


Abstract
Restriction endonucleases (REases) with 8-base specificity are rare specimens in nature. NotI from Nocardia otitidis-caviarum (recognition sequence 5′-GCGGCCGC-3′) has been cloned, thus allowing for mutagenesis and screening for enzymes with altered 8-base recognition and cleavage activity. Variants possessing altered specificity have been isolated by the application of two genetic methods. In step 1, variant E156K was isolated by its ability to induce DNA-damage in an indicator strain expressing M.EagI (to protect 5′-NCGGCCGN-3′ sites). In step 2, the E156K allele was mutagenized with the objective of increasing enzyme activity towards the alternative substrate site: 5′-GCTGCCGC-3′. In this procedure, clones of interest were selected by their ability to eliminate a conditionally toxic substrate vector and induce the SOS response. Thus, specific DNA cleavage was linked to cell survival. The secondary substitutions M91V, F157C and V348M were each found to have a positive effect on specific activity when paired with E156K. For example, variant M91V/E156K cleaves 5′-GCTGCCGC-3′ with a specific activity of 8.2 × 104 U/mg, a 32-fold increase over variant E156K. A comprehensive analysis indicates that the cleavage specificity of M91V/E156K is relaxed to a small set of 8 bp substrates while retaining activity towards the NotI sequence.
INTRODUCTION
Considerable progress has been made in isolating restriction endonucleases (REases) with altered substrate specificity. Many of the efforts have employed a DNA-damage indicator strain as pioneered by Heitman and Model (1,2) who developed an SOS induction assay to screen for relaxed variants of EcoRI in order to identify amino acids that make specific contacts to cognate DNA. More recently, a genetic selection and screening procedure lead to the discovery of two important side chains for BstYI specificity (wt specificity is 5′-RGATCY-3′ where R = A or G, Y = C or T). The BstYI evolution procedure yielded a variant with a marked increase in substrate specificity (3). The variant K133N/S172N possessed a 12-fold preference for AGATCT over AGATCC or GGATCT and cleavage of GGATCC was no longer detectable. In subsequent X-ray structural studies of wt BstYI, only two amino acid residues were found making base-specific contacts to the 5′-AGATCT-3′ sequence (4). Remarkably, the two residues were K133 and S172. In a structure-guided study of BsoBI (specificity = 5′-CYCGRG-3′), variant D246A was found to display a preference for cleavage of 5′-CCCGGG-3′ (5). Allele D246A was then subjected to random mutagenesis and in vivo screening to isolate five unique variants with increased activity. This study of BsoBI is the first example where a DNA-damage indicator strain was used for the primary purpose of increasing REase specific activity.
A novel approach was applied to Eco57I (5′-CTGAAG-3′) to obtain a Type IIG enzyme with altered specificity (6,7). A cleavage minus variant was randomly mutated and subjected to the methylase selection procedure. Challenging the mutant library in vitro with GsuI (5′-CTGGAG-3′) enabled isolation of an Eco57I variant with methylation specificity for 5′-CTGRAG-3′ and corresponding endonuclease activity upon restoration of the catalytic residue by site-directed mutagenesis.
Other progress has been made with altering the specificity of the homing endonuclease I-CreI (8). After determining the structure of I-CreI bound to its native pseudo-palindromic 22 bp site, several base-contacting positions were chosen forrandomization (9–11). Using a series of two genetic assays (12), I-CreI variants were analyzed for their ability to cleave mutant DNA target sites. This in vivo test for cleavage activity reliably predicted a specificity shift in several cases and the approach should allow rapid identification of novel endonucleases potentially useful as gene-specific reagents. The homing endonuclease PI-SceI was also studied to achieve altered target site recognition (13). Protein variants were assessed in vivo using a bacterial two-hybrid system where cell growth was linked to the ability of protein variants to bind to wt or mutant target sequences and turn on expression of the HIS3 and aadA genes. These in vivo homing endonuclease studies were possible due to the infrequency or absence of recognition sites within Escherichia coli. This feature allowed Gruen et al. (14) to design a genetic selection system where homing endonuclease cleavage is linked to cell survival. The authors proposed that this system may be used to isolate endonucleases with altered specificity. The proof of principle study was carried out with I-SceI, which recognizes an 18 bp sequence. Upon cell transformation, active endonuclease eliminated a conditionally toxic substrate vector carrying either two or four copies of the I-SceI recognition sequence without causing genomic DNA degradation. Conditional-toxicity was accomplished by activating the toxic gene product (Barnase Gln2amb/Asp44amb) by introducing a suppressor tRNA (supE) at the same time as the endonuclease gene. Recently, Chen and Zhao (15) reported a cell survival selection method for evolution of homing endonucleases using a substrate vector carrying the ccdB gene under control of the araBAD promoter and Chames et al. (16) reported a method for homing endonuclease selection in yeast based on the restoration of an auxotrophic marker via double-strand break induced homologous recombination.
The systems developed for selection of homing endonucleases inspired us to test the feasibility of using in vivo methods to alter the specificity of a rare-cutting REase. We chose to study NotI from Nocardia otitidis-caviarum (ATCC 14630) with the recognition sequence 5′-GCGGCCGC-3′. This sequence is present only twenty-three times in the E.coli K12 genome (17). Despite the low frequency, the NotI recognition sequence must be modified to prevent cell death even when using tightly controlled expression systems. So initial genetic procedures to alter NotI substrate specificity were carried out with methylation of the cognate recognition sequence, in this case provided by M.EagI (5′-NCGGCCGN-3′).
NotI is widely used in molecular biology applications, yet the protein itself has not been studied in detail. Early work reported purification of NotI from the native strain, and a gel filtration analysis estimated the molecular mass at 85 000 Da (18). When the notIR gene was cloned by R. Morgan (NEB), the sequence revealed a calculated molecular weight of 42 419 Da for the gene product. Therefore, the primary form of this Type IIP enzyme is a dimer in the absence of DNA substrate. Type IIP enzymes are so classified according to their homodimeric recognition of palindromic DNA sequences (19). The eventual objective of this project is to isolate a NotI variant capable of recognizing and cleaving an alternative 8-base palindromic sequence. Two parts of this project will be described. Part 1 consisted of a genetic screen for variants with altered specificity and this work yielded variant E156K. In the second part, variant E156K was further mutagenized and improved variants were isolated using a selection/screening system where cell survival is linked to elimination of a conditionally toxic substrate vector.
