two FGF1 and two FGFR2 protomers can form on a single chain of the co-receptor heparin. Using size-exclusion chromatography, we show that two complexes can form on heparin chains as small as 16 saccharide units. We also show by MS that discrete complexes containing exactly two copies of the minimal signalling unit are formed. However, the doublet of complexes appears to be less co-operative than the formation of the 2:2:1 FGF1:FGFR2:heparin complex, suggesting that this mechanism is one of a number of weaker interactions that might be involved in the formation of a focal complex on the cell surface.
Keywords: fibroblast growth factor (FGF), focal complex, heparin, heparan sulphate, receptor clustering
Abbreviations: FGF, fibroblast growth factor; FGFR, FGF receptor; HS, heparan sulphate
INTRODUCTION
Members of the FGF (fibroblast growth factor) family of proteins are found in all animals from the nematode worm Caenorhabditis elegans to mammals, and are required for the correct development of almost all tissues, organs and limbs [1,2]. FGFs are expressed by a wide range of cells to induce a signal in neighbouring or distant cells, modulating a spectrum of cell behaviours. Responsive cells express at least one member of the FGFR (FGF receptor) family of tyrosine kinase receptors. HS (heparan sulphate), a polysulphated glucosaminoglycan found associated with virtually all mammalian cells and the extracellular matrix, binds to both FGFs and FGFRs, and is required for the full activity of the FGF signalling cascade [3].
The extracellular domains of mammalian FGFRs are composed of two or three Ig-like superfamily domains (Ig domains I, II and III). Vertebrates have up to five FGFR paralogues [1,4]. FGFRs 1–4 have an intracellular tyrosine kinase domain of the split kinase family, whereas the intracellular domain of FGFR5 is unrelated to other signalling domains. Studies of FGFRs expressed in vivo [5] and in vitro [6] have shown that receptors lacking Ig domain I are fully competent to bind to FGFs and have enhanced FGF binding activities, suggesting that the first domain may have some inhibitory effect on FGF binding [7,8]. Crystallographic studies of FGF–FGFR complexes have confirmed that the FGF binding activity is located in Ig domains II and III, with interactions with Ig domain III providing specificity [8–13]. HS binding is also located in Ig domain II, in a long loop containing many positively charged residues [14].
HS consists of a polysaccharide backbone of 100–300 saccharide units in length composed of repeating disaccharides of N-acetylglucosamine and uronic acid [15,16], which is physiologically attached to cell-surface proteins of the syndecan and glypican families. HS involvement has been implicated in a variety of signalling systems [17,18]. The HS chains are altered by a range of enzymes that (i) replace N-acetyl with N-sulphate, (ii) epimerize glucuronic acid to iduronic acid, (iii) add sulphate groups to the 2-O position of iduronic acid, and (iv) add sulphate groups to the 6-O, and more rarely, the 3-O positions of glucosamine. Although these modifications are carried out incompletely, each activity is co-ordinated with previous steps in the biosynthesis, leading to the formation of domains of up to 12–14 saccharide units with heavy modification (S-domains) separated by stretches of 14–18 saccharide units with intermediate or low levels of modification [19]. The S-domains are believed to be the principal binding sites for heparin-binding growth factors. The overall level of modification is determined, in an as yet undefined manner, by the enzyme complement of the expressing cells. Heparin is a mast-cell-derived chemical analogue of the S-domains of HS. Fragments of heparin produced by chemical or enzymatic scission have been used for most experiments investigating the interaction of FGFs with N-sulphated glycosaminoglycans. Heparin is sufficient to replace HS for FGF signalling on cells where HS has been removed by chlorate treatment [3,20,21]. However, it should be emphasized that heparin is more highly sulphated than the majority of the S-domains in HS which have complex and highly variable sulphation patterns [22].
Deeper insights into the precise arrangement of the FGF–FGFR–HS complex have been provided by a series of crystal structures. These include the structures of seven members of the FGF family [23–29], a structure of two FGF1 molecules bound to heparin [30] and of FGF–FGFR heterodimers [8,10–13]. Two structures have been solved for complexes of the two prototypic FGFs, FGF1 and FGF2, in complex with FGFRs 1 and 2 and with heparin [9,31]. These structures suggest two different architectures for the complex with the dimerized receptor. Further crystallographic [13] and biochemical [32,33] evidence suggests that both of these architectures are biologically valid and that they are likely to play a role in the physiological signalling of FGFR.
The lack of certainty as to whether one or both of these complexes play a role in the physiological signalling of the FGFs highlights one of the major shortcomings of our understanding of this system. The majority of studies focused on the role of glucosaminoglycans in the FGF–FGFR complex have examined the minimum length of heparin that is required to form an active signalling complex. A recent trend has been to investigate also the minimum sulphation patterns required for FGF binding to heparin. However, evidence is growing that signalling complexes tend to involve a considerable number of receptor molecules clustered at discrete foci. This has been demonstrated for a wide range of receptor systems [34–37].
As the physiological HS molecules are 100–300 saccharide units long [15], the genuine FGFR interaction is likely to involve these long saccharide chains, rather than the minimal units that have hitherto been studied. Therefore we formed complexes of FGF1 and FGFR2 with intact HS chains and long heparin fragments. We observed multiple complexes of FGF1 and FGFR2 forming upon saccharide chains of physiological length. Using MS and analytical ultracentrifugation, we established that four copies each of FGF1 and FGFR2 can form upon a single heparin 24-mer in a reproducible and stable fashion. These data establish that multiple FGF complexes can form on a single saccharide chain, suggesting that multimerization upon HS chains at the cell surface could be one mechanism by which FGF signalling focal complexes are built.
EXPERIMENTAL
Chemicals
Chemicals were obtained from Sigma or from Melford Laboratories.
