

Abstract
Insulin resistance and β-cell dysfunction, two factors central to the pathogenesis of type 2 diabetes, were studied in relation to the development of diabetes in a group of participants with impaired glucose tolerance in the Diabetes Prevention Program (DPP) at baseline and after specific interventions designed to prevent diabetes. Participants were randomly assigned to placebo (n = 1,082), metformin (850 mg twice a day) (n = 1,073), or intensive lifestyle intervention (n = 1,079). The diabetes hazard rate was negatively associated with baseline insulin sensitivity (hazard rate ratio = 0.62–0.94 per SD difference, depending on treatment group and measure of sensitivity) and with baseline insulin secretion (hazard rate ratio = 0.57–0.76 per SD). Improvements in insulin secretion and insulin sensitivity were associated with lower hazard rates in all treatment arms (hazard rate ratio = 0.46–0.95 per SD increase and 0.29–0.79 per SD increase, respectively). In multivariate models that included the three metabolic variables (changes in body weight, insulin sensitivity, and insulin secretion) each significantly and independently predicted progression to diabetes when adjusted for the other two variables. The intensive lifestyle intervention, which elicited the greatest reduction in diabetes incidence, produced the greatest improvement in insulin sensitivity and the best preservation of β-cell function after 1 year, whereas the placebo group, which had the highest diabetes incidence, had no significant change in insulin sensitivity and β-cell function after 1 year. In the metformin group, diabetes risk, insulin sensitivity, and β-cell function at 1 year were intermediate between those in the intensive lifestyle and placebo groups. In conclusion, higher insulin secretion and sensitivity at baseline and improvements in response to treatment were associated with lower diabetes risk in the DPP. The better preventive effectiveness of intensive lifestyle may be due to improved insulin sensitivity concomitant with preservation of β-cell function.
Insulin resistance and β-cell dysfunction are two well-established characteristics of type 2 diabetes, impaired fasting glycemia, and impaired glucose tolerance (IGT). The latter two are strong risk factors for type 2 diabetes (1-6). Other risk factors for type 2 diabetes include obesity, sedentary lifestyle, prior gestational diabetes, ethnicity, hypertension, dyslipidemia, and family history of diabetes (7-18). The Diabetes Prevention Program (DPP), a multicenter, randomized controlled trial, examined the effect of two active interventions to prevent or delay type 2 diabetes in people at high risk and found that the risk for developing type 2 diabetes was reduced by 58 and 31% in the intensive lifestyle and metformin-treated groups, respectively, compared with the placebo-treated group (1). The DPP provided a unique opportunity to investigate various metabolic variables not only as determinants for the transition from IGT to type 2 diabetes but also as potential mechanisms whereby these two different treatment interventions prevent or delay diabetes. Thus, we evaluated indexes of insulin sensitivity and insulin secretion to assess whether progressive defects in these variables contributed to the development of diabetes.
RESEARCH DESIGN AND METHODS
The eligibility criteria, design, and methods of the DPP have been reported elsewhere (19), and the DPP protocol is available at www.bsc.gwu.edu/dpp. Briefly, eligibility criteria included ≥25 years of age, BMI ≥24 kg/m2 (≥22 kg/m2 in Asian Americans), and fasting plasma glucose levels between 95 and 125 mg/dl in addition to IGT (2-h postload glucose of 140–199 mg/dl). However, the lower limit of the fasting plasma glucose criteria did not apply to American Indians. Participants were excluded if they had conditions or took medicines that would impair their ability to participate. All participants gave informed consent and signed documents approved by the institutional review board at each center. Eligible participants received standard advice on healthy diet and physical activity and were randomly assigned to one of three additional interventions: 1) intensive lifestyle intervention with a goal of ≥150 min/week of activity and ≥7% loss of body weight; 2) metformin, 850 mg twice a day; or 3) matching placebo.
Outcomes. Development of diabetes was determined by an annual oral glucose tolerance test (OGTT) or by a semiannual fasting plasma glucose level with confirmation by a second test (19), using the criteria of the American Diabetes Association (20) and the World Health Organization (21). Body weight was measured to the nearest 0.1 kg semiannually.
Metabolic variables. Blood was drawn semiannually for fasting glucose determination. Annual blood draws were also done for fasting measurements of HbA1c (A1C) and insulin and for 75-g OGTT measurements of insulin (30 min postload) and glucose (30 and 120 min postload). Participants did not take metformin/placebo on the morning of the blood draw. Immunoreactive insulin and proinsulin were measured in plasma. Measurement methods for glucose, A1C, insulin, and proinsulin have been previously published (22). Because OGTTs were no longer performed once diabetes was diagnosed, we only present data in participants who had 30- and 120-min OGTT values at baseline and at year 1. Follow-up data for development of diabetes are presented through 31 July 2001 (see data analysis).
Insulin secretion and sensitivity were expressed using glucose and insulin measured in conventional units (milligrams per decaliter and microunits per milliliter, respectively). Insulin secretion was estimated by two methods (23,24): 1) the corrected insulin response (CIR) = (100 × 30-min insulin)/(30-min glucose × [30-min glucose − 70 mg/dl]) and 2) the insulin-to-glucose ratio (IGR) = (30-min insulin − fasting insulin)/(30-min glucose − fasting glucose). IGR and CIR were highly correlated at baseline (Spearman r = 0.95). Insulin sensitivity was estimated by two methods (25): 1) 1/fasting insulin and 2) the insulin sensitivity index (ISI), which is 22.5/(fasting insulin × [fasting glucose/18.01]), whose reciprocal is the homeostasis model assessment of insulin resistance (26). ISI and 1/fasting insulin were, as expected, highly correlated at baseline (Spearman r = 0.99).
Data analysis. Participants were followed for an average of 3.2 years from the start of the study (June 1996) through 31 July 2001. This period of 4 months, longer than that reported previously (1), allows use of all data from the masked phase of the DPP, because unmasking occurred in early August 2001. Encompassing of the additional follow-up data yielded risk reductions (95% CI) of developing diabetes of 30% (16–41%) for metformin and 55% (45–63%) for lifestyle, each compared with placebo. The diabetes risk reduction associated with lifestyle exceeded that with metformin by 37% (22–49%).