MATERIALS AND METHODS
Construction of selection vector ptoxBAC832
A conditionally toxic substrate vector was designed to possess ten 5′-GCTGCCGC-3′ cleavage sites (Figure 3). The vector carries a modified E.coli yidC gene and the product acts as a bacteriostat when overproduced by Isopropyl-β-d-thiogalactopyranoside (IPTG) induction of gene expression and vector copy number. In earlier studies, overproduction of 6His-YidC from pPROEX-HTb (Invitrogen) was found to arrest the growth of E.coli in exponential phase (20). Likewise, cells carrying pPROEX-6hisyidC do not form colonies when plated on LB-agar containing ≥50 µM IPTG. The wt yidC gene was transferred to vector pR976 (P. Riggs, NEB) using the cloning sites NcoI and EcoRI. A DNA linker was ligated into the NcoI site of pR976-yidC to provide multiple 5′-GCTGCCGC-3′ substrate sites and to provide coding sequence for additional N-terminal amino acids, notably histidine residues. The insertion clones were tested for conditional-toxicity in strain ER1992 by streaking on LB-agar containing IPTG. Clone p15A-TC1E imposed lethality at 1 mM IPTG and was subsequently sequenced to find 2 linkers and thus four 5′-GCTGCCGC-3′ substrate sites at the beginning of the yidC gene. The coding strand linker sequence is 5′-P-CATGGCTGCCGCGTACTACCACCACCACCTCGATTACGATATCCCAACGACCGAAAACCTGTATTTTGCTGCCGC-3′. The antisense strand linker sequence is 5′P-CATGGCGGCAGCAAAATACAGGTTTTCGGTCGTTGGGATATCGTAATCGAGGTGGTGGTGGTAGTACGCGGCAGC-3′ (substrate sites are underlined). Insertion of 2 linkers results in the following extension to the N-terminus of YidC: NH2-MAAAYYHHHLDYDIPTTENLYFAAAMAAAYYHHHLDYDIPTTENLYFAAA-. The lacI-yidC gene pair was amplified from p15A-TC1E by KOD HiFi DNA polymerase (Novagen) using forward and reverse primers both containing one 5′-GCTGCCGC-3′ substrate site. The lacI primer anneals to the lacI promoter sequence and creates the lacIq mutation (C→T). The lacIq primer sequence is 5′-GTGACTGAATTCGCTGCCGCCACCATCGAATGGTGCAAAACCTTTCGCG-3′. The yidC reverse primer sequence is 5′-GAGCTCGAATTCGAAGCTGCCGCTTCCAATTGGTGTGGCGGATGC-3′. The toxic operon (lacIq-Ptac-yidC) was cloned into vector pBAClac832, whose copy number is controlled by LacI repression/IPTG induction. Vector pBAClac832 was created by combining seqments of pFOS1 (NEB) and a modified pUC18. Vector pUC18 was first modified by replacing the ori RNAII promoter with the lacZ operator/promoter sequence. Next, the BspHI site at position 2534 was destroyed, which enabled reversal of the ampicillin resistance gene (bla) by digestion at BspHI sites 1526 and 2639. Reversal of the bla gene eliminates read-through transcription from priming plasmid replication. After bla reversal, IPTG-dependent replication of this pUC18 derivative was confirmed in strain ER2502 (LacI+). Further modification to pUC18 consisted of ligating two substrate linkers into the PciI site to insert four 5′-GCTGCCGC-3′ sites. The modified pUC origin and bla gene were excised by SfoI–SalI digestion and ligated into the HpaI site and the SalI site (at position 2560) of pFOS1 to create pBAClac832. The toxic operon (lacIq-Ptac-yidC) was EcoRI-digested and cloned into the EcoRI sites of pBAClac832 to create the final selection vector ptoxBAC832 (Figure 3). Single copy replication (without IPTG) was verified by comparing DNA yields of ptoxBAC832 to a low-copy reference plasmid after co-propagation in Luria–Bertani (LB) media containing 0.2% glucose. The addition of 0.2% glucose is reported to limit the copy number of BAC clones to one copy (21). The selection strain was derived from ER1992 (22), a DNA-damage indicator strain. In addition to carrying the toxic selection vector, the selection strain also carried a plasmid expressing M.EagI (23) to protect the NotI and EagI sites within the E.coli chromosome. ER1992 was first transformed with pSYX20-eagIM (TetR) followed by transformation with ptoxFOS832 to yield the final selection strain. The selection strain was propagated in LB (0.2% glucose) plus 15 µg/ml tetracycline and 50 µg/ml carbenicillin (Cb). Note that the selection strain does not grow at higher levels of carbenicillin or ampicillin as a result of the bla promoter modification (BspHI site mutagenesis). Electro-competent cells ER1992 [pSYX20-eagIM, ptoxBAC832] were prepared by washing two times with ice-cold 10% glycerol after growth to OD600 = 0.8. After growth to OD600 = 0.8, toxic vector maintenance was analyzed by diluting the cell culture and plating on LB, LB + IPTG, LB + Cb and LB + Cb + IPTG. Under conditions used in the selection (LB + IPTG), loss of toxicity was found to occur at a frequency of 0.7% primarily due to loss of the entire vector. When plated on LB + Cb + IPTG, loss of toxicity occurs at a frequency of 1 × 10−4. This indicates that mutation/deletion of tox-yidC is rare since toxicity is maintained when the bla gene is maintained.
Transformation of the selection strain
After electroporation, SOC media containing 0.2% glucose and 0.1 M MgCl2 and 0.1 M MgSO4 was added for cell recovery and the culture was incubated for 2 h at 37°C to allow for library expression and possible elimination of the single copy ptoxBAC832. After the 2 h incubation, each transformation mix was plated on 6 LB-agar plates (150 × 15 mm) containing 15 µg/ml Tet, 30 µg/ml Cam, 1 mM IPTG and 80 µg/ml X-gal. The presence of 1 mM IPTG in the agar serves to induce high-copy toxic vector replication from the pUC origin and, at the same time, the tox-yidC gene product is over-expressed from Ptac. Therefore, cells retaining the selection vector after the 2 h incubation period are prevented from growing on the selection plates. Increased blue color development is a report of increased DNA cleavage of substrates differing within the middle six base pairs of the NotI recognition sequence (i.e. 5′-GCTGCCGC-3′).
Bacterial strains
All strains were from the NEB collection. ER2744 [fhuA2 glnV44 e14− rfbD1? relA1? spoT1? endA1 thi-1 Δ(mcrC-mrr)114::IS10 lacZ::T7gene1]. ER2848 [ER2744 with lacIq on F′ (TetR)]. ER2169 {E.coli B Δ(mcrC-mrr)102::Tn10 gal ompT [lon] [dcm]λDE3 = lambda sBamH1o ΔEcoRI-B int::lacI::PlacUV5::T7 gene1 i21 Δ nin5}. ER1992 [endA1 thi-1 glnV44 mcr-67 Δ(mcrC-mrr)114::IS10 Δ(argF-lac)U169 dinD1::MudI1734 (KanR, LacZ+)].
Oligonucleotides for cloning of NotI
The forward degenerate primer used to isolate a fragment of the NotI gene: 5′-GAGCCSGAGGGSGCSAAGTTCAT-3′ anneals at codons 8 through 14. The reverse degenerate primer 5′-GASGCGAACTTGTASGCCAT-3′ anneals at codons 248 through 242. The forward NotI cloning primer is: 5′-GACGCATATGCGTTCCGATACGTCGGTGGAGCCAGAG-3′. The second and third codons of the gene were changed to the preferred E.coli codons (underlined). The reverse cloning primer is: 5′-GAATGTCGACCATCTCCACCCACG-3′.