Preparation of materials
FGF1 and the ligand-binding domain of FGFR2-IIIc were purified separately as described [9]. Size-defined heparin fragments were prepared from porcine mucosa by controlled heparin lyase I depolymerization (Flavobacterium heparinum; EC 4.2.2.7), followed by size-exclusion chromatography as described previously [38].
Size-exclusion chromatography
Size-exclusion chromatography was performed using a Superdex 200 HR column (Amersham Biosciences) of height 30 cm and diameter 10 mm. Samples were eluted isocratically with a buffer of 10 mM Hepes (pH 7.2) and 150 mM NaCl at a flow rate of 0.5 ml/min. All experiments were run on an Akta Explorer chromatography system (Amersham Biosciences), and the absorbance at 280 and 235 nm was recorded. The components of each experiment were added in the order: buffer, heparin, FGF and FGFR and mixed only after addition of the final component. To calculate apparent molecular masses, 100 μg each of thyroglobulin (667 kDa), ferritin (440 kDa), catalase (252 kDa), aldolase (168 kDa), BSA (67 kDa), ovalbumin (44 kDa), chymotrypsinogen (25 kDa) and ribonuclease A (14.2 kDa) (Amersham Biosciences) were applied to the same column and an identical elution was performed. The molecular mass was calculated according to the formula:
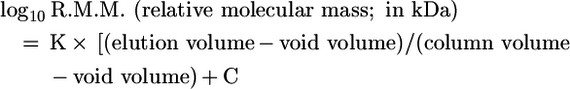 |
Values for K, C and the void volume were calculated from multiple values of the standards. Complex spectra were deconvoluted by assuming that they are the sum of two Gaussian curves. The parameters for the Gaussian curves were optimized using least-squares fitting over the range of the peak, restraining σ to 0.5.
Analytical ultracentrifugation
Samples for analytical ultracentrifugation were concentrated using a Vivaspin 6 centrifugal concentrator with a molecular mass cut-off of 5 kDa (Vivascience). Sedimentation velocity experiments were performed using an Optima XL-I (Beckman Coulter), with a double Epon fitted centrepiece. Sample volumes of 400 μl and reference volumes of 410 μl were used. The sample was centrifuged at 243500 g, with data taken every 4 min for 2 h. Data were processed using SEDFIT [39].
MS
Samples for MS were prepared using the size-exclusion chromatography system in 0.2 M ammonium acetate. After elution, fractions at the peak corresponding to the complex were isolated and pooled. The samples were extensively diafiltered to remove non-volatile ions using a concentrator as above.
Mass spectra were collected on an LCT and a QToF-2 mass spectrometer modified for the transmission and isolation of high-mass ions [40], which is equipped with a nanoflow Z-spraysource (Micromass). Nano-ESI capillaries were prepared in-house from borosilicate glass tubes of 1 mm outer diameter and 0.78 mm inner diameter (Harvard Apparatus) using a Flaming/Brown P-97 micropipette puller (Sutter Instruments), and gold-coated using an SEM sputter coater (Polaron). The capillary tips were cut under a stereo-microscope to give inner diameters of 1–5 μm, and typically 2–3 μl of solution was loaded for sampling. The pressures and accelerating potentials in the mass spectrometer were adjusted to strip away buffer adducts while preserving non-covalent interactions. The following experimental parameters were typically used (positive ion mode): capillary voltage, 1.7 kV; cone gas, 100 litres/h; sample cone, 90–200 V; extractor cone, 0–10 V; collision energy, 4 V; ion transfer stage pressure, 8.0×10−3 mbar; quadrupole analyser pressure, 1.8×10−5 mbar; ToF (time of flight) analyser pressure, 4.7×10−7 mbar.
External calibration of the spectra was achieved using solutions of caesium iodide. Data acquisition and processing were done using the MassLynx software (Micromass). All spectra are shown with minimal smoothing and without background subtraction.
RESULTS
FGF1 and FGFR2 bind to intact HS chains to form very-large-molecular-mass species
Full-length HS from porcine mucosa was split into three fractions according to the level of sulphation. The high- and low-sulphation fractions were added to FGF1 and FGFR2 and the complexes produced were analysed by size-exclusion chromatography. Both samples formed very large complexes, with a broad peak indicating heterogeneity in the complexes (Figure 1A). The size-exclusion peaks were symmetrical, indicating that the peak value can be used as a mean. The average sizes of these complexes were 465 kDa and 296 kDa respectively, for the highest and lowest sulphation levels. This suggests that multimers of the 2:2 FGF1:FGFR2 complex are forming on these HS chains.
Figure 1. Multiple complexes of FGF1 and FGFR2 form on long saccharide chains.

(A) Size-exclusion chromatography of long heterogeneous HS fragments. HS, FGF1 and FGFR2 (10 nmol of each) were mixed, incubated for 10 min and then applied on to a 24 ml Superdex 200 column. The absorbance of the eluant at 280 nm was measured. Results of a 2:2:1 FGF:FGFR:heparin decamer complex [32] and FGF1 are shown for comparison. (B) Size-exclusion chromatography of heparin fragments of intermediate length. Saccharide (5 nmol of each), FGF1 (10 nmol) and FGFR2 (10 nmol) were mixed and treated as above. The 2:2:1 FGF:FGFR:heparin decamer result is again shown for comparison. Figures were prepared using Microsoft Excel. (C) Acrylamide gel confirms that both FGF1 and FGFR2 bind to heparin fragments. The 24-mer heparin fragment (20 nmol) was mixed with 40 nmol of FGF1 and FGFR2. The mixture was incubated for 10 min and then added on to a 24 ml Superdex 200 column. Fractions (1 ml) were collected after a delay of 1.5 ml. Samples (20 μl) from the elution peak (fractions 10–14) were applied to a 15% (w/v) acrylamide gel, with standard markers. The gel was stained with Coomassie stain.