The SAS analysis system version 8.2 was used for all analyses (SAS Institute, Cary, NC). Means and overall comparisons over time were computed using repeated-measures ANOVA, adjusted for values at baseline. P values for comparisons between any two treatment arms were adjusted for multiple comparisons using the Holm procedure (27). Pairwise associations of the metabolic variables are described by Spearman rank-order correlations adjusted for sex, ethnic group, and baseline age.
Cox proportional hazards models (28) were used to assess the effect of baseline and time-dependent metabolic variables on the development of diabetes throughout the study with adjustment for baseline demographics (age, sex, and ethnicity). Hazard ratios were assumed to be constant over time. Models were run separately for each treatment group, and a test of heterogeneity was used to see if the effect of a metabolic variable differed across treatment groups. Metabolic variables in the time-dependent proportional hazards analyses were entered as the average change from baseline up to, but not including, each visit when diabetes was assessed.
Linear regression was used to estimate the relationship between log-transformed insulin secretion versus log-transformed insulin sensitivity at baseline (all groups combined) and at year 1 (for each treatment group) based on the known nonlinear relationship between these two variables (29). A constant of 3.0 was added before the log of IGR was calculated because of negative values in this measure of insulin secretion. The relationship between these two variables is thus a composite referred to as “β-cell function.”
RESULTS
Table 1 presents baseline characteristics by treatment group. There were no significant differences in any variables among the three intervention groups. Ninety-five percent of participants completed their first annual visit.
TABLE 1.
Baseline characteristics by treatment group
| Baseline characteristics* | Overall | Placebo | Metformin | Lifestyle |
|---|---|---|---|---|
| n | 3,234 | 1,082 | 1,073 | 1079 |
| Female | 2,191 (67.7) | 747 (69.0) | 710 (66.2) | 734 (68.0) |
| Race/ethnicity | ||||
| Caucasian | 1,768 (54.7) | 586 (54.2) | 602 (56.1) | 580 (53.8) |
| African American | 645 (19.9) | 220 (20.3) | 221 (20.6) | 204 (18.9) |
| Hispanic | 508 (15.7) | 168 (15.5) | 162 (15.1) | 178 (16.5) |
| American Indian | 171 (5.3) | 59 (5.5) | 52 (4.8) | 60 (5.6) |
| Asian American | 142 (4.4) | 49 (4.5) | 36 (3.4) | 57 (5.3) |
| Age (years) | 50.6 ± 10.7 | 50.3 ± 10.4 | 50.9 ± 10.3 | 50.6 ± 11.3 |
| Weight (kg) | 94.2 ± 20.3 | 94.3 ± 20.2 | 94.3 ± 19.9 | 94.1 ± 20.8 |
| BMI (kg/m2) | 34.0 ± 6.7 | 34.2 ± 6.7 | 33.9 ± 6.6 | 33.9 ± 6.8 |
| Waist (cm) | 105.1 ± 14.5 | 105.2 ± 14.3 | 104.9 ± 14.4 | 105.1 ± 14.8 |
| Glucose (mmol/l) | ||||
| Fasting | 5.9 ± 0.5 | 5.9 ± 0.5 | 5.9 ± 0.5 | 5.1 ± 0.4 |
| 30 min | 9.4 ± 1.4 | 9.4 ± 1.3 | 9.5 ± 1.4 | 9.4 ± 1.4 |
| 120 min | 9.1 ± 0.9 | 9.1 ± 1.0 | 9.2 ± 1.0 | 9.1 ± 0.9 |
| A1C (%) | 5.9 ± 0.5 | 5.9 ± 0.5 | 5.9 ± 0.5 | 5.9 ± 0.5 |
| Insulin (pmol/l) | ||||
| Fasting | 186 ± 105 | 185 ± 104 | 188 ± 103 | 184 ± 108 |
| 30 min | 698 ± 445 | 692 ± 413 | 713 ± 491 | 690 ± 429 |
| ISI | 0.195 ± 0.132 | 0.192 ± 0.128 | 0.191 ± 0.125 | 0.201 ± 0.142 |
| IGR | 124.3 ± 92.9 | 122.9 ± 85.8 | 124.9 ± 99.2 | 125.0 ± 93.4 |
| CIR | 0.634 ± 0.428 | 0.622 ± 0.370 | 0.643 ± 0.468 | 0.638 ± 0.440 |
| Fasting proinsulin (pmol/l) | 18.2 ± 13.9 | 18.2 ± 13.9 | 18.3 ± 14.6 | 18.0 ± 13.3 |
Data are means ± SD or n (%).
No significant differences among the three treatment groups.
Changes in metabolic variables over time.
Figure 1 depicts the mean and SE of the plasma glucose values during the OGTT at baseline and year 1 by treatment group excluding the 61 participants who developed diabetes at 6 months. The greatest reduction in glucose at year 1 occurred in the lifestyle group. Fasting and 30- and 120-min glucose values decreased significantly in the lifestyle group, whereas in the metformin group, only fasting and 120-min glucose levels decreased. In the placebo group, however, there was a significant increase in 30-min and a significant decrease in the 120-min glucose levels. At year 1, the decreases in 120-min glucose levels were not significantly different between the placebo and metformin groups but were significantly greater in the lifestyle group.
FIG. 1.
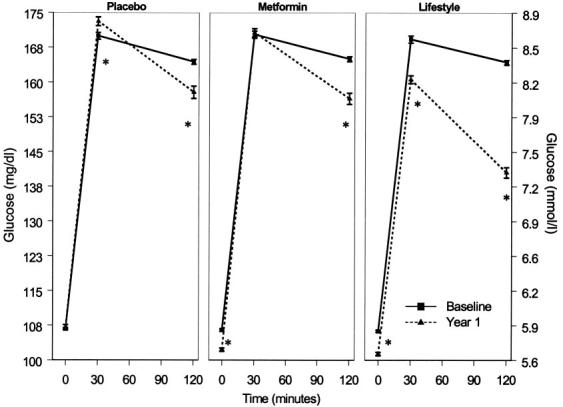
Plasma glucose during OGTT at baseline and year 1 by treatment group. Data are means ± SE. *P < 0.05 for test of difference between baseline and year 1 glucose value. Only participants who underwent OGTT testing at year 1 are included (i.e., those who did not develop diabetes at 6 months).