Plasmid substrates
Substrate pXba was created by ligating an XbaI fragment of Adenovirus-2 (nt 10 579 to 30 455) into the XbaI site of pUC19 (R. Morgan, NEB). The complete sequence of pXba is available via NEBcutter (http://tools.neb.com/NEBcutter2/index.php). All single site substrates were created by ligating annealed oligo duplexes into pUC19 digested with SalI and BamHI. For example, the pUC-GCT polylinker insert is: 5′-P-TCGACTCTAGAGCTGCCGCTG-3′ (top strand).
Mutagenesis
Random mutations were created by notIR gene amplification with Taq DNA polymerase using 25 cycles: (30 s at 94°C, 45 s at 58°C, 1:45 min at 72°C). Each reaction contained 7 mM MgSO4 and an unequal ratio of dNTPs (0.2 mM dATP and dGTP, 1.0 mM dCTP and dTTP). The T7 promoter primer (NEB S1248S) was used as the forward primer and the reverse primer was #279-125 (5′-CACGATCTCGAGGGCCGGTCCGACACCAGG-3′). Site-directed mutagenesis was used to create allele E156K. The forward primer was #286-184 (5′-CGATAGCTCGCCGAAGTTTTCATTCGA-3′) and the reverse primer was #286-185 (5′-TCGAATGAAAACTTCGGCGAGCTATCG-3′).
Protein purification
The IMPACT-CN purification system (NEB) was used to express and purify wt NotI and the variants of interest. Each notIR gene was cloned into the NdeI–SapI sites of pTYB1. As a result, each REase was expressed as a C-terminal fusion to the Sce intein and chitin binding domains. The host for over-expression was E.coli strain ER2848 (24) carrying pACYC-eagIM and pSYX20-bbvIM. The cells were grown at 30°C to OD600 = 0.65 and then induced overnight at 30°C with 0.4 mM IPTG. Cell extract was prepared using chitin column buffer: Tris–HCl (pH 7.8), 500 mM NaCl, 0.1 mM EDTA, 5% glycerol and 0.1% Triton X-100. After loading and washing, the chitin resin was flushed with column buffer containing 40 mM DTT. After overnight incubation at 4°C, REase was eluted with chitin column buffer containing 200 mM NaCl. The sodium chloride concentration was then reduced to 50 mM for DEAE column loading. REase was eluted from the DEAE column using a 0.05–0.8 M NaCl gradient. Fraction purity was assessed by SDS–PAGE and REase concentration was determined by the Bradford assay. Purified REase was dialyzed into NotI storage buffer [200 mM NaCl, 20 mM Tris–HCl (pH 7.4), 0.1 mM EDTA, 1 mM DTT and 50% glycerol] and stored at −20°C.
RESULTS
Cloning and characterization of wt NotI
The NotI endonuclease gene was cloned by first isolating a fragment of the gene by degenerate primer PCR. The NotI endonuclease was purified from N.otitidis-caviarum in order to perform protein sequencing (25). The protein was digested with cyanogen bromide, which produced four major peptide fragments of ∼24, 10, 4 and 3 kDa in size. Unambiguous amino acid sequence was determined for the 24, 10 and 4 kDa fragments. The N-terminal sequence of the 24 kDa peptide was found to match the N-terminal sequence of the endonuclease (GenBank accession no. DQ242471). Degenerate DNA primers based on the amino acid sequence of the N-terminal region and the 10 kDa fragment were synthesized and used to amplify this portion of the endonuclease gene from N.otitidis-caviarum genomic DNA by PCR. The primer sequences are listed in Materials and Methods. The amplified DNA fragment of ∼680 bp was cloned into pUC19 and sequenced. The amino acid sequence deduced from the DNA sequence of this PCR fragment matched the amino acid sequence of the NotI endonuclease determined by protein sequencing, confirming that this DNA fragment represented a portion of the NotI endonuclease gene. A BamHI library of N.otitidis-caviarum DNA was constructed in a Lambda Dash vector system to generate clones containing 9 to 23 kb inserts. The 680 bp fragment of the endonuclease gene was used as a probe to identify Lambda Dash clones containing this portion of the endonuclease gene. These lambda clones were purified and all were found to contain a common 14.5 kb fragment. The DNA flanking the probe was sequenced to identify the entire NotI endonuclease gene. DNA primers were designed to amplify the entire NotI endonuclease gene with an NdeI site at the 5′end and a SalI site ∼500 bp downstream of the 3′ end. The amplified DNA was ligated into the T7 expression vector pSYX22 and transformed into E.coli ER2169 competent cells pre-modified by M.EagI expressed from pACYC184. Clones expressing NotI activity were sequenced to confirm the presence of a wt NotI endonuclease gene. NotI was purified in order to determine the specific activity of the enzyme on a pUC19-derived single site substrate (data not shown). When using NEB buffer 3, the specific activity of NotI is 5 × 105 U per mg protein. (One unit is defined as the amount of enzyme required to linearize 1 µg of pUC-NotI in 1 h at 37°C in a reaction volume of 50 µl).
Selection of a NotI variant with altered cleavage specificity
An SOS induction assay (1,2,22,26,27) was employed to isolate a variant of NotI with altered cleavage specificity. The DNA-damage indicator strain ER1992 (22) was transformed with pACYC184-eagIM (CamR) to pre-modify and protect all 5′-CGGCCG-3′ sites within the genome. Therefore, limited endonuclease cleavage at sites other than 5′-NCGGCCGN-3′ were expected to induce the SOS response and result in expression of a dinD1::lacZ gene fusion. A randomly mutated notIR gene library was ligated into pAGR3 (3) immediately downstream of the tac promoter to allow low-level constitutive expression of the library. After electroporation into ER1992 [pACYC184-eagIM], transformants were selected on LB-agar plates containing chloramphenicol, ampicillin and X-gal. Of approximately 60 000 transformants, 10 blue colonies were isolated. The notIR gene of each clone was amplified by colony PCR and each was subcloned into the T7 expression vector pAII17 (28) for analysis of variant endonuclease activity. Expression of the NotI variants was conducted in ER2744 [pACYC184-eagIM]. Cell extracts were incubated with the GC-rich substrate pXba (22 562 bp) in order to maximize the likelihood of observing cleavage of one or more 8 bp sites related to the NotI recognition sequence. Cell extract from clone 44-2A produced a cleavage pattern similar to that shown in lane 7 of Figure 1. Sequencing clone 44-2A revealed the amino acid substitutions P9S and E156K. Site-directed mutagenesis was used to create the E156K allele and cleavage activity of this single variant was indistinguishable from the originally selected clone 44-2A (data not shown). NotI cleaves pXba six times leaving a large fragment of 16 749 bp. By inspection of the pXba sequence, it appeared that variant E156K was cleaving at 5′-GCTGCCGC-3′ in addition to the NotI site. Digestion of pBR322 (linearized with AflIII) with cell extract also indicated cleavage at 5′-TCGGCCGC-3′ (Figure 4, lane 5).
Figure 1.
