

Abstract
In this study, we examined the role of protein kinase C (PKC)-ɛ in the apoptosis and survival of glioma cells using tumor necrosis factor–related apoptosis inducing ligand (TRAIL)- stimulated cells and silencing of PKCɛ expression. Treatment of glioma cells with TRAIL induced activation, caspase-dependent cleavage, and down-regulation of PKCɛ within 3 to 5 hours of treatment. Overexpression of PKCɛ inhibited the apoptosis induced by TRAIL, acting downstream of caspase 8 and upstream of Bid cleavage and cytochrome c release from the mitochondria. A caspase-resistant PKCɛ mutant (D383A) was more protective than PKCɛ, suggesting that both the cleavage of PKCɛ and its down-regulation contributed to the apoptotic effect of TRAIL. To further study the role of PKCɛ in glioma cell apoptosis, we employed short interfering RNAs directed against the mRNA of PKCɛ and found that silencing of PKCɛ expression induced apoptosis of various glioma cell lines and primary glioma cultures. To delineate the molecular mechanisms involved in the apoptosis induced by silencing of PKCɛ, we examined the expression and phosphorylation of various apoptosis-related proteins. We found that knockdown of PKCɛ did not affect the expression of Bcl2 and Bax or the phosphorylation and expression of Erk1/2, c-Jun-NH2-kinase, p38, or STAT, whereas it selectively reduced the expression of AKT. Similarly, TRAIL reduced the expression of AKT in glioma cells and this decrease was abolished in cells overexpressing PKCɛ. Our results suggest that the cleavage of PKCɛ and its down-regulation play important roles in the apoptotic effect of TRAIL. Moreover, PKCɛ regulates AKT expression and is essential for the survival of glioma cells.
Introduction
Protein kinase C (PKC), a family of phospholipid-dependent serine-threonine kinases plays important roles in various cellular functions (1). PKC consists of at least 10 isoforms that are divided into the classic PKCs (α, β1, β2, and γ), the novel PKCs (δ, ɛ, η, and θ), and the atypical PKCs (PKCζ and PKCι/λ; ref. 2). Various PKC isoforms have been reported to regulate cell apoptosis in a stimulus- and isoform-dependent manner. Thus, PKCα and PKCι have been mainly associated with antiapoptotic effects in various systems (3, 4), whereas PKCδ, θ, and μ have been implicated as proapoptotic kinases (5, 6). The novel and atypical PKC isoforms have been reported to undergo caspase-dependent cleavage in response to various apoptotic stimuli and the accumulation of their constitutively active catalytic fragments has been associated with the regulation of cell apoptosis (7, 8).
PKCɛ has been implicated in the regulation of both cell survival and apoptosis in various cellular systems. Thus, overexpression of PKCɛ protected MCF-7 cells from tumor necrosis factor (TNF)-α-induced apoptosis (9) and promoted the survival of lung cancer cells (10). In contrast, PKCɛ has been shown to mediate neuronal death induced by oxidative stress (11) and the apoptosis of macrophages in response to lipopolysaccharide via c-Jun NH2- terminal kinase (JNK) activation (12). PKCɛ is overexpressed in gliomas (13, 14); however, its role in the regulation of glioma cell apoptosis has not been extensively studied.
TNF-related apoptosis inducing ligand (TRAIL; Apo2 ligand) belongs to the TNF superfamily (15). TRAIL induces apoptosis in transformed cells via binding to the death receptors TRAIL-R1 and TRAIL-R2 (16, 17). The mechanisms underlying TRAIL-induced apoptosis consist of the formation of the death-inducing signaling complex that is also common to other members of the death receptors (18). This leads to activation of caspase 8 at the death-inducing signaling complex followed by either activation of a mitochondrial-independent pathway via caspase 3 and 7 or activation of a mitochondrial-dependent pathway by activation of caspase 9 (19). In addition, recent studies reported that TRAIL activates the transcriptional nuclear factor κB (NF-κB) and JNK in various cellular systems (20) and that NF-κB (21) and phosphoinositide-3-kinase/AKT (22) are involved in the resistance of some transformed cells to the apoptotic effect of TRAIL. PKC signaling has also been shown to modulate TRAIL-induced apoptosis by inhibiting the recruitment of key DD-containing adaptor proteins to their membrane associated signaling complexes (23, 24).
Here, we studied the role of PKCɛ in the apoptosis and survival of glioma cells using the apoptotic stimulus TRAIL and silencing of PKCɛ. We found that TRAIL induced caspase-dependent cleavage and down-regulation of PKCɛ and that both the loss of full-length PKCɛ and its cleavage play important roles in the apoptotic function of TRAIL. Moreover, our results using short interfering RNAs (siRNA) further indicate that the expression of PKCɛ is essential for the survival of glioma cells and implicate AKT in this response.