Higher-order FGF:FGFR complexes form on heparin fragments of intermediate length
To investigate these multimeric complexes further, we examined the effects on complex formation of using fragments of heparin of intermediate length. We prepared complexes with fractions of heparin with average lengths of 16, 20 and 24 saccharide units. Sufficient heparin was added for one heparin molecule per two molecules of FGF1 and FGFR2. Size-exclusion of these complexes showed an intriguing pattern (Figure 1B and Table 1). Each heparin sample had two discrete peaks superimposed on to one another. Deconvolution of the spectra into the component peaks (see Supplementary Figure 1 at http://www.BiochemJ.org/bj/393/bj3930741add.htm) demonstrated that the 24-mer sample consisted predominantly of a large complex (Complex A), with a small amount of a complex closer in size to the 2:2:1 FGF:FGFR:heparin complex; the 20-mer had approximately equal amounts of a species slightly smaller than Complex A and the lower-molecular-mass species, whereas the 16-mer had predominantly the lower-molecular-mass species. The increase in the apparent mass of Complex A and the 2:2:1 FGF:FGFR:heparin complex, with the change in heparin length, is likely to be due to an increase in the Stokes' radius caused by heparin extending beyond the protein complex and a greater separation between components in the case of Complex A. The presence of two discrete peaks in all of these samples suggests that these two species are qualitatively different and that the apparent shift in mass between the 12-mer and 24-mer cannot be due to the effect of overhanging heparin on the Stokes' radius. Analyses of fractions corresponding to the larger peak show approximately equal amounts of FGFR2 and FGF1 (Figure 1C), suggesting a 1:1 stoichiometry of FGF1:FGFR2 on this heparin sample, consistent with complexes observed previously.
Table 1. Peak elution volumes and calculated molecular masses for FGF1, FGFR2 and heparin complexes.
The elution volume for each complex was taken as the volume at which the reading at 280 nm was highest. For the heparin 16-, 20- and 24-mer samples, the observed chromatogram was deconvoluted into the two overlapping peaks (see Supplementary Figure 1 at http://www.BiochemJ.org/bj/393/bj3930741add.htm). Values are the average of at least two experiments. Values for FGFR2 and FGF1 (taken from [32]) are given for comparison.
| Elution volume | Apparent molecular | Molecular mass of proposed | |
|---|---|---|---|
| Heparin sample | (ml) | mass (kDa) | majority species (kDa) |
| 10-Mer | 14.0 | 107 | 84 (2:2:1 FGF:FGFR:heparin) |
| 12-Mer | 14.0 | 104 | 84 (2:2:1 FGF:FGFR:heparin) |
| 16-Mer | |||
| Peak 1 | 13.0 | 171 | 166 (4:4:1 FGF:FGFR:heparin) |
| Peak 2 | 13.8 | 116 | 85 (2:2:1 FGF:FGFR:heparin) |
| 20-Mer | |||
| Peak 1 | 12.8 | 189 | 167 (4:4:1 FGF:FGFR:heparin) |
| Peak 2 | 13.7 | 123 | 86 (2:2:1 FGF:FGFR:heparin) |
| 24-Mer | |||
| Peak 1 | 12.5 | 227 | 168 (4:4:1 FGF:FGFR:heparin) |
| Peak 2 | 13.5 | 138 | 87 (2:2:1 FGF:FGFR:heparin) |
| FGFR2 | 15.9 | 39.9 | 25 |
| FGF1 | 17.7 | 16.3 | 16 |
The higher-order complexes can be competed out by the use of excess heparin
The interactions required to form the 2:2:1 FGF1:FGFR2:heparin complex seem to be co-operative, as the radius of the observed complex was unaffected even when an excess of heparin was added to the sample (Figure 2A). To test whether the interactions involved in the formation of Complex A are similarly co-operative, further experiments were performed with either excess or insufficient heparin 24-mer (Figure 2B). When insufficient heparin was present, the excess FGF1 and FGFR2 did not form a complex, confirming that the complexes formed were specific. However, when excess heparin was present, the peak observed remained symmetrical, but the average mass was reduced. This suggests that Complex A is non-co-operative and will form wherever there is insufficient heparin. The mass of the observed peak decreased as greater excesses of heparin were added. The addition of an excess of 10-mer heparin from bovine lung (which showed a higher level of sulphation; Figure 2C) resulted in the formation of species with a mass similar to that of the complex prepared as described by Pellegrini et al. [9]. This confirms that Complex A is less co-operative than the 2:2:1 FGF1:FGFR2:heparin complex.
Figure 2. Effects of excess heparin on the formation of larger FGF–FGFR complexes.

Mixtures of heparin, FGF1 and FGFR2 were incubated for 10 min and then applied on to a 24 ml Superdex 200 column. The absorbance of the eluant at 280 nm was measured. (A) Heparin decamer (5 or 25 nmol) was mixed with 10 nmol each of FGF1 and FGFR2. (B) FGF1, FGFR2 and heparin 24-mer or 12-mer were mixed in the amounts shown (in nmol). (C) FGF1, FGFR2, heparin 24-mer and heparin decamer were mixed in the amounts show (in nmol). The result obtained using 5 nmol of heparin in (A) is shown for comparison.
Analytical ultracentrifugation confirms that higher-order complexes are present
Following these observations, a sample of Complex A with twice the required concentration of heparin 24-mer (Figure 2B) was characterized by analytical ultracentrifugation. This analysis (Figure 3) shows four complexes at significant abundances. These correspond to masses of 34, 64, 98 and 146 kDa. It is notable that the abundances of the smaller peaks were approximately equal, with a lower abundance for the 146 kDa peak.