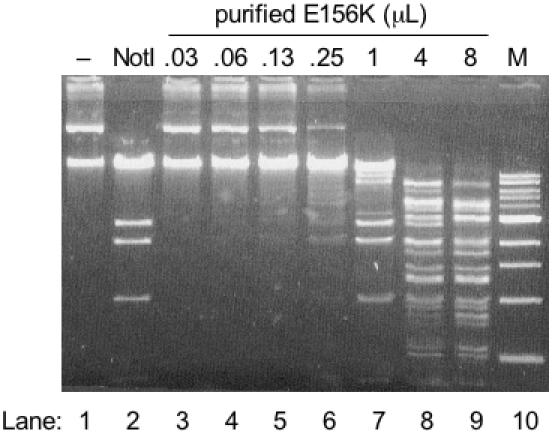
Altered cleavage characteristics of NotI variant E156K determined by digestion of plasmid substrate pXba. Lane 1, no enzyme addition (−); Lane 2, digestion with 100 U wt NotI; Lanes 3–9, increasing amounts of variant E156K. Lane M, 1 kb DNA ladder (NEB). All reactions were incubated at 37°C for 60 min in 1× NEB buffer 3.
Variant E156K was purified using the IMPACT-CN system (as described in Materials and Methods). Overproduction in E.coli was accomplished by providing genomic modification with the C5 methylases M.EagI and M.BbvI [see REBASE (29), http://rebase.neb.com]. M.BbvI methylates the first cytosine in the 5′-GCWGC-3′ sequence and this allowed for greater E156K production as compared to M.EagI modification alone (data not shown). Figure 1 displays the results of incubating purified E156K with substrate pXba. Lane 7 corresponds to an enzyme to plasmid ratio of 2 (enzyme as amount of dimer). Near complete digestion of pXba is observed in lane 9 where the enzyme excess is 16-fold. The substrate pXba contains nine 5′-GCTGCCGC-3′ sites and one 5′-TCGGCCGC-3′ site. A virtual digest of pXba at these sites plus the NotI sites gave a pattern similar but not identical to that in lane 9 of Figure 1. To confirm cleavage at 5′-GCTGCCGC-3′ a pUC19-derived substrate containing a single site was incubated with purified E156K and complete linearization was achieved with an 8-fold excess of enzyme/substrate (Figure 2). The expected cut site was verified by run-off sequencing of the pUC-GCT linear product (data not shown). Variant E156K was also incubated with a substrate containing the closely related symmetric sequence 5′-GCTGCAGC-3′ but cleavage at this site could not be detected. Considering these results, the next objective was to increase the specific activity of this variant by selecting clones with secondary mutations.
Figure 2.

Variant E156K digestion of a substrate containing a single 5′-GCTGCCGC-3′ site. Plasmid pUC-GCT was derived from pUC19. The enzyme/substrate (E/S) molar ratio is given above each lane. Lane M, 1 kb DNA ladder. All reactions were incubated at 37°C for 60 min in 1× NEB BamHI buffer.
Selection of variants with enhanced cleavage of 5′-GCTGCCGC-3′
The second stage of this project was designed to increase the activity of variant E156K. A novel modification to the SOS induction assay was designed to link cell survival to cleavage at 5′-GCTGCCGC-3′. This modification was inspired by the work of Gruen et al. (14). In our selection/screening procedure, strain ER1992 carried the conditionally toxic substrate vector ptoxBAC832 (Figure 3). IPTG induction of tox-yidC expression and vector copy number halts cell growth, thus colony formation is linked to elimination of the toxic vector. The frequency of colony formation without introduction of a variant endonuclease library was found to be 0.7% (see Materials and Methods). The selection strain reports enhanced cleavage of any non-protected substrate sites (as blue colony color) but by design only after cleavage of 5′-GCTGCCGC-3′ results in toxic vector elimination. The selection strain was modified by M.EagI, so all 8 bp sites conforming to 5′-NCGGCCGN-3′ were protected from endonucleolytic cleavage. Therefore, blue color development is a report of increased cleavage of substrates differing within the middle 6 bp of the NotI recognition sequence (i.e. 5′-GCTGCCGC-3′). Although blue color development may reflect a general relaxation of substrate specificity, the in vivo screen places a strict constraint on the degree of relaxation allowed. As enzyme specificity is relaxed, the cell becomes increasingly susceptible to lethal levels of DNA-damage. Therefore, NotI variants selected by this in vivo procedure were expected to fall within a narrow range with regard to substrate specificity and specific activity.
Figure 3.
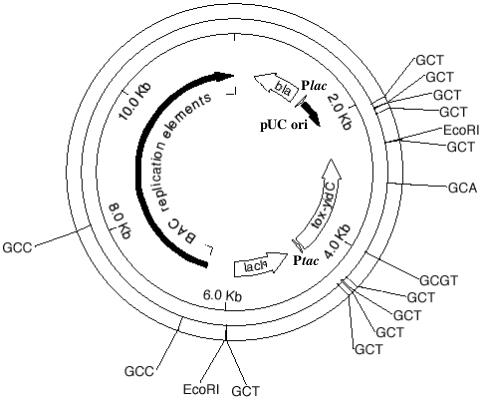
An illustration of the conditionally toxic selection vector ptoxBAC832 prepared by the MapDraw program of DNASTAR Lasergene. GCT denotes the sequence 5′-GCTGCCGC-3′. GCA denotes the sequence 5′-GCAGCCGC-3′. GCC denotes the sequence 5′-GCCGCCGC-3′. GCGT denotes the sequence 5′-GCGTCCGC-3′.
A randomly mutated gene library (derived from the E156K allele) was cloned into three versions of pACYC-T7ter (30) to determine which vector would provide the appropriate level of expression for the selection procedure. The appropriate level of E156K expression would inflict a minor level of DNA-damage on the selection strain so that the dinD1::lacZ reporter gene would be minimally expressed. In such a scenario, ‘up-mutants’ can be selected. The appropriate expression vector was determined by comparing the parent vector pACYC-T7ter with two other versions where deletions were made upstream of the T7 promoter. Vector pACYC-T7ΔSP was modified by a SmaI–PshAI deletion, which removes the lacI gene and the four rrnb transcriptional terminators upstream of the T7 promoter. Vector pACYC-T7terΔEP was modified by an EcoNI–PshAI deletion, which removes only the lacI gene. The net effect of each deletion is increased expression of the notI library gene from read-through transcription of the chloramphenicol resistance gene (cat). Since the selection strain ER1992 does not express the T7 RNA polymerase, cat read-through transcripts serve as the primary source of notIR mRNA from pACYC-T7terΔEP and pACYC-T7ΔSP. Vector pACYC-T7terΔEP was chosen as the library vector according to the phenotype presented by the parent clone encoding E156K. The phenotype of E156K was light-blue when transformed into the selection strain.
The mutated library was prepared from the notIR E156K allele by error-prone PCR mutagenesis and the product was cloned into pACYC-T7terΔEP. Approximately 10 ng of ligation mix was transformed into the selection strain in each of two electroporation reactions. The normal plating efficiency (without IPTG) was tested and determined to be 1 × 107 c.f.u./µg. Under selection conditions (with IPTG), each transformation yielded ∼1000 colonies (a 100-fold reduction). A total of 12 medium to dark blue colonies were obtained and the plasmid DNA from each single colony was isolated by the QIAprep procedure. The notIR insert of each isolate was amplified by PCR using a T7 forward primer (NEB S1248S) and reverse primer 279-125. Each PCR fragment was sequenced to determine the NotI variant amino acid sequence.