Materials and Methods
Materials
Polyclonal anti-PKCɛ antibodies were purchased from Santa Cruz (Santa Cruz, CA) or from Upstate Inc. (Charlottesville, VA). Both antibodies were directed against the COOH-terminal (V5 region) of PKCɛ. Human TRAIL was from Peprotech (Rocky Hill, NJ), and anti-active caspase 3, AKT, p38, JNK, Erk, STAT, AKT, Bax and Bcl2 antibodies were obtained from Cell Signaling Technology (Beverly, MA). Leupeptin, aprotinin, phenylmethylsulfonyl fluoride (PMSF) and sodium vanadate were obtained from Sigma Chemical Co. (St. Louis, MO). The caspase inhibitors, Z-DEVD-FMK, Z-VAD-FMK, Z-IETD-FMK, Z-LEHD-FMK and the PKCɛ peptide (ERMRPRKRQGSVRRRV) were obtained from Calbiochem (La Jolla, CA).
Glioma cells and cell transfection
The glioma cell lines, A172, U87, U251 and LN-229, were grown on tissue culture dishes in medium consisting of DMEM containing 10% FCS, 2 mmol/L glutamine, penicillin (50 units/mL), and streptomycin (0.05 mg/mL).
Primary cultures were obtained from freshly resected tissues following 1 hour of surgical removal. Institutional Review Board–approved informed consent was obtained from all patients or from the patient’s guardian for use of tumor tissue collected at the time of tumor resection. Samples were first washed in PBS and then minced into small pieces in DMEM with 10% FCS and were further triturated to obtain maximal cell dispersion. Cells were plated in 25 cm2 tissue culture flasks and were grown for 7 to 10 days. Cultures were used up to passage 7.
Cells were transfected either with the control vectors or with the different PKCɛ expression vectors by electroporation using the Nucleofector device (Amaxa Biosystems, Germany). Transfection efficiency using nucleofection was about 80% to 90%.
Site-directed mutagenesis of PKCɛ
PKCɛ cloned into the pCMVtag2B plasmid served as a template vector for the site-directed mutagenesis. The caspase cleavage site of PKCɛ (D383A) was mutated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and the following primers: sense, (5′) GTCGGCCACCGCTGGCCAGCTGG (3′); antisense, (5′) CCAGCTGGCCAGCGGTGGCCGAC (3′). The mutation was confirmed by DNA sequencing.
Construction of PKCɛ green fluorescent protein fusion protein
cDNA encoding PKCɛ was fused into the NH2-terminal–enhanced green fluorescent protein (GFP) vector pEGFP-N1 (Clontech, Palo Alto, CA). The original pEGFP-N1 vector was modified by the insertion of a MluI site in the plasmid polylinker as previously described (25). The clone containing the GFP-PKCɛ was constructed by the excision of PKCɛ from MTH-PKC plasmids by digestion with XhoI and MluI. The insert was then ligated into the modified GFP vector using the same restriction sites. DNA sequencing of the GFP-PKC constructs confirmed the intended reading frame.
Adenovirus preparation and infection
The AdEasy system was kindly provided by Dr. Vogelstein (The Johns Hopkins University School of Medicine, MD; ref. 26). PKCɛ and PKCɛ kinase–dead mutants were first cloned into the pShuttle-CMV vector as previously described for PKCδ (27). Cells were incubated with a multiplicity of infection of 5 at the appropriate recombinant adenovirus vectors for 1 hour. The medium was then replaced with fresh medium and the cells were used 24 to 48 hours post-infection.
Short interfering RNA transfection
siRNA duplexes were synthesized and purified by Dharmacon (Lafayette, CO). The siRNA sequence for targeting PKCɛ mRNA was 5′-GAUGAAGGAGGCGCUCAGTT-3′. A scrambled sequence was used as a negative control. In addition, we used a pool of four PKCɛ siRNA duplexes which were also obtained from Dharmacon. Transfection of siRNAs was done using 50 nmol/L PKCɛ or scrambled siRNAs and OligofectAMINE (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. PKCɛ protein levels were determined using Western blot analysis.
Measurements of cell apoptosis
Cell apoptosis was measured using propidium iodide staining and analysis by flow cytometry as previously described (25). Briefly, detached cells and trypsinized adherent cells were pooled, fixed in 70% ethanol for 1 hour on ice, washed with PBS and treated for 15 minutes with RNase (50 μmol/L) at room temperature. Cells were then stained with propidium iodide (5 μg/mL) and analyzed on a Becton Dickinson (Mountain View, CA) cell sorter. Cell apoptosis was also examined by Western blot analysis of PARP cleavage using anti-PARP antibody (BD PharMingen, San Diego, CA) and by trypan blue exclusion assay.