Among all participants, including those who developed diabetes, fasting glucose, insulin, and proinsulin concentrations were significantly lower than placebo at the first annual visit in the metformin and the lifestyle groups and increased during the 2nd and 3rd years, with the levels remaining significantly lower than in the placebo group (Fig. 2). At 1 year, there was less reduction in fasting insulin in the metformin than in the lifestyle group, with the lifestyle participants having the greatest reduction throughout all years of follow-up.
FIG. 2.
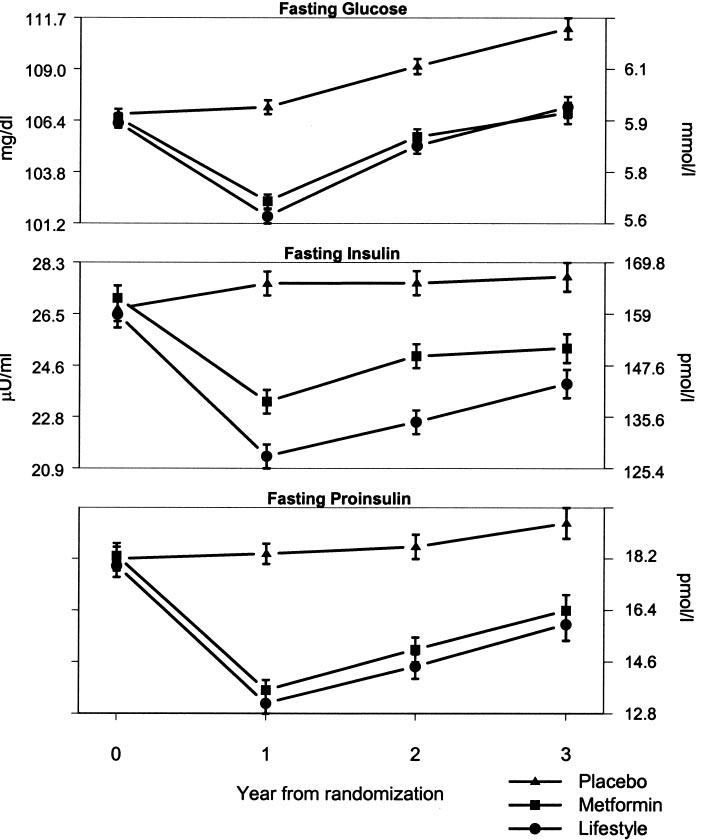
Metabolic variables over time by treatment group. Data are means ± SE in conventional and SI units. All tests of mean changes over time between any two groups are statistically significant (P < 0.001) except in fasting glucose and fasting insulin between the lifestyle and metformin groups. The number of participants decreased over time because of the variable length of time that individuals were in the study. For example, data on fasting glucose were available for 3,065 at 1 year, 3,015 at 2 years, and 1,910 at 3 years.
Table 2 shows the partial correlations (adjusted for age, sex, and race/ethnicity) at baseline for glycemic indexes (fasting plasma glucose and A1C) with weight, fasting insulin, proinsulin, IGR, and CIR. All these correlations were significant in this large sample. As expected, there were strong correlations between 1/fasting insulin and ISI and between CIR and IGR.
TABLE 2.
Partial Spearman correlations at baseline adjusted for age, sex, and race/ethnicity (n = 3,160)
| Weight | Proinsulin | 1/FI | ISI | CIR | IGR | Fasting glucose | A1C | |
|---|---|---|---|---|---|---|---|---|
| Weight | 1 | |||||||
| Fasting proinsulin | 0.39 | 1 | ||||||
| 1/FI | −0.40 | −0.64 | 1 | |||||
| ISI | −0.41 | −0.66 | 0.99 | 1 | ||||
| CIR | 0.21 | 0.27 | −0.50 | −0.44 | 1 | |||
| IGR | 0.17 | 0.24 | −0.41 | −0.37 | 0.95 | 1 | ||
| Fasting glucose | 0.18 | 0.32 | −0.21 | −0.33 | −0.29 | −0.15 | 1 | |
| A1C | 0.12 | 0.22 | −0.12 | −0.16 | −0.09 | −0.06 | 0.31 | 1 |
1/FI, 1/fasting insulin.
Table 3 shows partial correlations of the changes from baseline to year 1 for the same measurements by treatment group, adjusted for baseline age, sex, and race/ethnicity. Correlations between the year 1 changes in metabolic variables were similar among the treatment groups, except for the correlations between 1/fasting insulin and either CIR or IGR, which were higher in the metformin group.
TABLE 3.
Partial correlations of changes from baseline to year 1, adjusted for baseline age, sex, and race/ethnicity
| Weight | Proinsulin | 1/FI | ISI | CIR | IGR | Fasting glucose | A1C | |
|---|---|---|---|---|---|---|---|---|
| Placebo (n = 933) | ||||||||
| Weight | 1 | |||||||
| Fasting proinsulin | 0.36 | 1 | ||||||
| 1/FI | −0.30 | −0.39 | 1 | |||||
| ISI | −0.33 | −0.41 | 0.98 | 1 | ||||
| CIR | −0.02 | 0.02 | −0.14 | −0.08 | 1 | |||
| IGR | 0.01 | 0.03 | −0.07 | −0.06 | 0.89 | 1 | ||
| Fasting glucose | 0.30 | 0.32 | −0.34 | −0.49 | −0.28 | −0.05 | 1 | |
| A1C | 0.22 | 0.19 | −0.11 | −0.13 | −0.07 | −0.02 | 0.22 | 1 |
| Metformin (n = 937) | ||||||||
| Weight | 1 | |||||||
| Fasting proinsulin | 0.33 | 1 | ||||||
| 1/FI | −0.29 | −0.34 | 1 | |||||
| ISI | −0.31 | −0.35 | 0.98 | 1 | ||||
| CIR | 0.00 | 0.03 | −0.22 | −0.17 | 1 | |||
| IGR | 0.02 | 0.05 | −0.16 | −0.15 | 0.89 | 1 | ||
| Fasting glucose | 0.26 | 0.31 | −0.34 | −0.46 | −0.24 | −0.06 | 1 | |
| A1C | 0.26 | 0.24 | −0.16 | −0.18 | 0.00 | 0.04 | 0.20 | 1 |
| Lifestyle (n = 946) | ||||||||
| Weight | 1 | |||||||
| Fasting proinsulin | 0.37 | 1 | ||||||
| 1/FI | −0.38 | −0.38 | 1 | |||||
| ISI | −0.40 | −0.37 | 0.98 | 1 | ||||
| CIR | −0.07 | 0.03 | −0.15 | −0.10 | 1 | |||
| IGR | −0.02 | 0.03 | −0.12 | −0.10 | 0.89 | 1 | ||
| Fasting glucose | 0.34 | 0.31 | −0.41 | −0.53 | −0.26 | −0.06 | 1 | |
| A1C | 0.35 | 0.29 | −0.23 | −0.24 | −0.08 | −0.04 | 0.25 | 1 |
Correlations with an absolute value >0.07 (placebo), >0.06 (metformin), and >0.06 (lifestyle) are significant at the 0.05 level, and correlations are adjusted for age, sex, and race/ethnicity. 1/FI, 1/fasting insulin.