Figure 3. Analytical ultracentrifugation of the larger FGF–FGFR–heparin complex.

FGF1, FGFR2 and heparin 24-mer (40 nmol of each) were mixed and the complex purified by size-exclusion chromatography as described in the legend for Figure 1. The purified sample was concentrated to 0.5 mg/ml and centrifuged at 243500 g for 60 min. Scans were taken every 4 min, starting at 12 min. (A) Raw data from analytical ultracentrifugation. Inset, residuals from the calculated solution. (B) Continuous sedimentation coefficient distribution for the calculated solution. (C) Continuous mass distribution for the calculated solution.
MS confirms that two 2:2 FGF1:FGFR2 complexes bind to the heparin 24-mer
The FGF1–FGFR2–heparin 24-mer complex was also stable when size-exclusion chromatography was performed using an ammonium acetate buffer (results not shown). The purified complex was analysed by MS (Figure 4). This shows three significant species (Table 2), which correspond to complexes of FGF:FGFR:heparin with stoichiometries of 1:1:1 (Species A), 2:2:1 (Species B) and 4:4:1 (Species C).
Figure 4. MS of larger FGF–FGFR–heparin complexes.

FGF1, FGFR2 and heparin 24-mer (40 nmol of each) were mixed and prepared as described in the legend to Figure 1 in 0.2 M ammonium acetate. The spectrum shows three major series of ions, corresponding to a 1:1:1 FGF:FGFR:heparin complex (A), a 2:2:1 FGF:FGFR:heparin complex (B) and a 4:4:1 FGF:FGFR:heparin complex (C).
Table 2. Theoretical molecular masses for the species observed in MS of the larger FGF:FGFR:heparin complexes compared with the masses for the principal component measured experimentally.
Predicted masses of the heparin 24-mer assume that the saccharide is fully sulphated at the N-, 2-O- and 6-O-positions.
| Complex stoichiometry | ||
|---|---|---|
| (FGF:FGFR:heparin) | Theoretical mass (Da) | Measured mass (Da) |
| 1:1:1 | 47196 | 47040±20 |
| 2:2:1 | 87476 | 87741±62 |
| 4:4:1 | 168036 | 167792±77 |
DISCUSSION
Multimers of the FGF1–FGFR2 complex apparently form on long saccharide chains
When full-length HS chains are added to FGF1 and FGFR2, the complexes observed are extremely large (up to 500 kDa; Figure 1A). It is not clear how many FGF1–FGFR2 pairs are assembled upon the HS; however, this appears to represent an extremely large aggregation of protein and saccharide. From this, it is clear that the binding of multiple units of the FGF1–FGFR2 complex observed by Pellegrini et al. [9] upon a single heparin chain is not prohibited. The use of shorter fragments of heparin, however, leads to the formation of complexes of a discrete size. The 16-mer of heparin forms two complexes (Figure 1B), one approximately the same as is observed with a 12-mer (corresponding to the 2:2:1 FGF1:FGFR2:heparin complex observed previously [9,32]) and one larger complex (Complex A). However, only a small proportion of the heparin chains is competent to form Complex A. More of the species from the 20-mer fraction are competent to form this larger complex, binding approximately half of the FGF1–FGFR2 pairs. When a 24-mer is used, the vast majority of the protein forms complexes with a molecular mass predicted to be 227 kDa. These observations suggest that some of the longer heparin species contain sufficient modifications (i.e. additions of sulphate groups to the heparin chain and epimerizations of the uronic acids) for two FGF1–FGFR2 complexes to form on them (Figure 5B). The data suggest that the minimum length required for this to occur could be as few as 16–18 saccharide units. The inability of all of the 20-mer heparin chains to carry out this function suggests that the pattern of modifications is likely to be significant in determining the capacity for the binding of multiple complexes. The 24-mer fraction appears to be strongly competent to form two complexes per chain.
Figure 5. Models of higher-order FGF–FGFR–saccharide complexes in solution and on the cell surface.

(A) Summary of in vivo and experimental species. Eukaryotic cells predominantly express FGFRs with three extracellular Ig domains and an intracellular tyrosine kinase (TK). HS chains are attached to glypican (or syndecan) proteins. For the present study, Ig domains II and III of FGFR2 and FGF1 were expressed. (B) Upper panel, representation of the complex of two FGF–FGFR pairs dimerized on a heparin decamer (based on [9], with Ig domain conformation modelled from [12]). Locations of Ig domains II and III are marked. Lower panel, model of four FGF–FGFR pairs associated upon a heparin 16-mer formed by the formation of two copies of the complex shown above upon contiguous binding sites on a single chain. One copy (rear) of the FGF1–FGFR2–heparin complex has proteins shaded darker. (C) Models of cell surface formation of multimeric FGFR complexes on a single chain of HS. Left-hand panel, two complexes binding to the regions flanking an S-domain. Right-hand panel, two complexes binding to adjacent S-domains separated by regions of intermediate sulphation and a short low-sulphation region.
The species in these fractions are derived from the partial enzymatic cleavage of heparin. As heparin lyase I acts upon GlcNS(±6S)–HexA(2S) linkages (where GlcNS is N-sulphated glucosamine, 6S is 6-O-sulphate, HexA is hexuronic acid, and 2S is 2-O-sulphate), it can cleave between most disaccharide units within the heparin polymer chain. We used sized fractions isolated from an incomplete endolytic cleavage of heparin (approx. 10% of maximum depolymerization). The internal sequences of the fragments will be dominated by the trisulphated disaccharide unit GlcNS(6S)–IdoA(2S) (where IdoA is L-iduronic acid) which comprises approx. 80% of the porcine mucosal heparin chain [41]. Variations in sequence that arise from differences in position and frequency of di- and mono-sulphated units could influence the organization and stability of multiprotein complexes that form on the saccharide backbone. In the case of the shorter heparin fragments, the FGF1–FGFR2 complexes will be in close proximity with a greater potential for steric clashes. In these cases, the precise pattern of the heparin modifications will be most important. There will be few, or only one, possible sites on which the two complexes can assemble and, if one of these sites lacks the required modifications, the complex is unlikely to form. Shorter heparin fragments will place stricter requirements for precise modifications.