Preparation of cell homogenates and immunoblot analysis
Cell pellets were resuspended in 100 μL of lysis buffer [25 mmol/L Tris-HCl (pH 7.4), 50 mmol/L NaCl, 0.5% Na deoxycholate, 2% NP40, 0.2% SDS, 1 mmol/L PMSF, 50 μg/mL aprotinin, 50 μmol/L leupeptin, 0.5 mmol/L Na3VO4] on ice for 15 minutes. Sample buffer (2×) was added and the samples were boiled for 5 minutes. Lysates were resolved by SDS-PAGE and were transferred to nitrocellulose membranes. Following incubation with the primary antibody, specific reactive bands were detected using a goat anti-rabbit or goat antimouse IgG conjugated to horseradish peroxidase (Bio-Rad, Hercules, CA) and by enhanced chemiluminescence Western blotting detection kit (Amersham, Arlington Heights, IL). Equal loading was verified by Ponceau S staining or by using anti-actin or anti-tubulin antibodies.
Cytochrome c release
Cytochrome c release from the mitochondria was determined in the cytosolic fraction. Mitochondrial and cytosolic fractions were isolated using the ApoAlert Cell Fractionation Kit (Clontech, BD Biosciences) according to the manufacturer’s instructions. Cytochrome c was identified in the cytosolic fraction using a rabbit anti–cytochrome c antibody.
Measurement of caspase 8 activity
Caspase 8 activity was measured using the QuantiPak assay kit obtained from Biomol (Plymouth Meeting, PA) using the fluorescent substrate Ac-IETD-AMC according to the manufacturer’s recommendations.
Immunoprecipitation and immune complex PKCɛ kinase assay
Immunoprecipitation of PKCɛ and the PKCɛ kinase assay were done as previously described (28). Briefly, cells treated with TRAIL were lysed in lysis buffer [10 mmol/L Tris-HCl (pH 7.5), 2 mmol/L EDTA and EGTA, 0.5 mmol/L DTT, 200 μmol/L PMSF, 1 μg/mL aprotinin, 2 μg/mL leupeptin, 100 μmol/L sodium orthovanadate, and 0.2% Triton X-100]. Lysates were centrifuged at 4°C and supernatants were incubated with 4 μg of anti-PKCɛ antibody for 1 hour at 4°C followed by incubation with 100 μL of protein A/G PLUS-Agarose beads for an additional 4 hours. Immunoprecipitates were then used in a kinase assay that was carried out in 200 μL of reaction mixture containing 20 mmol/L HEPES (pH 7.4), 10 mmol/L MgCl2, 0.1 mmol/L EGTA, 0.1 mg/mL PKCɛ-specific substrate (ERMRPRKRQGSVRRRV), 200 μg/mL phosphatidylserine, 20 μg/mL diacylglycerol, 0.1 mmol/L ATP, and 0.1 μCi/reaction of γ-P32-ATP. The reaction mixture was preincubated for 3 minutes in 30°C. Reactions were initiated by adding 25 μL of preincubated mixture to the immunoprecipitates and incubation at 30°C for 10 minutes. Reaction was terminated by spotting 10 μL of each supernatant onto the phosphocellulose filter papers (P-81). The filters were washed thrice in 0.5% phosphoric acid and counted for radioactivity. Cell pellets were separated by PAGE and immunoblotted for PKCɛ to normalize for the small differences in the amount of immunoprecipitated kinase.
Statistical analysis
The results are presented as the mean ±SE. Data were analyzed using ANOVA and a paired Student’s t test to determine the level of significance between the different groups.
Results
TRAIL induces caspase-dependent cleavage and down-regulation of PKCɛ in glioma cells
PKCɛ has been implicated in the regulation of cell apoptosis in various cellular systems (9–12). To examine the role of PKCɛ in glioma cell apoptosis, we first employed the apoptotic stimulus TRAIL and the A172 glioma cells which are highly sensitive to this ligand. TRAIL induced activation of PKCɛ, initial activation was observed after 15 minutes of treatment, and was further increased after 2 to 3 hours of treatment (Fig. 1A). The activation of PKCɛ was followed by translocation of PKCɛ to the plasma membrane within 15 minutes of treatment as was observed using PKCɛ tagged to GFP (Fig. 1B). In addition, TRAIL induced cleavage of PKCɛ and a gradual loss of the full-length isoforms (Fig. 1C). Low levels of the catalytic fragment of PKCɛ (43 kDa) were already observed after 1 hour, whereas higher levels of this fragment were observed after 2 to 3 hours of treatment. At this time, the expression of the full-length PKCɛ was significantly reduced, and by 5 hours, PKCɛ expression was barely detected (Fig. 1C). The accumulation of the catalytic fragment of PKCɛ preceded the cleavage of PARP, which was first detected after 3 hours of TRAIL treatment (Fig. 1C) and the onset of cell apoptosis as measured using propidium iodide staining and fluorescence-activated cell sorting analysis (data not shown).