Table 1 summarizes the NotI clones isolated from the ptoxBAC832-mediated selection. The parent clone (variant E156K) was selected twice. This is a result of processing all blue survivors regardless of color intensity since the visual distinction between light or medium blue is not clear. The other ten selected clones each contained one mutation resulting in an additional amino acid substitution. Most striking is the repeated selection of double mutants E156K/F157C and M91V/E156K.
Table 1.
NotI clones isolated from the ptoxBAC832-mediated selection procedure
| Clone name | Amino acid substitutions |
|---|---|
| EP1 | E156K/V348M |
| EP2 | E156K only |
| EP3 | E156K/T352A |
| EP4 | E156K/E74V |
| EP5 | E156K/F157C |
| EP6 | E156K/F157C |
| EP7 | E156K/F157C |
| EP8 | E156K/F157C |
| EP9 | E156K only |
| EP10 | M91V/E156K |
| EP11 | M91V/E156K |
| EP12 | E156K/A243V |
All mutant alleles were cloned into expression vector pLT7K (31) to enable analysis of activity using cell extracts. The expression strain ER2848 (24) co-expressed M.EagI and M.BbvI (5′-GCWGC-3′) to protect NotI sites and the desired alternative sites within the host genome. The most active variants were EP1 (E156K/V348M), EP5-EP8 (E156K/F157C) and EP10-EP11 (M91V/E156K) as revealed by λ DNA digestion (data not shown). Note that λ DNA does not encode any NotI sites but does possess five 5′-GCTGCCGC sites. The digestion patterns of the three most active double variants were nearly indistinguishable and similar to the pattern observed for purified E156K. Thus, all second site mutations appeared to primarily enhance specific activity. Cell extracts from EP1, EP5 and EP10 were analyzed in more detail by assessing cleavage of pBR322 (Figure 4). In this assay, variant EP10 (lane 4) appeared to possess the highest activity (assuming an equal expression level). The expression level of each variant is governed by whether host DNA-damage is prevented by co-expression of a site-specific MTase. Therefore, a REase variant possessing a desired range of specificity is allowed to accumulate within the modified host cells while variants with undesirable substrate specificity are lethal or poorly expressed. This fact is relevant when screening cell extracts for REase activity. If considerable cleavage activity is observed, then one may assume that the substrate specificity falls primarily within the desired range.
Figure 4.
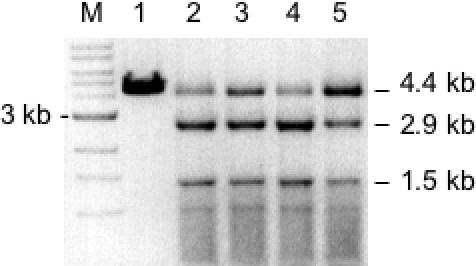
Evaluation of double variants by digestion of pBR322 (linearized by AflIII). Lane M, 1 kb DNA ladder; Lane 1, incubation with 100 U wt NotI to confirm no cleavage of pBR322; Lane 2, incubation with cell extract from clone EP1 (E156K/V348M); Lane 3, incubation with cell extract from clone EP5 (E156K/F157C); Lane 4, incubation with cell extract from clone EP10 (M91V/E156K); Lane 5, incubation with cell extract containing variant E156K. All reactions were at 37°C for 60 min in 1× NEB buffer 3. Production of 2.9 and 1.5 kb fragments is consistent with cleavage at 5′-TCGGCCGC-3′.
The cleavage specificity of M91V/E156K was determined by overdigesting (E/S > 100) the XbaI fragment from nt 10 579 to 30 455 of Adenovirus-2 DNA. This digestion and all subsequent reactions were carried out at 37°C in NEB BamHI buffer [150 mM NaCl, 10 mM Tris–HCl, 10 mM MgCl2, 1 mM DTT (pH 7.9 at 25°C)]. After digestion, the cohesive ends were made blunt by T4 DNA polymerase. The DNA was purified by QIAprep spin column and ligated to pUC19 (linearized with HincII and dephosphorylated by CIP). The ligation mix was transformed into E.cloni® electro-competent cells (Lucigen). Forty clones containing insert were isolated by blue/white screening and the ligation junctions were sequenced with M13/pUC primers S1224S and S1233S (NEB). By this procedure, DNA cleavage was confirmed at the sites listed in Table 2 (excluding 5′-GAGGCCGC-3′ due to the absence of this site in the substrate and excluding 5′-GGGGCCGC-3′). The junction sequences also verified that double-strand (ds) cleavage is occurring at the expected positions to leave 4-base 5′ overhangs. The specific activity data in Table 2 is summarized from reactions where purified enzyme was incubated with pUC19-derived single site substrates as exemplified in Figures 2 and 5. A control digest confirmed that wt pUC19 is resistant to cleavage by variant M91V/E156K (data not shown). The complete analysis indicates that variant M91V/E156K is able to cleave seven miscognate sites with nearly the same efficiency as cleavage of the cognate NotI site. In contrast, nearly exclusive nicking was found to occur at 5′-GCGTCCGC-3′ (data not shown). It appears that ds cleavage at the other asymmetric sites may proceed through a nicked intermediate as indicated by transient formation of a slower mobility species in Figures 2 and 5. Considering the homodimeric nature of wt NotI, we expected that variant M91V/E156K would be able to act upon a symmetric 8 bp site. However, cleavage of the closely related 5′-GCTGCAGC-3′ sequence was not detectable.
Table 2.
Summary of alternative cleavage sites and specific activity of variant M91V/E156K
| Site name | Sequence | Specific activitya | Comments |
|---|---|---|---|
| Notl | GCGGCCGC | 2.1 × 104 | |
| AEagC | ACGGCCGC | 1.6 × 105 | |
| CEagC | CCGGCCGC | 1.6 × 105 | |
| TEagC | TCGGCCGC | 2.1 × 104 | Impaired by overlapping Dam |
| GCA | GCAGCCGC | – | Slow cleavage with E/S >1 |
| GCC | GCCGCCGC | – | Slight cleavage with E/S >1 |
| GCT | GCTGCCGC | 8.2 × 104 | |
| GCGA | GCGACCGC | 1.0 × 104 | |
| GCGC | GCGCCCGC | – | Slow cleavage with E/S >1 |
| GCGT | GCGTCCGC | 2.1 × 104 | Units refer to nicking activity |
| GAGG | GAGGCCGC | 8.2 × 104 | |
| GTGG | GTGGCCGC | 1.0 × 104 | |
| GGGG | GGGGCCGC | – | Slow nicking with E/S >1 |
| ATGG | ATGGCCGC | – | Slight cleavage with E/S >1 |
aSpecific activity is given as units per mg enzyme.
Specific activity was determined by linearizing single site substrates derived from pUC19. One unit is defined as the amount of enzyme required to linearize 1 µg of a pUC 19 derivative in 1 h at 37°C in a reaction volume of 50 µl.
Figure 5.