The results with excess heparin suggest that these complexes are not strongly co-operative (Figure 2). Whereas excess heparin will not lead to significant breakdown of the 2:2:1 FGF1:FGFR2:heparin complex, an excess of heparin leads to a reduction in the apparent molecular mass of Complex A. This suggests that there is a low energetic gain in forming the second FGF1–FGFR2 complex on heparin in this manner. It may be that there are still some steric hindrances in the case of the 24-mer and that the excess heparin allows more favourable formation of complexes. Alternatively, it may be that there are few interactions favouring the formation of the second complex on the same heparin molecule as the first, leading to the formation of a distribution of complexes. This contrasts with the formation of the 2:2:1 FGF1:FGFR2:heparin complex (observed by Pellegrini et al. [9]), where there is very little protein–protein interaction between the two FGF1–FGFR2 dimers. The binding of heparin to an FGF1–FGFR2 dimer imposes a considerable restriction upon the motion of the heparin. Whereas the sugars are largely free to rotate in solution, the intimate binding of the sulphate groups to FGF1 and FGFR2 necessitates that the conformational flexibility of the heparin is considerably reduced. Therefore we suggest that an entropic penalty for the loss of heparin flexibility is paid for in binding one FGF1–FGFR2 heterodimer, so the binding of a second heterodimer on the opposite face of the same heparin is likely to be more favoured than binding to another heparin molecule. This is also suggested by the finding that breaking the ring structure of one sugar creates an inhibitor of FGF signalling and angiogenesis [42]. In the case of the double complexes of Complex A, there is no equivalent loss of flexibility further along the heparin molecule from the binding of the first 2:2:1 FGF1:FGFR2:heparin complex.
MS and analytical ultracentrifugation establish that the species observed in size-exclusion chromatography represent higher-order complexes
The identity of Complex A was confirmed by the results of MS and analytical ultracentrifugation. The analytical ultracentrifugation results suggest that there are four significant species (Figure 3), including one of a higher mass than the 2:2:1 FGF1:FGFR2:heparin complex described by crystallography [9]. The mass of this larger species is approx. 150 kDa, which corresponds approximately to a complex with three or four copies of each of the protein units. However, the precise composition of the complexes, and in particular the stoichiometry with respect to heparin, cannot be deduced from the analytical ultracentrifugation data as the resolution is not high enough.
MS results give more accurate masses for three species (Figure 4 and Table 2). The observed peaks are somewhat broad: this is not unexpected, given that heparin samples from porcine mucosa are somewhat heterogeneous in their sulphation and that the heparin component of this sample is likely to show some variability in length. Nevertheless, the level of accuracy observed is extremely high and gives a firm indication of the masses of the principal components of the sample. The masses obtained are all within a few hundred Daltons of the predicted values for complexes with a stoichiometry of 1:1:1, 2:2:1 and 4:4:1 of FGF1:FGFR2:heparin. The lower observed molecular masses for the complexes are not unexpected: porcine mucosal heparin is typically approx. 81.9% sulphated [41], whereas the predicted values are for 100% sulphation.
The most significant result from the MS is the identification of a species with an average mass of 167792 Da. This strongly suggests that the species is a 4:4:1 FGF1:FGFR2:heparin complex, as the predicted mass for such a complex is just 244 Da larger at 168036 Da. The possibility of two heparin chains being present is excluded, as the increase in mass associated with this would be 6–7 kDa. This result firmly establishes that two FGF1–FGFR2 complexes can form upon a single saccharide chain of sufficient length. This suggests a mechanism by which clusters of FGFRs might form on the cell surface in response to exposure to an FGF signal (Figure 5C).
The masses of the principal component of the 4:4:1 FGF1:FGFR2:heparin complex observed suggest that the heparin chains that are involved in these interactions will be very sulphate rich. The average mass for the 4:4:1 complex is close to that predicted for full sulphation, suggesting that heparin fragments with higher sulphation levels are selected for binding in these experiments. The difference between the average observed and the expected masses for the 4:4:1 complex is 244 Da. This could be accounted for by the loss of, on average, three sulphates (change of 79 Da per sulphate group gained or lost). The mean sulphation of a heparin 24-mer is 29–30 sulphates (out of the possible 36) [41], suggesting that there may possibly be an enrichment of higher sulphated species in Complex A. It is possible that the true loss of sulphation will be higher than this, as the species observed may also include small amounts of bound solvent. The data suggest that, of the 36 sulphate groups that could be accommodated on a fully sulphated heparin fragment of 24 saccharide units (excluding the possibility of the rare 3-O-sulphate group), the majority must be present in the species that form the 4:4:1 complex.
Conclusions and perspective
Our data suggest that longer fragments of heparin are capable of forming larger complexes of FGFs and FGFRs than the 2:2 FGF:FGFR complexes described by X-ray crystallography [9,31]. Indeed, a subset of species with lengths as short as 16 or 18 saccharide units may be competent to form 4:4:1 FGF:FGFR:heparin complexes. Further increases in length increase the proportion of heparin chains that are competent to form such a complex. Longer heparin fragments should be capable of binding even more FGF and FGFR units. In addition, the data suggest that the higher-order complexes are not as stable as some of the 2:2 complexes described previously, as, in solution, an excess of heparin can reduce (but not abolish) the formation of these complexes. Finally, it appears that the saccharides involved in the formation of these interactions are the more highly sulphated chains from the pool of heparin fragments used to prepare the samples.