Figure 1.
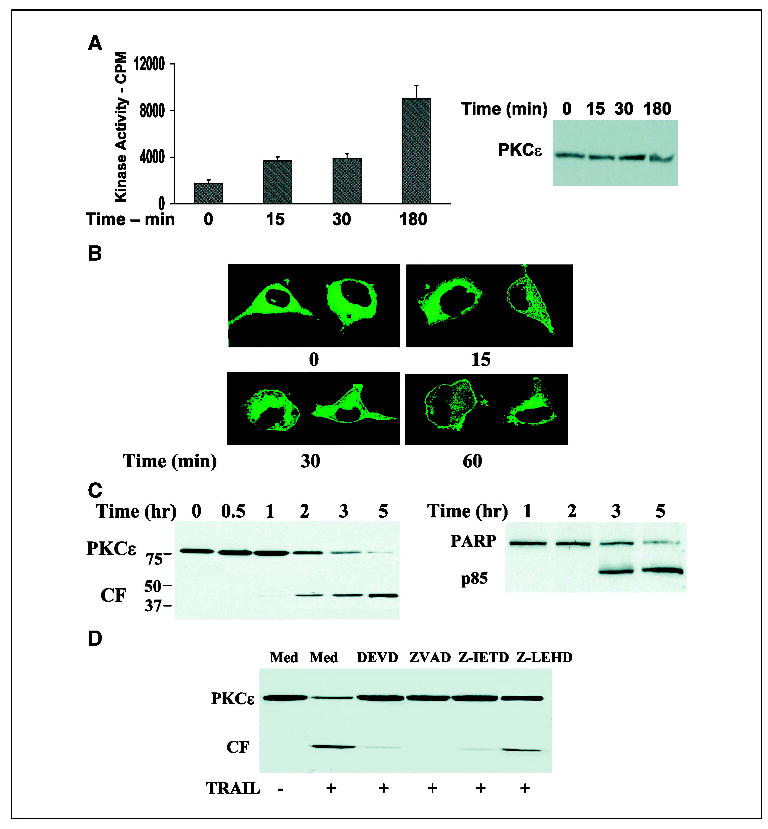
TRAIL induces activation, cleavage, and down-regulation of PKCɛ in glioma cells. A172 cells were treated with TRAIL for 0 to 3 hours and the activity of PKCɛ was determined using an immune complex PKCɛ kinase assay. Samples were also subjected to immunoblot analysis for the determination of total PKCɛ levels (A). Translocation of PKCɛ in response to TRAIL was determined in A172 cells transfected with PKCɛ-GFP. Cells were stimulated with TRAIL (100 ng/mL) for various periods of time and were then visualized by confocal microscopy (B). Cleavage and expression of PKCɛ was determined in A172 cells treated with TRAIL for 0 to 5 hours by Western blot using an anti-PKCɛ antibody that recognizes the catalytic domain [anti-PKCɛ (C-15), Santa Cruz] and cell apoptosis was measured in parallel using PARP cleavage (C). The role of caspase 3, 8, and 9 in the cleavage of PKCɛ was determined using pretreatment of the cells with DEVD, ZVAD, Z-IRTD, or Z-LEHD (10 μmol/L) for 30 minutes followed by incubation with TRAIL for an additional 3 hours (D). Columns, means; bars, ±SE (A); results from one of four separate experiments which gave similar results (B, C, and D).
Pretreatment of the cells with the caspase inhibitors Z-VAD (pan-caspase), DEVD (caspases 3), and Z-IETD (caspase 8) for 30 minutes prior to TRAIL administration inhibited the cleavage of PKCɛ by TRAIL, whereas the caspase 9 inhibitor, Z-LEHD elicited a partial inhibitory effect (Fig. 1D). Similarly, the caspase inhibitors significantly reduced the apoptosis induced by TRAIL as evidenced by PARP cleavage (data not shown).
Cleavage and down-regulation of PKCɛ in TRAIL-sensitive and resistant glioma cells
The cleavage and down-regulation of PKCɛ were further studied in various TRAIL-sensitive and resistant glioma cell lines and in primary glioma cultures (Fig. 2). TRAIL induced a decrease in the expression of the full-length PKCɛ and accumulation of the PKCɛ catalytic fragment in the TRAIL-sensitive cell lines (A172, U251, and U87) albeit to a different degree (Fig. 2B). Thus, the full-length PKCɛ was significantly decreased and high levels of the 43 kDa fragment accumulated in the A172 and U251 cells that exhibited high sensitivity to the apoptotic effect of TRAIL, whereas smaller changes were observed in the U87 cells that exhibited lower sensitivity to TRAIL (Fig. 2A and B). In contrast, no cleavage of PKCɛ was observed in the TRAIL-resistant cell line, LN-229 (Fig. 2A and B), even when the cells were examined after 24 hours of TRAIL treatment (data not shown), suggesting a role of the cleaved form of PKCɛ in the apoptotic effect of TRAIL.