Prediction of diabetes by individual metabolic factors Baseline glycemic variables. Cox proportional hazards models were used to predict progression to diabetes for baseline levels of each metabolic factor adjusted for baseline demographics (age, sex, and race/ethnicity). The hazard rates for developing diabetes were positively associated with baseline fasting plasma glucose, 120-min plasma glucose, and A1C (Fig. 3). Estimates of the absolute risk gradient associated with a given value of a covariate using the range of covariate values (5th–95th percentiles) were used to describe the hazard rate for a participant with a covariate value equal to the group mean. The point on each line indicates the expected hazard rate for a subject with a metabolic variable equal to the mean value for the group as estimated in the life table analysis. The risk of developing diabetes was lowest among participants in the lifestyle arm, intermediate among those assigned to metformin, and highest among placebo participants across the range of baseline glycemic variables. Diabetes incidence was more strongly associated with baseline fasting glucose in the placebo group than in the others (P < 0.001).
FIG. 3.

Diabetes hazard rates by baseline metabolic variables. Cox proportional hazards models were used to estimate the risk of developing diabetes. Estimates of the absolute risk gradient associated with a given value of a covariate using the range of covariate values (5th–95th percentiles) were used to describe the hazard rate for a participant with a covariate value equal to the group mean. The baseline values were standardized for comparability of the hazard rates among the covariates. The point on each line indicates the expected hazard rate for a subject with a metabolic variable equal to the mean value for the group as estimated in the life table analysis. In separate models using the baseline metabolic variables adjusted for baseline age, sex, and race/ethnicity, all three baseline metabolic variables significantly predicted progression to diabetes in all treatment groups (P < 0.001).
Baseline and changes in body weight, insulin sensitivity, and insulin secretion. The hazard rates for diabetes were positively associated with baseline weight in the lifestyle and placebo groups when adjusted for baseline demographics (age, sex, and race/ethnicity) (Fig. 4, left panels). In all treatment groups, hazard rates were negatively associated with one estimate of insulin sensitivity (ISI) and both estimates of insulin secretion (CIR and IGR). In the lifestyle and placebo groups, hazard rates were negatively associated with baseline 1/fasting insulin.
FIG. 4.

Hazard rates for diabetes by metabolic variables at baseline and mean changes from baseline. In separate models using the baseline metabolic variables adjusted for baseline age, sex, race/ethnicity (left panels), all baseline metabolic variables significantly predicted progression to diabetes in all treatment groups (P = 0.016 in the metformin group for ISI and P < 0.001 for the rest of the variables) except for baseline weight and 1/fasting insulin in the metformin group. The time-dependent models using changes in the metabolic variables from baseline up to but not including the values at the time of progression to diabetes were adjusted for the baseline metabolic variable, baseline age, sex, and race/ethnicity (right panels). All changes in metabolic variables significantly predicted progression to diabetes (P = 0.007 for IGR in the placebo group and P < 0.001 for the rest of the variables). The metabolic variables changed from baseline at a significance level of 0.05 for all metabolic variables except for weight in the placebo group.
Proportional hazards models were used to estimate the effects of changes in metabolic variables on diabetes incidence with time-dependent covariates adjusted for baseline demographics and the baseline value (Fig. 4, right panels). Changes in weight and proinsulin over time were strong predictors of diabetes, with decreased weight and decreased proinsulin predicting a lower hazard of diabetes in all treatment arms (Fig. 4B and D). A greater improvement in either estimate of insulin sensitivity (1/fasting insulin or ISI) was also associated with lower diabetes risk in all treatment arms (Fig. 4F and H). Finally, an increase in estimated insulin secretion (CIR or IGR) also predicted lower diabetes risk (Fig. 4J and L). In multivariate models that included all three metabolic variables, changes in weight, either estimate of insulin sensitivity, and either estimate of insulin secretion significantly and independently predicted progression to diabetes when adjusted for the other two variables in all three treatment arms (data not shown).
Prediction of diabetes by multiple metabolic factors Baseline insulin sensitivity and insulin secretion. Figure 5 shows the hazard rates for progression to diabetes when participants were grouped by tertiles of baseline insulin secretion (using either CIR or IGR) and sensitivity (using 1/fasting insulin or ISI). Hazard rates for diabetes were lowest among the lifestyle participants, intermediate among the metformin participants, and highest among the placebo participants. Using either set of estimates, low insulin sensitivity and low insulin secretion at baseline predicted higher diabetes risk in all treatment arms. In general, these treatment effects were seen regardless of the categories of sensitivity and secretion, except in the lowest risk categories (high insulin sensitivity and secretion), in which hazard rates were uniformly low.
FIG. 5.