These observations suggest that the binding of multiple FGF complexes to a single heparin chain is favourable in solution. The conditions used differ from those on the cell surface in several important manners. First, on the cell surface, FGFRs will be inserted into the membrane. This has the effect of reducing the dimensionality of the interaction space from three to two dimensions, which tends to increase affinities considerably. Secondly, HS is not limiting on the cell; indeed, it is present in sufficiently large quantities that most FGFRs are likely to be complexed to different molecules in the resting state [43]. Finally, HS differs in structure from heparin. Heparin has a very high density of sulphation throughout its entire length; in contrast, the corresponding regions in HS, the S-domains, are spaced at intervals of approx. 14–16 disaccharide units [44]. Our present findings indicate that FGF1–FGFR2 pairs will form stable complexes on these domains only if they are extensively modified by O-sulphation. Sequencing data on HS from NIH 3T3 cells (which are responsive to FGF1) indicate that the degree of O-sulphation of the S-domains increases with S-domain length [22] and that this should enhance their capacity for assembling FGF–FGFR signalling complexes. In principle, two such complexes could form by docking on to two spatially discrete long S-domains analogous with those observed on the heparin 24-mer (12 disaccharide units) fragments in the present study. However, they would be likely to be separated by a greater distance in HS, thus minimizing steric clashes. Loo and co-workers [45] have suggested that an FGF8–FGFR complex could form on regions of intermediate or low sulphation in HS, but our findings indicate that when FGF1 is the ligand there is a requirement for high sulphation for complexing with FGFR. This correlates with the data of Lindahl and co-workers [46], who have shown that high-affinity binding of FGF1 requires a cluster of N-, 2-O- and 6-O-sulphate groups, and with bioactivity studies that have shown that FGF1-induced mitogenesis in HS-deficient cells requires highly sulphated HS saccharides [47].
It is clear that re-distribution in the cell to form discrete aggregates upon activation is a feature of receptor systems [34–37], although no direct evidence of this has yet been shown for FGFRs. This implies that FGFRs will alter their binding to HS upon activation. As weak interactions are observed between FGFR molecules themselves [9] and as FGFRs are known to bind to multiple other proteins, including N-cadherin [48], N-CAM (neural cell-adhesion molecule) [49] and XFLRT3 (fibronectin-leucine-rich transmembrane protein 3) [50], as well as their intracellular binding partners, there are multiple possibilities for molecules that drive the clustering of FGFRs. This clustering, in response to the FGF signal, will involve changes in the interactions with HS. At the cluster site, the considerable concentration of biomolecules on the membrane will drive additional interactions between the aggregating species. The formation of multiple units of the FGF–FGFR–HS complex upon a single chain of HS might be such an interaction, as the concentration of HS relative to protein is likely to be reduced in the cluster (even given the tendency of HS bearing proteins to join in clusters), and the distance between S-domains on the physiological HS will be sufficient to prevent steric hindrances from disfavouring such complexes from forming. The possibility of multiple FGF complexes involving a single HS chain forming at clustering sites must therefore be considered when building models of the mature FGF signalling complex.
Online Data
Acknowledgments
We thank Ralph Bradshaw for critical reading of the manuscript prior to submission. We are grateful to Dr Sue Malcolm (Institute of Child Health, London, U.K.) for providing the FGFR2 cDNA, and to Dr Dave Fernig (School of Biosciences, University of Liverpool, Liverpool, U.K.) for providing the FGF1 clone. We thank Barbara Mulloy (NIBSC, Potters Bar, Herts., U.K.) for providing samples of bovine lung heparin for comparison, Martin Moncrieffe for assistance with analytical ultracentrifugation, and Ralf Torgrip and Leonard Csenki for helpful discussions regarding the deconvolution of chromatographs. N.J.H. is funded by the Wellcome Trust and the BBSRC (Biotechnology and Biological Sciences Research Counci). L.E.A. and L.L.I. are funded by the BBSRC. C.J.R. and J.T.G. are funded by Cancer Research U.K. C.V.R. is funded by the Royal Society.