Figure 2.
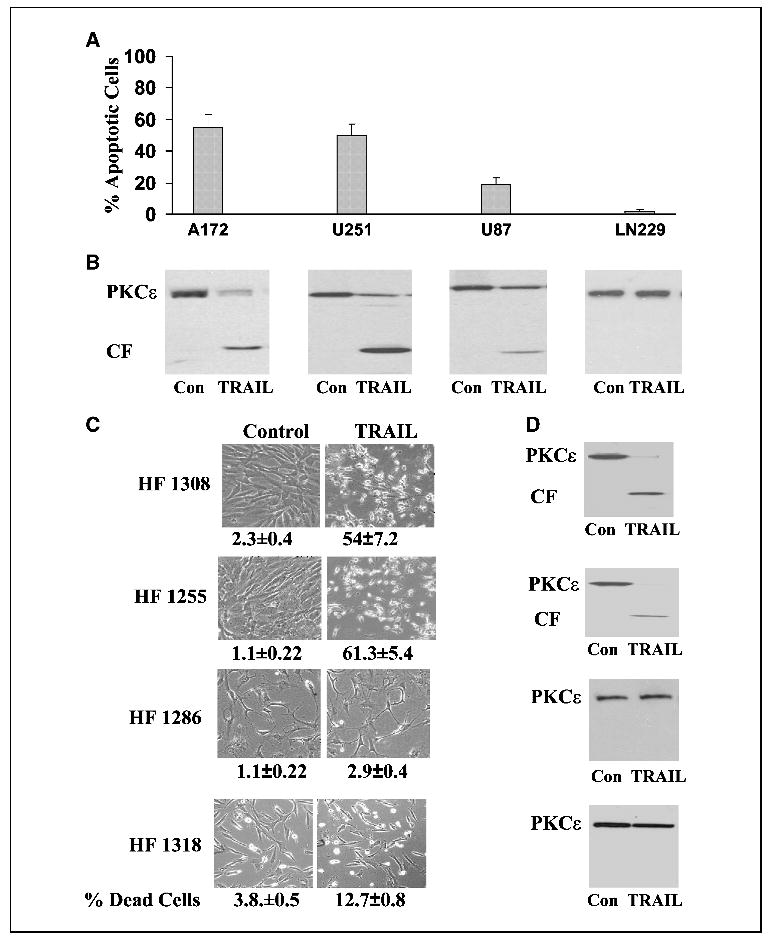
Cleavage and down-regulation of PKCɛ in TRAIL-sensitive and -resistant glioma cells. Various glioma cell lines (A and B) and primary glioma cultures (C and D) were treated with 100 ng/mL TRAIL for 5 hours. Cell apoptosis was determined using propidium iodide staining and fluorescence-activated cell sorting analysis (A) or by trypan blue exclusion assay (C) and the expression and cleavage of PKCɛ was determined using Western blot analysis (B and D). The morphology of the cells was monitored under a phase contrast light microscope (C). Columns, means; bars, ±SE (A); results from one representative experiment out of four similar experiments (B, C, and D).
Similar results were observed with the primary glioma cells. An increase in the catalytic fragment and a decrease in the full-length PKCɛ were observed in the TRAIL-sensitive glioma cultures (HF 1308 and HF 1255), whereas no changes were observed in the TRAIL-resistant primary glioma cells (HF 1286 and HF1318) following 5 hours (Fig. 2C and D) or 24 hours of treatment (data not shown).
Overexpression of PKCɛ protects glioma cells from the apoptosis induced by TRAIL
Because the expression of PKCɛ was dramatically decreased in TRAIL-treated cells, we examined whether overexpression of PKCɛ can protect the A172 cells from apoptosis induced by TRAIL. For these experiments, we used both an adenovirus vector expressing PKCɛ and the tg2b-PKCɛ expression vector. As presented in Fig. 3A, both infection of the A172 cells with an adenovirus vector expressing PKCɛ and transfection of the cells resulted in overexpression of PKCɛ and treatment of the cells with TRAIL induced cleavage of the PKCɛ (Fig. 3A). Overexpression of PKCɛ decreased the apoptosis of the A172 cells in response to TRAIL as compared with control LacZAd- infected cells or as compared with the control vector– transfected cells (Fig. 3B). Thus, PKCɛ decreased cell apoptosis by about 50% as compared with control vector cells.
Figure 3.