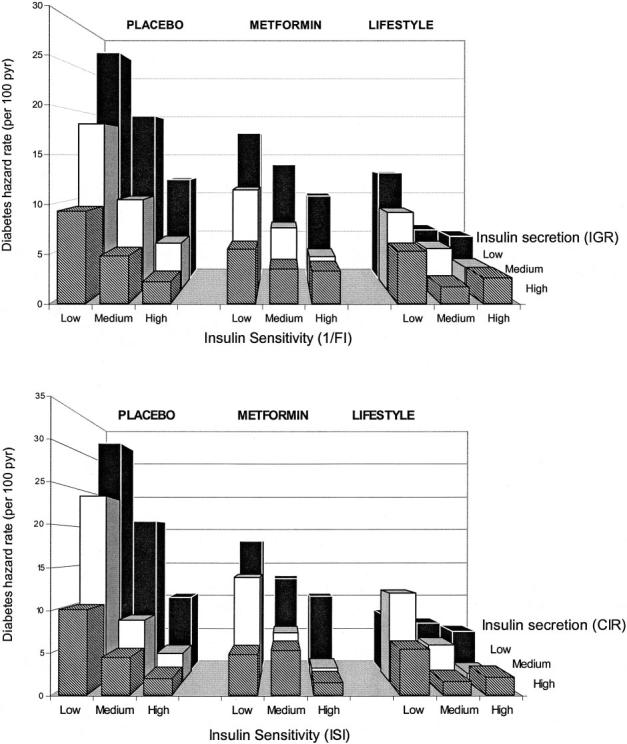
Hazard rate for developing diabetes by tertiles of baseline insulin sensitivity and baseline insulin secretion. The tertiles are defined using the percentiles (33.3rd and 66.7th) of 1/fasting insulin (0.033 and 0.053), ISI (0.123 and 0.205), CIR (0.416 and 0.700), and IGR (80.0 and 136.0).
Changes in insulin secretion and sensitivity. The relationship between insulin secretion and sensitivity is shown in Fig. 6 at baseline overall and at year 1 for each treatment group. Lines were fit by linear regression of the log-transformed variables and then transformed back to the original scale for plotting. For simplicity, the figure only displays all groups combined at baseline (thick line for the 10th–90th percentiles of the insulin sensitivity), and arrows to depict the change in β-cell function for each group are shown. Again, we compared two combinations of insulin sensitivity and insulin secretion (IGR versus 1/fasting insulin and CIR versus ISI), and the results were similar. In both comparisons, the relationship between insulin secretion and sensitivity was curved (i.e., linear on the log-log scale). This relationship describes β-cell function because insulin sensitivity is known to modulate insulin secretion (29). The arrows depict the change in β-cell function from baseline to year 1 for each group at their respective median insulin sensitivity. Using the Wilcoxon's signed-rank test for paired observations from baseline and year 1, we examined the changes in insulin sensitivity and secretion. The lifestyle group had the greatest improvement in insulin sensitivity (ISI and 1/fasting insulin, P < 0.001), and this was associated with a significant decrease in insulin secretion (IGR, P = 0.008). When relating insulin sensitivity to insulin secretion, the changes resulted in a rightward shift from the baseline curve and are compatible with an enhancement of β-cell function relative to that at baseline (i.e., improved secretion relative to sensitivity). The metformin group showed a more modest improvement in insulin sensitivity (ISI and 1/fasting insulin, P < 0.001) with a significant decrease in insulin secretion (IGR, P < 0.001); but whereas the absolute response declined, this decrease was not as large as might be expected from the concomitant change in insulin sensitivity and therefore can be interpreted as improved secretion relative to sensitivity. Finally, in the placebo group there were no significant changes in either estimate of insulin sensitivity, a significant decrease in insulin secretion (IGR, P = 0.03) but no significant change in β-cell function in contrast to the evidence for improved β-cell function seen in the metformin and lifestyle groups.
FIG. 6.

Insulin secretion versus insulin sensitivity at baseline and year 1 by treatment group. Only participants who underwent OGTT testing at year 1 are included (i.e., those who had not previously been diagnosed with diabetes). Insulin secretion and sensitivity were calculated using glucose and insulin measured in conventional units (milligrams per deciliter and microunits per milliliter, respectively). β-Cell function is described by the relationship between insulin secretion (IGR and CIR) and insulin sensitivity (1/fasting insulin and ISI) at baseline and year 1 by treatment group. The curve represents the regression line of the logarithm of estimated insulin secretion as a linear function of the logarithm of estimated insulin sensitivity for all participants at baseline. The arrows that connect the estimated insulin secretion for the median insulin sensitivity at baseline and year 1 illustrate the effects of the interventions after 1 year.
DISCUSSION
The fasting and 2-h glucose levels at baseline predict the development of diabetes in the DPP, as expected. In addition, we found that A1C, proinsulin, insulin sensitivity, and insulin secretion also predicted the development of diabetes in these high-risk individuals. Furthermore, analysis of the changes in plasma glucose and insulin during the 1st year of the study suggests that development of diabetes in the placebo group resulted from continued decreases in insulin sensitivity and β-cell function (i.e., insulin secretion relative to sensitivity), whereas reduction in the incidence of diabetes observed in the two active interventions was due to their ability to increase insulin sensitivity and improve β-cell function.
It is well recognized that fasting and 2-h OGTT glucose levels are positively associated with risk of progression to diabetes (5). This was also observed in this large cohort of DPP participants who were at high risk for diabetes. That these two measures were the strongest predictors of diabetes is not surprising, because the diagnosis of diabetes was based on these same measures. The interventions chosen for the DPP were selected for their ability to lower glucose levels and proved to have differential effects on reducing the incidence of diabetes. Although the risk of diabetes was associated with glucose variables within each treatment group, the risk of developing diabetes based on these variables at baseline was lowest for participants in the lifestyle arm, intermediate for those taking metformin, and greatest for those receiving placebo.
The development of diabetes was also positively related to baseline body weight and negatively to estimates of insulin sensitivity and secretion at baseline, in keeping with the findings of previous studies performed in a variety of populations (3-5). As with the glucose variables, body weight and estimated insulin secretion and sensitivity predicted diabetes in a similar fashion in each treatment group. We further assessed the impact of changes in these three measures on the hazard rate for developing diabetes. Interestingly, we found that change in weight was an important factor, with greater weight loss translating to lower risk. In addition, improved insulin sensitivity was also associated with a decrease in the risk of developing diabetes, with the lifestyle intervention again being the most effective. On the other hand, a beneficial effect of the intervention was indicated by an improvement in β-cell function over the 1st year of intervention. Thus it appears that whereas change in body weight was a strong predictor of the risk of developing diabetes in the DPP, the interventions probably had differential effects on other variables or direct effects in the liver or muscle even after accounting for insulin secretion.