References
- 1.Powers C., McLeskey S., Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr. Rel. Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 2.Ornitz D., Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:reviews3005.1–3005.12. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ornitz D., Yayon A., Flanagan J., Svahn C., Levi E., Leder P. Heparin is required for cell-free binding of fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol. Cell. Biol. 1992;12:240–247. doi: 10.1128/mcb.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sleeman M., Fraser J., McDonald M., Yuan S., White D., Grandison P., Kumble K., Watson J., Murison J. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 2001;271:171–182. doi: 10.1016/s0378-1119(01)00518-2. [DOI] [PubMed] [Google Scholar]
- 5.Johnson D., Lee P., Lu J., Williams L. Diverse forms of a receptor for acidic and basic fibroblast growth factors. Mol. Cell. Biol. 1990;10:4728–4736. doi: 10.1128/mcb.10.9.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crumley G., Bellot F., Kaplow J., Schlessinger J., Jaye M., Dionne C. High-affinity binding and activation of a truncated FGF receptor by both aFGF and bFGF. Oncogene. 1991;6:2255–2262. [PubMed] [Google Scholar]
- 7.Wang F., Kan M., Xu J., Yan G., McKeehan W. Ligand-specific structural domains in the fibroblast growth factor receptor. J. Biol. Chem. 1995;270:10222–10230. doi: 10.1074/jbc.270.17.10222. [DOI] [PubMed] [Google Scholar]
- 8.Olsen S., Ibrahimi O., Raucci A., Zhang F., Eliseenikova A., Yayon A., Basilico C., Linhardt R., Schlessinger J., Mohammadi M. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc. Natl. Acad. Sci. U.S.A. 2004;101:935–940. doi: 10.1073/pnas.0307287101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellegrini L., Burke D., von Delft F., Mulloy B., Blundell T. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature (London) 2000;407:1029–1034. doi: 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- 10.Plotnikov A., Schlessinger J., Hubbard S., Mohammadi M. Structural basis for FGF receptor dimerization and activation. Cell. 1999;98:641–650. doi: 10.1016/s0092-8674(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 11.Plotnikov A., Hubbard S., Schlessinger J., Mohammadi M. Crystal structures of two FGF–FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101:413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 12.Stauber D., DiGabriele A., Hendrickson W. Structural interactions of fibroblast growth factor receptor with its ligands. Proc. Natl. Acad. Sci. U.S.A. 2000;97:49–54. doi: 10.1073/pnas.97.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeh B., Igarashi M., Eliseenkova A., Plotnikov A., Sher I., Ron D., Aaronson S., Mohammadi M. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2266–2271. doi: 10.1073/pnas.0436500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kan M., Wang F., Xu J., Crabb J., Hou J., McKeehan W. An essential heparin-binding domain in the fibroblast growth factor receptor kinase. Science. 1993;259:1918–1921. doi: 10.1126/science.8456318. [DOI] [PubMed] [Google Scholar]
- 15.Varki A., Cummings R., Esko J., Freeze H., Hart G., Marth J. New York: Cold Spring Harbor Laboratory Press; 1999. Essentials of Glycobiology. [PubMed] [Google Scholar]
- 16.Sugahara K., Kitagawa H. Heparin and heparan sulfate biosynthesis. IUBMB Life. 2002;54:163–175. doi: 10.1080/15216540214928. [DOI] [PubMed] [Google Scholar]
- 17.Harmer N., Chirgadze D., Kim K., Pellegrini L., Blundell T. The structural biology of growth factor receptor activation. Biophys. Chem. 2003;100:545–553. doi: 10.1016/s0301-4622(02)00305-8. [DOI] [PubMed] [Google Scholar]
- 18.Lin X. Functions of heparan sulphate proteoglycans in cell signaling during development. Development. 2004;131:6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 19.Murphy K., Merry C., Lyon M., Roberts I., Thompson J., Gallagher J. A new model for the domain structure of heparan sulphate based on the novel specificity of K5 lyase. J. Biol. Chem. 2004;279:27239–27245. doi: 10.1074/jbc.M401774200. [DOI] [PubMed] [Google Scholar]
- 20.Rapraeger A., Krufka A., Olwin B. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- 21.Walker A., Turnbull J., Gallagher J. Specific heparan sulfate saccharides mediate the activity of basic fibroblast growth factor. J. Biol. Chem. 1994;269:931–935. [PubMed] [Google Scholar]
- 22.Merry C., Lyon M., Deakin J., Hopwood J., Gallagher J. Highly-sensitive sequencing of the sulphated domains of heparan sulphate. J. Biol. Chem. 1999;274:18455–18462. doi: 10.1074/jbc.274.26.18455. [DOI] [PubMed] [Google Scholar]
- 23.Zhu X., Komiya H., Chirino A., Faham S., Fox G., Arakawa T., Hsu B., Rees D. Three-dimensional structures of acidic and basic fibroblast growth factors. Science. 1991;251:90–93. doi: 10.1126/science.1702556. [DOI] [PubMed] [Google Scholar]
- 24.Eriksson A., Cousens L., Weaver L., Matthews B. Three-dimensional structure of human basic fibroblast growth factor. Proc. Natl. Acad. Sci. U.S.A. 1991;88:3441–3445. doi: 10.1073/pnas.88.8.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellosta P., Iwahori A., Plotnikov A., Eliseenkova A., Basilico C., Mohammadi M. Identification of receptor and heparin binding sites in fibroblast growth factor 4 by structure-based mutagenesis. Mol. Cell. Biol. 2001;21:5946–5957. doi: 10.1128/MCB.21.17.5946-5957.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye S., Luo Y., Lu W., Jones R., Linhardt R., Capila I., Toida T., Kan M., Pelletier H., McKeehan W. Structural basis for interaction of FGF-1, FGF-2, and FGF-7 with different heparan sulfate motifs. Biochemistry. 2001;40:14429–14439. doi: 10.1021/bi011000u. [DOI] [PubMed] [Google Scholar]