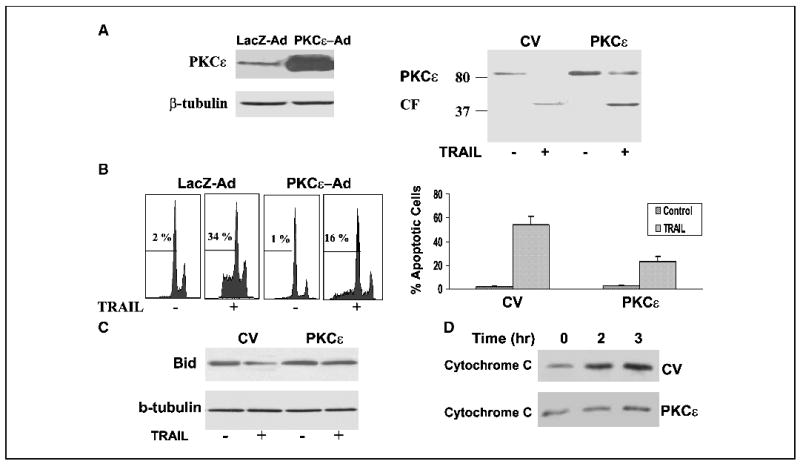
Overexpression of PKCɛ protects A172 cells from TRAIL-induced apoptosis. A172 cells were infected with adenovirus vectors expressing PKCɛ (PKCɛ-AdV) and LacZ (CV, LacZ-AdV; A and B) or transfected with control vector or tg2b-PKCɛ (A and B). Following 24 hours, the cells were treated with TRAIL for 5 hours, cleavage of PKCɛ was determined using Western blot analysis (A) and cell apoptosis was determined using propidium iodide staining and fluorescence-activated cell sorting analysis (B). Bid expression was determined after 90 minutes (C) and cytochrome c release was determined after 2 hours of treatment (D) as described in Materials and Methods. The results are representative of four similar experiments (A, B, C, and D); columns, means; bars, ±SE (B).
Overexpression of PKCɛ did not inhibit the activation of caspase 8 by TRAIL (data not shown); however, it abolished the decrease in Bid expression (Fig. 3C) and the release of cytochrome c from the mitochondria to the cytosol (Fig. 3D), suggesting that PKCɛ acted downstream of caspase 8 activation and upstream of Bid cleavage and activation of the mitochondria pathway.
The PKCɛ caspase-resistant mutant (D383A) is more protective than PKCɛ against TRAIL-induced apoptosis
The partial protection of the exogenous PKCɛ against the apoptosis induced by TRAIL could be due to an apoptotic function of the cleaved overexpressed PKCɛ. To examine this possibility, we constructed a PKCɛ mutant in which the aspartic acid at the SSPD site was mutated to alanine (D383A mutant). Following transfection, the A172 cells expressed comparable levels of PKCɛ and the PKCɛ D383A mutant (Fig. 4A). Similar to the results described in Fig. 3, the wild-type PKCɛ underwent cleavage in response to TRAIL (Fig. 4A) and decreased the apoptosis of the cells by about 40% to 50% cells, as shown by measurements of cell apoptosis (Fig. 4B) and by the morphologic appearance of the cells (Fig. 4C). In contrast, the PKCɛ D383A did not undergo cleavage in response to TRAIL treatment (Fig. 4A) and overexpression of this mutant exerted a stronger protective effect against the apoptosis induced by TRAIL. Thus, in these cells, only 5% to 10% of the cells were apoptotic as compared with 55% to 60% apoptotic cells in the control vector cells (Fig. 4B and C). These results suggest that the PKCɛ D383A acted as a dominant-negative of PKCɛ and that the cleavage of PKCɛ contributed to the apoptosis induced by TRAIL. Similarly, we found that the expression of active caspase 3 induced by TRAIL was inhibited by PKCɛ and to a larger degree by the PKCɛ D338A mutant (Fig. 4D).
Figure 4.