Although the relative importance of decreased insulin sensitivity and insulin secretion in the pathogenesis of type 2 diabetes has been debated, it is generally accepted that deterioration of either or both is critical in determining the progression of hyperglycemia. In this large cohort of individuals with IGT, our results clearly confirm this relationship because each variable had an independent effect on the risk of developing diabetes. Furthermore, when each treatment group was divided into tertiles of insulin sensitivity and secretion at baseline (Fig. 5), those with the greatest risk of developing diabetes ranked in the lowest tertiles for both insulin sensitivity and secretion. The longitudinal analyses (Fig. 4) confirm that progressive loss of insulin secretion or worsening of insulin sensitivity was associated with greater diabetes risk. Whereas lifestyle and metformin treatments reduced the incidence of diabetes, they did not change the independent effects of insulin sensitivity and secretion on the risk for diabetes, so that those individuals with the lowest sensitivity and secretion within each treatment group still had the greatest risk, whereas those with the highest insulin sensitivity and secretion had uniformly low risk regardless of intervention group.
Another approach to assessing the effects of insulin sensitivity and secretion on progression to diabetes involved the known nonlinear relationship between these two variables (29). By logarithmically transforming both secretion and sensitivity, we used linear models of secretion as a function of sensitivity. This results in a curve when plotted on the untransformed scales, as in Fig. 6, and allows an assessment of insulin secretion relative to the prevailing insulin sensitivity, thus providing a measure of β-cell function. A special case of such curves is a hyperbola, in which the product of the two variables (secretion and sensitivity) is a constant. This hyperbolic relationship has previously been described when more direct measures of insulin secretion and sensitivity were used (29). The relationships in the current data were not hyperbolic but were well described as nonlinear. Based on this relationship of insulin sensitivity and insulin secretion, changes in insulin secretion may reflect one of three scenarios regarding β-cell function. The first would be compatible with no change in β-cell function and would occur if changes in the insulin response and insulin sensitivity were such that the relationship remained the same. This would result in β-cell function moving along the curve describing the relationship for a given group of participants. The second scenario would be one in which β-cell function was improved. This would be characterized by a change in the relationship between insulin sensitivity and the insulin response with a shift to the right of or above the curve. Finally, a decrease in β-cell function would be characterized by a shift to the left of or below the curve.
The effects of both metformin and lifestyle in reducing the incidence of diabetes were associated with rightward shifts in the points representing median insulin secretion and sensitivity (Fig. 6, arrows), compatible with improved insulin sensitivity and β-cell function. With metformin, there was a small improvement in insulin sensitivity. Moreover, although the insulin secretory response appeared to decline in absolute terms, this change was relatively less than the change in insulin sensitivity, hence suggesting improved β-cell function. The lifestyle intervention resulted in the greatest improvement in insulin sensitivity, and the absolute insulin secretory response also appears to have remained unchanged. These findings are compatible, with lifestyle having the greatest effect to preserve β-cell function and it being the more effective intervention to reduce the rate of development of diabetes. Lifestyle intervention and, to a lesser extent, metformin enhanced β-cell function and improved insulin sensitivity, leading to a reduced risk for developing diabetes. Similar findings of the effect of lifestyle intervention have been reported for the Finnish Diabetes Prevention Study (30). In the lifestyle intervention and control groups, insulin sensitivity did not change significantly over 4 years, but at the end of this period, it tended to be greater in the intervention group. Insulin secretion did not change in the lifestyle intervention group but decreased significantly in the control group. Thus, it would appear that β-cell function was unchanged in the lifestyle intervention group while declining in the control group. Finally, the DPP findings are also consistent with a recently reported study that demonstrated that a weight loss intervention in older individuals was associated with improved insulin sensitivity together with a relative preservation of the insulin response reflective of improved β-cell function (31) and provide a plausible mechanism by which lifestyle changes reduced the incidence of diabetes in other diabetes prevention studies (32-34).
We also measured fasting proinsulin because production of insulin is dependent on the proteolytic processing in the β-cell secretory granule of proinsulin to insulin and C-peptide (35). However, this process is never complete so that not all proinsulin molecules are cleaved, resulting in a small quantity of proinsulin being secreted along with insulin and C-peptide (36-38). As with fasting insulin, fasting proinsulin levels increase with declining insulin sensitivity, probably as a result of the increased secretory demand on the β-cell (39). It has thus been suggested that the fasting proinsulin level represents a measure of insulin sensitivity and not β-cell function (40,41). Because of the strong relationship between the fasting proinsulin and fasting insulin levels, proinsulin appeared to be a good marker of risk of progression to diabetes, with higher proinsulin levels at baseline resulting in the greater risk.
In summary, an examination of insulin sensitivity and secretion at baseline and during follow-up in the DPP highlights the fact that reduction in both insulin sensitivity and β-cell function underlie the progression from IGT to diabetes. Those individuals who were most insulin resistant and had the poorest insulin secretory responses were at greatest risk of developing diabetes in the DPP. Although the magnitude of weight reduction was a predictor of a beneficial response to the intervention, changes in insulin sensitivity and insulin secretion were also independent predictors of a beneficial outcome. The effect of metformin and intensive lifestyle in reducing the incidence of diabetes is related in part to their ability to shift the relationship between insulin sensitivity and insulin secretion in a direction compatible with an improvement in β-cell function, with lifestyle being more effective in doing so. These observations strongly suggest that future approaches to diabetes prevention should preferably include approaches that enhance both insulin sensitivity and β-cell function and if combined with weight loss, may be even more beneficial.
ACKNOWLEDGMENTS
Funding was provided by the National Institutes of Health through the National Institute of Diabetes and Digestive and Kidney Diseases, the Office of Research on Minority Health, the National Institute of Child Health and Human Development, the Office of Women's Health, and the National Institute on Aging. In addition, the Indian Health Service, the Centers for Disease Control and Prevention, the American Diabetes Association, and two pharmaceutical companies, Bristol-Myers Squibb and Parke-Davis, contributed support. The General Clinical Research Center Program, National Center for Research Resources, supported many of the clinical centers. Support to the clinical centers and the coordinating center was provided by the National Institute of Diabetes and Digestive and Kidney Diseases through a Cooperative Agreement, except for the Southwestern American Indian Centers, which were supported directly by the National Institute of Diabetes and Digestive and Kidney Diseases and the Indian Health Service.
We gratefully acknowledge the commitment and dedication of the participants of the DPP.