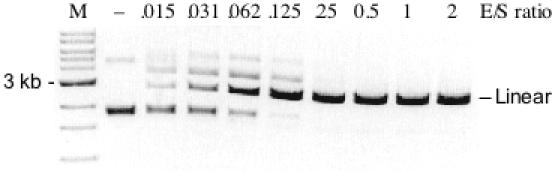
Variant M91V/E156K digestion of plasmid substrate pUC-GCT. The enzyme/substrate (E/S) molar ratio is given above each lane. Lane M, 1 kb DNA ladder. All reactions were incubated at 37°C for 60 min in 1 × NEB BamHI buffer. (−) indicates no enzyme addition.
The data of Figures 2 and 5 provide evidence for the activity enhancement resulting from the M91V secondary substitution. Remarkably, this seemingly minor side chain modification results in a 32-fold increase in specific activity towards 5′-GCTGCCGC-3′. In fact, M91V/E156K performs ds cleavage at several 8 bp sequences with activities ranging from 1.6 × 105 to 1.0 × 104 U per mg protein, which is only 3- to 50-fold lower than the activity of wt NotI towards its cognate sequence.
A competition assay was conducted to evaluate cleavage of this alternative site in a reaction where the wt NotI recognition sequence is also present. Figure 6 shows the results of incubating variant M91V/E156K with equimolar amounts of two linear substrates derived from pUC-NotI and pUC-GCT. Complete cleavage of both substrates occurs at nearly the same enzyme level indicating a slight preference for the wt recognition sequence. The practicality of using variant M91V/E156K in extended DNA digestion reactions was tested by incubating excess enzyme (E/S = 30) with 1 µg T7 genomic DNA for times ranging up to 16 h. A virtual digest by NEBcutter (32) is given in Figure 7A while the actual results are displayed in Figure 7B. (Note that ds cleavage at 5′-GCGTCCGC-3′ and was excluded in the virtual digest since this site is primarily nicked). The cleavage pattern at 1 h is nearly identical to the cleavage pattern at 16 h. As indicated by the data in Table 2, cleavage at 5′-GCAGCCGC-3′ is slow even with excess enzyme. This leads to slow production of fragments 3 and 5 as listed in the table within Figure 7A. However, anomalous DNA digestion is not observed after the 16 h incubation time. These results and the results of digesting various other substrates in this study provide a high degree of certainty that the substrate specificity of M91V/E156K has been fully characterized.
Figure 6.
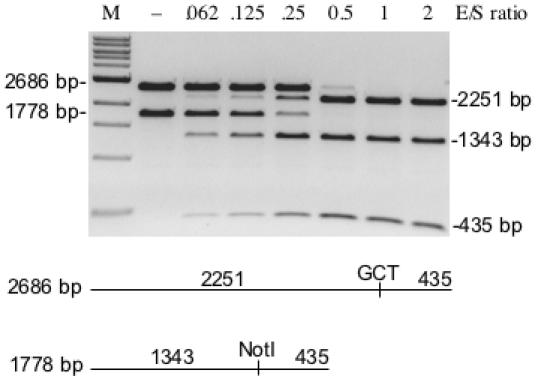
Competitive cleavage of two linear substrates by variant E156K/M91V. The 2686 bp substrate was derived from pUC-GCT and the 1778 bp substrate was derived from pUC-NotI. Equimolar amounts of each substrate were mixed and incubated with increasing concentrations of enzyme. All reactions were incubated at 37°C for 60 min in 1× NEB BamHI buffer. (−) indicates no enzyme addition.
Figure 7.
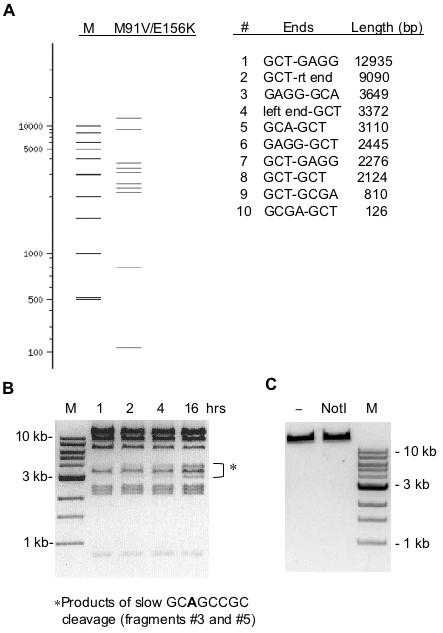
Digestion of T7 genomic DNA by variant M91V/E156K. (A) A virtual digest of T7 DNA using NEBcutter (29). Since site 5′-GCGTCCGC-3′ is nicked by this enzyme it was not included in the virtual digest. M is a size marker lane. (B) Actual results of digesting T7 DNA in 1× NEB BamHI buffer for various times at 37°C. A 30-fold molar excess of enzyme was used in each reaction. (C) A control showing no digestion of T7 DNA by 200 U wt NotI.
Finally, we confirmed that the activity of M91V/E156K is not an exaggerated star activity of the wt enzyme. A thorough study of wt NotI was conducted using the plasmid substrates listed in Table 2 so that non-specific nicking or ds cleavage would be revealed. Limited ds cleavage was only detected at ACGGCCGC when the E/S ratio exceeded one and when low salt conditions were used (NEB buffer 2 with 50 mM NaCl). When using a recommended buffer (i.e. NEB buffer 3 containing 100 mM NaCl) no star activity was detected with a molar excess of wt enzyme.
DISCUSSION
REases exhibiting 8 bp substrate specificity are rarely isolated from nature (see REBASE). This may be partly due to screening methods but, most likely, such enzymes are indeed rare specimens. The objective of this project was to alter the specificity of the native REase NotI to obtain an enzyme with novel specificity towards one or more alternative 8 bp sites. The absence of a three dimensional structure of NotI led us to design an approach based on gene randomization and in vivo screening and selection. The initial stage of the project was designed to isolate variants with altered specificity. This was achieved by screening NotI variants within a DNA-damage indicator strain modified by a cognate DNA MTase (1). In this procedure, M.EagI substituted for M.NotI but it may be considered as cognate since it modifies 5′-GCGGCCGC-3′ as well as all other 8 bp sequences conforming to 5′-NCGGCCGN-3′. Cells reporting DNA-damage were expected to contain NotI variants able to cleave one or more sites differing within the middle 6 bp of the NotI recognition sequence. Stage 1 yielded variant E156K with additional cleavage activity at the asymmetric site 5′-GCTGCCGC-3′ (arbitrarily designated as the top strand). Overproduction of variant E156K was enabled by providing a methylation state to prevent genomic DNA cleavage at sites containing 5′-GCWGC-3′ (M.BbvI). Upon purification of variant E156K, cleavage at the alternative recognition sequences was found to be very inefficient. Thus, a second stage of the project was designed to isolate variants with enhanced cleavage of the 5′-GCTGCCGC-3′ target sequence.