- 27.Plotnikov A., Eliseenkova A., Ibrahimi O., Shriver Z., Sasisekharan R., Lemmon M., Mohammadi M. Crystal structure of fibroblast growth factor 9 reveals regions implicated in dimerization and autoinhibition. J. Biol. Chem. 2001;276:4322–4329. doi: 10.1074/jbc.M006502200. [DOI] [PubMed] [Google Scholar]
- 28.Olsen S., Garbi M., Zampieri N., Eliseenkova A., Ornitz D., Goldfarb M., Mohammadi M. FHFs share structural but not functional homology to FGFs. J. Biol. Chem. 2003;278:34226–34236. doi: 10.1074/jbc.M303183200. [DOI] [PubMed] [Google Scholar]
- 29.Harmer N., Chirgadze D., Pellegrini L., Fernandez-Recio J., Blundell T. The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry. 2004;43:629–640. doi: 10.1021/bi035320k. [DOI] [PubMed] [Google Scholar]
- 30.DiGabriele A., Lax I., Chen D., Svahn C., Jaye M., Schlessinger J., Hendrickson W. Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature (London) 1998;393:812–817. doi: 10.1038/31741. [DOI] [PubMed] [Google Scholar]
- 31.Schlessinger J., Plotnikov A., Ibrahimi O., Eliseenkova A., Yeh B., Yayon A., Linhardt R., Mohammadi M. Crystal structure of a ternary FGF–FGFR–heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 32.Harmer N., Lung L., Pellegrini L., Mulloy B., Robinson C., Blundell T. Towards a resolution of the stoichiometry of the fibroblast growth factor (FGF)-FGF receptor-heparin complex. J. Mol. Biol. 2004;339:821–834. doi: 10.1016/j.jmb.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 33.Ibrahimi O., Yeh B., Eliseenikova A., Zhang F., Olsen S., Igarashi M., Aaronson S., Linhardt R., Mohammadi M. Analysis of mutations in fibroblast growth factor (FGF) and a pathogenic mutation in FGF receptor (FGFR) provides direct evidence for the symmetric two-end model for FGFR dimerization. Mol. Cell. Biol. 2005;25:671–684. doi: 10.1128/MCB.25.2.671-684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franco R., Canals M., Marcellino D., Ferre S., Agnati L., Mallol J., Casado V., Ciruela F., Fuxe K., Lluis C., Canela E. Regulation of heptaspanning-membrane-receptor function by dimerization and clustering. Trends Biochem. Sci. 2003;28:238–243. doi: 10.1016/S0968-0004(03)00065-3. [DOI] [PubMed] [Google Scholar]
- 35.Gillham H., Golding M., Pepperkok R., Gullick W. Intracellular movement of green fluorescent protein-tagged phosphatidylinositol 3-kinase in response to growth factor receptor signaling. J. Cell Biol. 1999;146:869–880. doi: 10.1083/jcb.146.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gulbins E., Grassme H. Ceramide and cell death receptor clustering. Biochim. Biophys. Acta. 2002;1585:139–145. doi: 10.1016/s1388-1981(02)00334-7. [DOI] [PubMed] [Google Scholar]
- 37.Sacchettini J., Baum L., Brewer C. Multivalent protein-carbohydrate interactions. A new paradigm for supermolecular assembly and signal transduction. Biochemistry. 2001;40:3009–3015. doi: 10.1021/bi002544j. [DOI] [PubMed] [Google Scholar]
- 38.Goger B., Halden Y., Rek A., Mosl R., Pye D., Gallagher J., Kungl A. Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: a model for chemokine regulation at inflammatory sites. Biochemistry. 2001;41:1640–1646. doi: 10.1021/bi011944j. [DOI] [PubMed] [Google Scholar]
- 39.Schuck P. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sobott F., Hernandez H., McCammon M., Tito M., Robinson C. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal. Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 41.Lyon M., Rushton G., Askari J., Humphries M., Gallagher J. Elucidation of the structural features of heparan sulfate important for interaction with the Hep-2 domain of fibronectin. J. Biol. Chem. 2000;275:4599–4606. doi: 10.1074/jbc.275.7.4599. [DOI] [PubMed] [Google Scholar]
- 42.Casu B., Guerrini M., Guglieri S., Naggi A., Perez M., Torri G., Cassinelli G., Ribatti D., Carminati P., et al. Undersulfated and glycol-split heparin endowed with antiangiogenic activity. J. Med. Chem. 2004;47:838–848. doi: 10.1021/jm030893g. [DOI] [PubMed] [Google Scholar]
- 43.Powell A., Fernig D., Turnbull J. Fibroblast growth factor receptors 1 and 2 interact differently with heparin/heparan sulfate. J. Biol. Chem. 2002;277:28554–28563. doi: 10.1074/jbc.M111754200. [DOI] [PubMed] [Google Scholar]
- 44.Turnbull J., Gallagher J. Distribution of iduronate-2-sulphate in heparan sulphate: evidence for an ordered polymeric structure. Biochem. J. 1991;273:553–559. doi: 10.1042/bj2730553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loo B.-M., Salmivirta M. Heparin/heparan sulfate domains in binding and signaling of fibroblast growth factor 8b. J. Biol. Chem. 2002;277:32616–32623. doi: 10.1074/jbc.M204961200. [DOI] [PubMed] [Google Scholar]
- 46.Kreuger J., Salmivirta M., Sturiale L., Giménez-Gallego G., Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J. Biol. Chem. 2001;276:30744–30754. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- 47.Pye D., Vives R., Hyde P., Gallagher J. Regulation of FGF-1 mitogenic activity by heparan sulfate oligosaccharides is dependent on specific structural features: differential requirements for the modulation of FGF-1 and FGF-2. Glycobiology. 2000;10:1183–1192. doi: 10.1093/glycob/10.11.1183. [DOI] [PubMed] [Google Scholar]
- 48.Utton M., Eickholt B., Howell F., Wallis J., Doherty P. Soluble N-cadherin stimulates fibroblast growth factor receptor dependent neurite outgrowth and N-cadherin and the fibroblast growth factor receptor co-cluster in cells. J. Neurochem. 2001;76:1421–1430. doi: 10.1046/j.1471-4159.2001.00140.x. [DOI] [PubMed] [Google Scholar]
- 49.Cavallaro U., Niedermeyer J., Fuxa M., Christofori G. N-CAM modulates tumour-cell adhesion to matrix by inducing FGF-receptor signalling. Nat. Cell Biol. 2001;3:650–657. doi: 10.1038/35083041. [DOI] [PubMed] [Google Scholar]
- 50.Bottcher R., Pollet N., Delius H., Niehrs C. The transmembrane protein XFLRT3 forms a complex with FGF receptors and promotes FGF signalling. Nat. Cell Biol. 2004;6:38–44. doi: 10.1038/ncb1082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.