Glossary
- CIR
corrected insulin response
- DPP
Diabetes Prevention Program
- IGR
insulin-to-glucose ratio
- IGT
impaired glucose tolerance
- ISI
insulin sensitivity index
- OGTT
oral glucose tolerance test
APPENDIX
Diabetes Prevention Program Writing Committee. Abbas E. Kitabchi, Marinella Temprosa, William C. Knowler, Steven E. Kahn, Sarah E. Fowler, Steven M. Haffner, Reuben Andres, Christopher Saudek, Sharon L. Edelstein, Richard Arakaki, Mary Beth Murphy, and Harry Shamoon.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.The Diabetes Prevention Program Research Group Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Modan M, Karasik A, Halkin H, Fuchs Z, Lusky A, Shitrit A, Modan B. Effect of past and concurrent body mass index on prevalence of glucose intolerance and type 2 (non-insulin-dependent) diabetes and on insulin response: the Israel study of glucose intolerance, obesity and hypertension. Diabetologia. 1986;29:82–89. doi: 10.1007/BF00456115. [DOI] [PubMed] [Google Scholar]
- 3.Saad MF, Knowler WC, Pettitt DJ, Nelson RG, Mott DM, Bennett PH. The natural history of impaired glucose tolerance in the Pima Indians. N Engl J Med. 1988;319:1500–1506. doi: 10.1056/NEJM198812083192302. [DOI] [PubMed] [Google Scholar]
- 4.Lillioja S, Mott DM, Howard BV, Bennett PH, Yki-Jarvinen H, Freymond D, Nyomba BL, Zurlo F, Swinburn B, Bogardus C. Impaired glucose tolerance as a disorder of insulin action: longitudinal and cross-sectional studies in Pima Indians. N Engl J Med. 1988;318:1217–1225. doi: 10.1056/NEJM198805123181901. [DOI] [PubMed] [Google Scholar]
- 5.Edelstein SL, Knowler WC, Bain RP, Andres R, Barrett-Connor EL, Dowse GK, Haffner SM, Pettitt DJ, Sorkin JD, Muller DC, Collins VR, Hamman RF. Predictors of progression from impaired glucose tolerance to NIDDM: an analysis of six prospective studies. Diabetes. 1997;46:701–710. doi: 10.2337/diab.46.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE, American Diabetes Association GENNID Study Group β-Cell function is the major determinant of oral glucose tolerance in four ethnic groups in the United States. Diabetes. 2002;51:2170–2178. doi: 10.2337/diabetes.51.7.2170. [DOI] [PubMed] [Google Scholar]
- 7.Medalie JH, Papier CM, Goldbourt U, Herman JB. Major factors in the development of diabetes mellitus in 10,000 men. Arch Intern Med. 1975;135:811–817. [PubMed] [Google Scholar]
- 8.Knowler WC, Pettitt DJ, Savage PJ, Bennett PH. Diabetes incidence in Pima Indians: contributions of obesity and parental diabetes. Am J Epidemiol. 1981;113:144–156. doi: 10.1093/oxfordjournals.aje.a113079. [DOI] [PubMed] [Google Scholar]
- 9.Jarrett RJ, Keen H, McCartney P. The Whitehall Study: ten year follow-up report on men with impaired glucose tolerance with reference to worsening to diabetes and predictors of death. Diabet Med. 1984;1:279–283. doi: 10.1111/j.1464-5491.1984.tb01973.x. [DOI] [PubMed] [Google Scholar]
- 10.Ohlson LO, Larsson B, Bjorntorp P, Eriksson H, Svardsudd K, Welin L, Tibblin G, Wilhelmsen L. Risk factors for type 2 (non-insulin-dependent) diabetes mellitus: thirteen and one-half years of follow-up of the participants in a study of Swedish men born in 1913. Diabetologia. 1988;31:798–805. doi: 10.1007/BF00277480. [DOI] [PubMed] [Google Scholar]
- 11.DeFronzo RA. The triumvirate: β-cell, muscle, liver: a collusion responsible for NIDDM. Diabetes. 1988;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- 12.Haffner SM, Stern MP, Mitchell BD, Hazuda HP, Patterson JK. Incidence of type II diabetes in Mexican Americans predicted by fasting insulin and glucose levels, obesity, and body-fat distribution. Diabetes. 1990;39:283–288. doi: 10.2337/diab.39.3.283. [DOI] [PubMed] [Google Scholar]
- 13.Harris MI. Epidemiological correlates of NIDDM in Hispanics, whites, and blacks in the U.S. population. Diabetes Care. 1991;14(Suppl 3):639–648. doi: 10.2337/diacare.14.7.639. [DOI] [PubMed] [Google Scholar]
- 14.Bergstrom RW, Newell Morris LL, Leonetti DL, Shuman WP, Wahl PW, Fujimoto WY. Association of elevated fasting C-peptide level and increased intra-abdominal fat distribution with development of NIDDM in Japanese-American men. Diabetes. 1990;39:104–111. doi: 10.2337/diacare.39.1.104. [DOI] [PubMed] [Google Scholar]
- 15.Martin BL, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn CR. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet. 1992;340:925–929. doi: 10.1016/0140-6736(92)92814-v. [DOI] [PubMed] [Google Scholar]
- 16.Cassano PA, Rosner B, Vokonas PS, Weiss ST. Obesity and body fat distribution in relation to the incidence of non-insulin-dependent diabetes mellitus: a prospective cohort study of men in the normative aging study. Am J Epidemiol. 1992;136:1474–1486. doi: 10.1093/oxfordjournals.aje.a116468. [DOI] [PubMed] [Google Scholar]
- 17.de Vegt F, Dekker JM, Jager A, Hienkens E, Kostense PJ, Stehouwer CD, Nijpels G, Bouter LM, Heine RJ. Relation of impaired fasting and postload glucose with incident type 2 diabetes in a Dutch population: the Hoorn Study. J Am Med Assoc. 2001;285:2109–2113. doi: 10.1001/jama.285.16.2109. [DOI] [PubMed] [Google Scholar]
- 18.Felber JP, Golay A. Pathways from obesity to diabetes. Int J Obes Relat Metab Disord. 2002;26(Suppl 2):S39–S45. doi: 10.1038/sj.ijo.0802126. [DOI] [PubMed] [Google Scholar]