Stage 2 was designed as a genetic selection where cell survival is linked to cleavage of the target sequence residing within a conditionally toxic vector. The proper level of endonuclease library expression was empirically established so that up-mutants could be isolated without inflicting a lethal level of genomic DNA-damage. The key consideration of this selection is the number of target sites within the vector versus the number within the E.coli genome. In this case, the vector was designed to encode ten target sequences all flanking the tox-yidC gene while the E.coli K12 genome possesses over 600 of the target sites (or an average of one per 7 kb). Regardless of the unfavorable ratio of sites, the selection procedure yielded several variants with enhanced specific activity towards 5′-GCTGCCGC-3′. The fact that multiple clones of the same genotype were selected out of a library of 1 × 107 is evidence that these blue colonies did not appear by chance. Spontaneous loss of the toxic vector (at a frequency of 0.7%) is a source of survivors but the additional requirement of blue colony phenotype nearly eliminated interference by false positives. In fact, only the two selected parent clones may be regarded as false positives. Further selection procedures will employ a vector with multiple copies of the final desired recognition sequence 5′-GCTGCAGC′-3′. This site occurs only 104 times within the E.coli genome (every 44 kb on average). We speculate that variant M91V/E156K may be further mutated to cleave this symmetric site.
The secondary amino acid substitutions present in the most active stage 2 variants were M91V, F157C and V348M. A suppressor substitution at position 157 might be expected in this situation and can be rationalized in terms of spatial constraints. Replacing phenylalanine with cysteine compensates for a larger side chain at position 156 in the parent E156K. However, the relevance of compensatory side chain functionality is difficult to discuss without an X-ray structure. Most likely, E156 is a DNA contacting residue due to the severe effect of inserting a lysine at this position. Supporting this idea is an observation that E156 falls in the middle of a putative Type II active site consensus motif: TD(X)12EVE (position T151 to E167). Another prediction based on alignment with EagI indicates that the active site residues may be FD(X)21EIQ (position F159 to Q184). In any case, E156 is expected to be in close proximity to the DNA substrate. In contrast, it is indeed possible that residue 91 and/or residue 348 be located some distance away from the protein–DNA interface. As observed in the study of BsoBI, most suppressor substitutions were not in direct contact with DNA or even in the vicinity of the original amino acid substitution (5). Subtle changes (such as M91V or V348M) are clearly important for acquiring efficient cleavage of a new DNA sequence. Many structure-guided endeavors have shown that REase specificity cannot be distinctly altered by simply substituting DNA contact residues (33–38).
One may imagine that NotI variant (M91V/E156K) is an ‘intermediate’ on an evolutionary pathway. The parent specificity 5′-GCGGCCGC-3′ is still retained; yet several new sequences yield to cleavage. The results of the competition assay confirm that the double variant is losing the ability to recognize the NotI site, which is an eventual goal of this project. Recognition of 5′-GCTGCCGC-3′ may be considered as one step toward recognition of the palindromic sequence 5′-GCTGCAGC-3′. This desired specificity is also compatible with M.BbvI (5′-GCWGC-3′) host protection so further laboratory evolution would employ this MTase for REase over-expression steps. Evolution of REase specificity in nature is not well characterized. A recent study argues that MboI is evolutionarily related to EcoRI, despite the fact of divergent substrate specificity (39). This proposition gains more credence if DNA MTases are brought into the discussion. The primary determinant of REase specificity may be, in fact, the partner MTase. Without adequate host protection, REase evolution may not be possible. One scenario is co-evolution of MTase and REase specificity. But equally likely (and possible in the case of MboI) is the scenario of REase evolution to match the specificity of an acquired or existing MTase. The laboratory evolution of BstYI (3) was based on evolving REase function to match an existing MTase specificity. Similarly, another step forward can be taken in the evolution of NotI by carrying out a procedure analogous to that applied to BstYI. Many recent advances in endonuclease engineering have one feature in common. Genetic methods were applied to enzymes or variants whose cytotoxicity is ‘manageable’. The selection system established for evolving NotI may be applied to other rare-cutting REases or homing endonucleases. Engineered endonucleases with specificity in the 8–18 bp range will bridge a gap left by the forces of nature.
Acknowledgments
The authors thank Ross Dalbey and Minyong Chen for providing the pPROEX-6hisyidC construct; Rich Roberts and Siu-hong Chan for critical reading of the manuscript; Laurie Mazzola and Jennifer Ware for DNA sequencing; Roger Knott for oligonucleotide synthesis and purification; Elisabeth Raleigh and Mern Sibley for bacterial strains; and Don Comb for support. S.L.P. was supported by the NEB summer student internship program. Funding to pay the Open Access publication charges for this article was provided by NEB.
Conflict of interest statement. None declared.
REFERENCES
- 1.Heitman J., Model P. Mutants of the EcoRI endonuclease with promiscuous substrate specificity implicate residues involved in substrate recognition. EMBO J. 1990;9:3369–3378. doi: 10.1002/j.1460-2075.1990.tb07538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heitman J., Model P. SOS induction as an in vivo assay of enzyme-DNA interactions. Gene. 1991;103:1–9. doi: 10.1016/0378-1119(91)90383-m. [DOI] [PubMed] [Google Scholar]
- 3.Samuelson J.C., Xu S.Y. Directed evolution of restriction endonuclease BstYI to achieve increased substrate specificity. J. Mol. Biol. 2002;319:673–683. doi: 10.1016/S0022-2836(02)00343-1. [DOI] [PubMed] [Google Scholar]
- 4.Townson S.A., Samuelson J.C., Xu S.Y., Aggarwal A.K. Implications for switching restriction enzyme specificities from the structure of BstYI bound to a BglII DNA sequence. Structure (Camb) 2005;13:791–801. doi: 10.1016/j.str.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Z., Zhou J., Friedman A.M., Xu S.Y. Isolation of BsoBI restriction endonuclease variants with altered substrate specificity. J. Mol. Biol. 2003;330:359–372. doi: 10.1016/s0022-2836(03)00595-3. [DOI] [PubMed] [Google Scholar]
- 6.Rimseliene R., Maneliene Z., Lubys A., Janulaitis A. Engineering of restriction endonucleases: using methylation activity of the bifunctional endonuclease Eco57I to select the mutant with a novel sequence specificity. J. Mol. Biol. 2003;327:383–391. doi: 10.1016/s0022-2836(03)00142-6. [DOI] [PubMed] [Google Scholar]
- 7.Janulaitis A., Stankevicius K., Lubys A., Markauskas A. Nuclease. 20030040614 A1. U.S. patent application. 2003