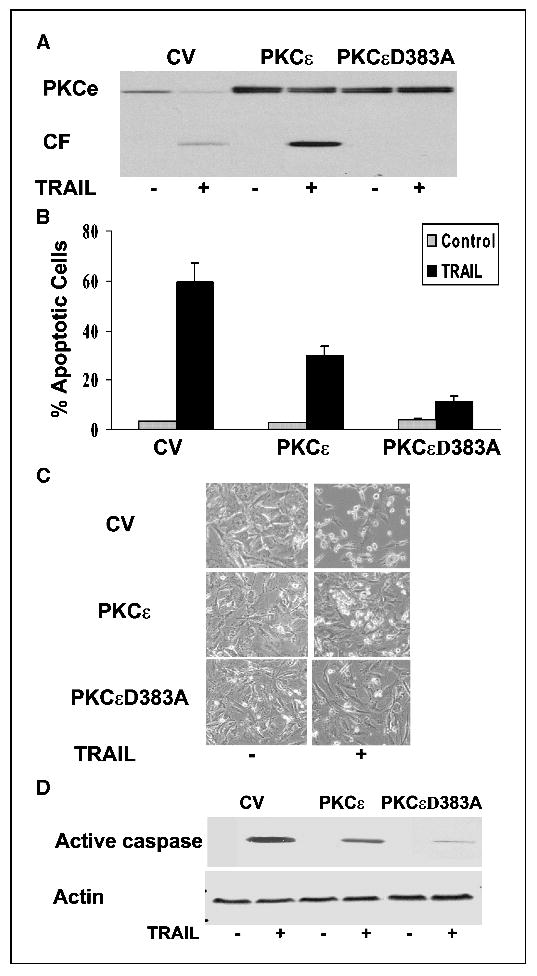
Cleavage of PKCɛ plays a role in the apoptotic effect of TRAIL. A172 cells were transfected with control vector, PKCɛ, or PKCɛD383A. Following 48 hours, the cells were treated with TRAIL for 5 hours and the cleavage of PKCɛ was determined using Western blot analysis (A). Cell apoptosis was determined using propidium iodide staining and fluorescence-activated cell sorting analysis (B). The cells were also visualized using a phase contrast microscope (C). The levels of active caspase 3 were determined using Western blot analysis (D). The results are representative of four similar experiments (A, C, and D); columns, means; bars, ±SE (B).
Silencing of PKCɛ induces apoptosis of glioma cells
Our results thus far suggest that the loss of PKCɛ contributes to the apoptosis induced by TRAIL. We therefore examined whether the expression of PKCɛ was essential for the survival of glioma cells. For these experiments, we designed a siRNA targeting the human PKCɛ mRNA (ɛ1 siRNA). In addition, we employed a pool of four PKCɛ siRNA duplexes (Dharmacon, ɛ2 siRNA). Transfection of the A172 cells with either PKCɛ siRNAs decreased the expression of PKCɛ in the cells by 90% after 3 days of transfection (Fig. 5A), whereas it did not affect the levels of the other PKC isoforms expressed in the A172 cells (PKCα, β, γ, δ, ζ and μ; data not shown). The PKCɛ siRNA transfected cells exhibited a high degree of cell apoptosis as compared with cells transfected with control scrambled siRNA as determined by propidium iodide staining and fluorescence-activated cell sorting analysis (Fig. 5A) or by histone ELISA (data not shown).
Figure 5.
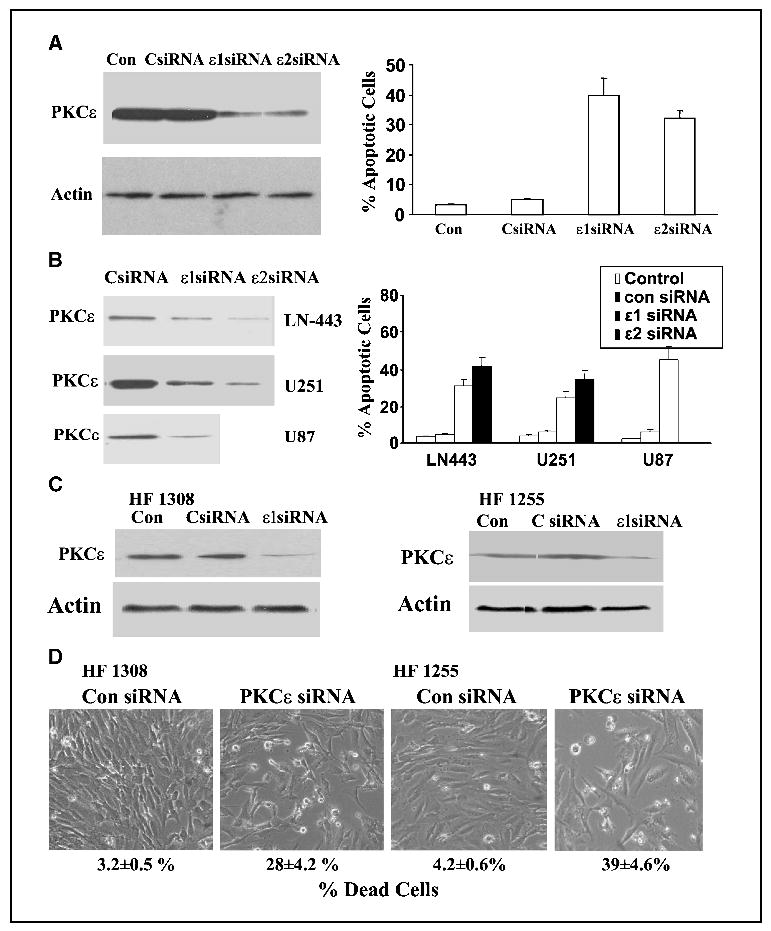
Silencing of PKCɛ expression induces apoptosis in glioma cells. The glioma cell lines, A172 (A) LN-443, U251, and U87 (B) and the primary glioma cultures HF1308 and HF1255 (C and D) were transfected with 50 nmol/L scrambled siRNA (CsiRNA) and siRNAs targeting the mRNA of PKCɛ (PKCɛ siRNAs). Following 72 hours, the expression of PKCɛ was determined using Western blot analysis (A, B, and C) and cell apoptosis was determined using propidium iodide staining and fluorescence-activated cell sorting analysis (A and B) or trypan blue exclusion and phase contrast microscopy (D). The results represent one of four separate experiments, which gave similar results (A and B) or are the means ±SE of five independent experiments (A and B; FACS analysis).