- 19.The Diabetes Prevention Program Research Group The Diabetes Prevention Program: design and methods for a clinical trial in the prevention of type 2 diabetes. Diabetes Care. 1999;22:623–634. doi: 10.2337/diacare.22.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Expert Committee on the Diagnosis and Classification of Diabetes Mellitus Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183–1197. doi: 10.2337/diacare.20.7.1183. [DOI] [PubMed] [Google Scholar]
- 21.Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1. Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 22.The Diabetes Prevention Program Research Group The Diabetes Prevention Program: baseline characteristics of the randomized cohort. Diabetes Care. 2000;23:1619–1629. doi: 10.2337/diacare.23.11.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sluiter WJ, Erkelens DW, Reitsma WD, Doorenbos H. Glucose tolerance and insulin release, a mathematical approach. I. Assay of the β-cell response after oral glucose loading. Diabetes. 1976;25:241–244. doi: 10.2337/diab.25.4.241. [DOI] [PubMed] [Google Scholar]
- 24.Phillips DI, Clark PM, Hales CN, Osmond C. Understanding oral glucose tolerance: comparison of glucose or insulin measurements during the oral glucose tolerance test with specific measurements of insulin resistance and insulin secretion. Diabet Med. 1994;11:286–292. doi: 10.1111/j.1464-5491.1994.tb00273.x. [DOI] [PubMed] [Google Scholar]
- 25.Hanson RL, Pratley RE, Bogardus C, Narayan KM, Roumain JM, Imperatore G, Fagot-Campagna A, Pettitt DJ, Bennett PH, Knowler WC. Evaluation of simple indices of insulin sensitivity and insulin secretion for use in epidemiologic studies. Am J Epidemiol. 2000;151:190–198. doi: 10.1093/oxfordjournals.aje.a010187. [DOI] [PubMed] [Google Scholar]
- 26.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 27.Holm S. A simple sequentially rejective Bonferroni test procedure. Scand J Stat. 1979;6:5–70. [Google Scholar]
- 28.Cox DR. Regression models in life tables. J R Stat Soc. 1972;34:187–220. [Google Scholar]
- 29.Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, Neifing JL, Ward WK, Beard JC, Palmer JP, Porte D., Jr Quantification of the relationship between insulin sensitivity and B-cell function in human subjects: evidence for a hyperbolic function. Diabetes. 1993;42:1663–1672. doi: 10.2337/diab.42.11.1663. [DOI] [PubMed] [Google Scholar]
- 30.Uusitupa M, Lindi V, Louheranta A, Salopuro T, Lindström J, Tuomilehto J, Finnish Diabetes Prevention Study Group Long-term improvement in insulin sensitivity by changing lifestyles of people with impaired glucose tolerance: 4-year results from the Finnish Diabetes Prevention Study. Diabetes. 2003;52:2532–2538. doi: 10.2337/diabetes.52.10.2532. [DOI] [PubMed] [Google Scholar]
- 31.Utzschneider KM, Carr DB, Barsness SM, Kahn SE, Schwartz RS. Diet-induced weight loss is associated with an improvement in β-cell function in older men. J Clin Endocrinol Metab. 2004;89:2704–2710. doi: 10.1210/jc.2003-031827. [DOI] [PubMed] [Google Scholar]
- 32.Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, IlanneParikka P, Keinanen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, Salminen V, Uusitupa M. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–1350. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 33.Eriksson K-F, Lindgarde F. Prevention of type 2 (non-insulin-dependent) diabetes mellitus by diet and physical exercise: the 6-year Malmo feasibility study. Diabetologia. 1991;34:891–898. doi: 10.1007/BF00400196. [DOI] [PubMed] [Google Scholar]
- 34.Pan XR, Li GW, Hu YH, Wang JX, Yang WY, An ZX, Hu ZX, Lin J, Xiao JZ, Cao HB, Liu PA, Jiang XG, Jiang YY, Wang JP, Zheng H, Zhang H, Bennett PH, Howard BV. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance: the Da Qing IGT and Diabetes Study. Diabetes Care. 1997;20:537–544. doi: 10.2337/diacare.20.4.537. [DOI] [PubMed] [Google Scholar]
- 35.Duckworth WC, Kitabchi AE, Heinemann M. Direct measurement of plasma proinsulin in normal and diabetic subjects. Am J Med. 1972;53:418–427. doi: 10.1016/0002-9343(72)90137-4. [DOI] [PubMed] [Google Scholar]
- 36.Kitabchi AE. Proinsulin and C-peptide a review. Metabolism. 1977;26:547–587. doi: 10.1016/0026-0495(77)90099-3. [DOI] [PubMed] [Google Scholar]
- 37.Mykkanen L, Haffner SM, Hales CN, Ronnemaa T, Laakso M. The relation of proinsulin, insulin, and proinsulin-to-insulin ratio to insulin sensitivity and acute insulin response in normoglycemic subjects. Diabetes. 1997;46:1990–1995. doi: 10.2337/diab.46.12.1990. [DOI] [PubMed] [Google Scholar]
- 38.Reaven GM, Chen Y-DI, Hollenbeck CB, Sheu WHH, Ostrega D, Polonsky KS. Plasma insulin, C-peptide, and proinsulin concentrations in obese and nonobese individuals with varying degrees of glucose tolerance. J Clin Endocrinol Metab. 1993;76:44–48. doi: 10.1210/jcem.76.1.8421101. [DOI] [PubMed] [Google Scholar]
- 39.Kahn SE, Leonetti DL, Prigeon RL, Bergstrom RW, Fujimoto WY. Proinsulin as a marker for the development of NIDDM in Japanese-American men. Diabetes. 1995;44:173–179. doi: 10.2337/diab.44.2.173. [DOI] [PubMed] [Google Scholar]
- 40.Ward WK, LaCava EC, Paquette TL, Beard JC, Wallum BJ, Porte D., Jr Disproportionate elevation of immunoreactive proinsulin in type 2 (non-insulin-dependent) diabetes mellitus and experimental insulin resistance. Diabetologia. 1987;30:698–702. doi: 10.1007/BF00296991. [DOI] [PubMed] [Google Scholar]
- 41.Kahn SE, Halban PA. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes. 1997;46:1725–1732. doi: 10.2337/diab.46.11.1725. [DOI] [PubMed] [Google Scholar]