- 8.Sussman D., Chadsey M., Fauce S., Engel A., Bruett A., Monnat R., Jr, Stoddard B.L., Seligman L.M. Isolation and characterization of new homing endonuclease specificities at individual target site positions. J. Mol. Biol. 2004;342:31–41. doi: 10.1016/j.jmb.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 9.Heath P.J., Stephens K.M., Monnat R.J.J., Stoddard B.L. The structure of I-Crel, a group I intron-encoded homing endonuclease. Nature Struct. Biol. 1997;4:468–476. doi: 10.1038/nsb0697-468. [DOI] [PubMed] [Google Scholar]
- 10.Jurica M.S., Monnat R.J., Jr, Stoddard B.L. DNA recognition and cleavage by the LAGLIDADG homing endonuclease I-CreI. Mol. Cell. 1998;2:469–476. doi: 10.1016/s1097-2765(00)80146-x. [DOI] [PubMed] [Google Scholar]
- 11.Chevalier B., Turmel M., Lemieux C., Monnat R.J., Jr, Stoddard B.L. Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J. Mol. Biol. 2003;329:253–269. doi: 10.1016/s0022-2836(03)00447-9. [DOI] [PubMed] [Google Scholar]
- 12.Seligman L.M., Chisholm K.M., Chevalier B.S., Chadsey M.S., Edwards S.T., Savage J.H., Veillet A.L. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res. 2002;30:3870–3879. doi: 10.1093/nar/gkf495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gimble F.S., Moure C.M., Posey K.L. Assessing the plasticity of DNA target site recognition of the PI-SceI homing endonuclease using a bacterial two-hybrid selection system. J. Mol. Biol. 2003;334:993–1008. doi: 10.1016/j.jmb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Gruen M., Chang K., Serbanescu I., Liu D.R. An in vivo selection system for homing endonuclease activity. Nucleic Acids Res. 2002;30:e29. doi: 10.1093/nar/30.7.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z., Zhao H. A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Res. 2005;33:e154. doi: 10.1093/nar/gni148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chames P., Epinat J.-C., Guillier S., Patin A., Lacroix E., Pâques F. In vivo selection of engineered homing endonucleases using double-strand break induced homologous recombination. Nucleic Acids Res. 2005;33:e178. doi: 10.1093/nar/gni175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blattner F.R., Plunkett G., IIIrd, Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 18.Sznyter L.A., Brooks J.E. The characterization and cloning of the NotI restriction-modification system. Heredity. 1988;61:308. doi: 10.1016/0378-1119(88)90250-8. [DOI] [PubMed] [Google Scholar]
- 19.Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev S., Dryden D.T., Dybvig K., et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samuelson J.C., Chen M., Jiang F., Moller I., Wiedmann M., Kuhn A., Phillips G.J., Dalbey R.E. YidC mediates membrane protein insertion in bacteria. Nature. 2000;406:637–641. doi: 10.1038/35020586. [DOI] [PubMed] [Google Scholar]
- 21.Wild J., Hradecna Z., Szybalski W. Conditionally amplifiable BACs: switching from single-copy to high-copy vectors and genomic clones. Genome Res. 2002;12:1434–1444. doi: 10.1101/gr.130502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fomenkov A., Xiao J.-P., Dila D., Raleigh E., Xu S.-Y. The endo-blue method for direct cloning of restriction endonuclease genes in E.coli. Nucleic Acids Res. 1994;22:2399–2403. doi: 10.1093/nar/22.12.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sznyter L.A., Marcus A.M., Brooks J.E. The cloning and characterization of the EagI restriction-modification system. ASM Abstracts. 1989;89:180. [Google Scholar]
- 24.Raleigh E.A. Host strain for low uninduced expression of foreign RNA polymerase genes. 6,569,669. U.S patent no. 2003
- 25.Morgan R.D., Benner J.S., Claus T.E. Isolated DNA encoding the NotI restriction endonuclease and related methods for producing the same. 5,371,006. U.S. patent no. 1994
- 26.Walker G.C. Mutagenesis and inducible responses to deoxyribonucleic acid damage in Escherichia coli. Microbiol. Rev. 1984;48:60–93. doi: 10.1128/mr.48.1.60-93.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker G.C. Inducible DNA repair systems. Annu. Rev. Biochem. 1985;54:425–457. doi: 10.1146/annurev.bi.54.070185.002233. [DOI] [PubMed] [Google Scholar]
- 28.Kong H., Kucera R.B., Jack W.E. Characterization of a DNA polymerase from the hyperthermophile archaea Thermococcus litoralis. Vent DNA polymerase, steady state kinetics, thermal stability, processivity, strand displacement, and exonuclease activities. J. Biol. Chem. 1993;268:1965–1975. [PubMed] [Google Scholar]
- 29.Roberts R.J., Vincze T., Posfai J., Macelis D. REBASE—restriction enzymes and DNA methyltransferases. Nucleic Acids Res. 2005;33:D230–D232. doi: 10.1093/nar/gki029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samuelson J.C., Zhu Z., Xu S.Y. The isolation of strand-specific nicking endonucleases form a randomized SapI expression library. Nucleic Acids Res. 2004;32:3661–3671. doi: 10.1093/nar/gkh674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong H., Lin L.F., Porter N., Stickel S., Byrd D., Posfai J., Roberts R.J. Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 2000;28:3216–3223. doi: 10.1093/nar/28.17.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vincze T., Posfai J., Roberts R.J. NEBcutter: a program to cleave DNA with restriction enzymes. Nucleic Acids Res. 2003;31:3688–3691. doi: 10.1093/nar/gkg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorner L.F., Bitinaite J., Whitaker R.D., Schildkraut I. Genetic analysis of the base-specific contacts of BamHI restriction endonuclease. J. Mol. Biol. 1999;285:1515–1523. doi: 10.1006/jmbi.1998.2408. [DOI] [PubMed] [Google Scholar]
- 34.Ivanenko T., Heitman J., Kiss A. Mutational analysis of the function of Met137 and Ile197, two amino acids implicated in sequence-specific DNA recognition by the EcoRI endonuclease. Biol. Chem. 1998;379:459–465. doi: 10.1515/bchm.1998.379.4-5.459. [DOI] [PubMed] [Google Scholar]
- 35.Schottler S., Wenz C., Lanio T., Jeltsch A., Pingoud A. Protein engineering of the restriction endonuclease EcoRV—structure-guided design of enzyme variants that recognize the base pairs flanking the recognition site. Eur. J. Biochem. 1998;258:184–191. doi: 10.1046/j.1432-1327.1998.2580184.x. [DOI] [PubMed] [Google Scholar]
- 36.Flores H., Osuna J., Heitman J., Soberon X. Saturation mutagenesis of His114 of EcoRI reveals relaxed-specificity mutants. Gene. 1995;157:295–301. doi: 10.1016/0378-1119(94)00863-n. [DOI] [PubMed] [Google Scholar]
- 37.Lanio T., Jeltsch A., Pingoud A. On the possibilities and limitations of rational protein design to expand the specificity of restriction enzymes: a case study employing EcoRV as the target. Protein Eng. 2000;13:275–281. doi: 10.1093/protein/13.4.275. [DOI] [PubMed] [Google Scholar]
- 38.Nastri H.G., Evans P.D., Walker I.H., Riggs P.D. Catalytic and DNA binding properties of PvuII restriction endonuclease mutants. J. Biol. Chem. 1997;272:25761–25767. doi: 10.1074/jbc.272.41.25761. [DOI] [PubMed] [Google Scholar]
- 39.Pingoud V., Sudina A., Geyer H., Bujnicki J.M., Lurz R., Luder G., Morgan R., Kubareva E., Pingoud A. Specificity changes in the evolution of type II restriction endonucleases: a biochemical and bioinformatic analysis of restriction enzymes that recognize unrelated sequences. J. Biol. Chem. 2005;280:4289–4298. doi: 10.1074/jbc.M409020200. [DOI] [PubMed] [Google Scholar]