Similar results were obtained with the LN-443, U251, and U87 glioma cell lines (Fig. 5B) and with the two primary glioma cell cultures, HF1308 and HF 1255 (Fig. 5C and D). Transfection of these cells with the PKCɛ siRNAs significantly reduced the expression of PKCɛ in these cells (Fig. 5B and C) and increased cell death of the transfected cells as shown by propidium iodide staining (Fig. 5B), the morphologic appearance of the cells and by trypan blue exclusion assay (Fig. 5D).
Loss of PKCɛ induces a decrease in the expression of Akt
To explore the mechanisms by which knockdown of PKCɛ induces cell apoptosis in glioma cells, we examined the expression and phosphorylation of various apoptosis-related proteins in the A172 cells transfected with the PKCɛ siRNA. As presented in Fig. 6A, knockdown of PKCɛ specifically decreased the expression of PKCɛ, whereas no changes were observed in the expression of PKCδ. The silencing of PKCɛ increased the expression of active caspase 3, whereas it did not affect the expression of the apoptosis-related proteins, Bax and BCl2, or the phosphorylation and expression of the kinases JNK, Erk, p38, and STAT1 (Fig. 6A). In contrast, the knockdown of PKCɛ expression significantly inhibited the phosphorylation and expression of AKT in these cells (Fig. 6A).
Figure 6.
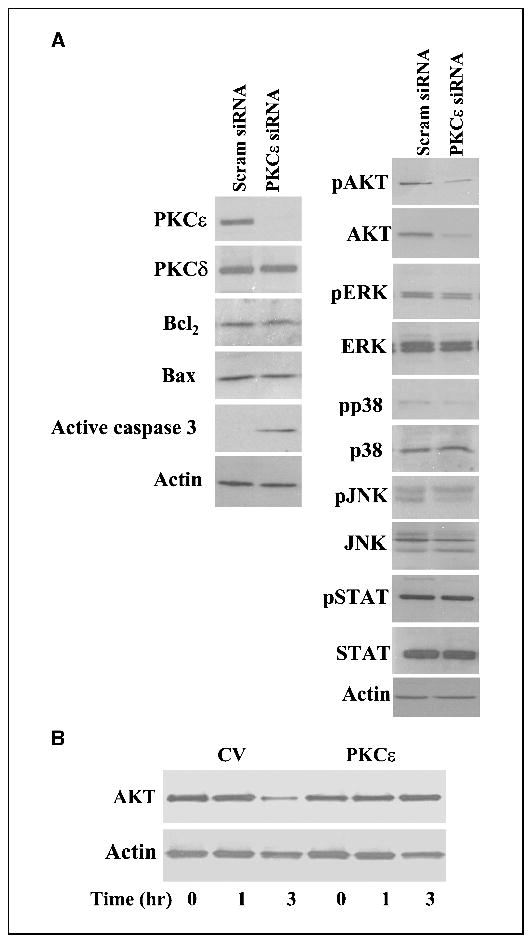
Loss of PKCɛ reduces the expression of AKT in glioma cells. A172 cells were transfected with 50 nmol/L scrambled siRNA or with siRNAs targeting the mRNA of PKCɛ (PKCɛ siRNA). Following 72 hours, the expression of PKCɛ, PKCδ, active caspase 3, Bax and Bcl2 as well as the expression and phosphorylation of AKT, Erk1/2, p38, JNK, and STAT1 were determined using Western blot analysis (A). To examine the role of PKCɛ in AKT expression in TRAIL-treated cells, A172 cells overexpressing control vector or PKCɛ were treated with TRAIL (100 mg/mL) for 1 to 3 hours and the expression of AKT was measured using Western blot analysis (B). The results represent one of four separate experiments, which gave similar results.
Because silencing of PKCɛ reduced the expression of AKT, we examined whether loss of PKCɛ expression in response to TRAIL treatment also reduced the expression of this protein. For these experiments, we used cells transfected with control vector and PKCɛ and treated them with TRAIL for 3 hours. As presented in Fig. 6B, treatment of control vector cells with TRAIL significantly decreased the expression of AKT in the cells after 3 hours of treatment (a time in which PKCɛ was cleaved and degraded). In contrast, no significant decrease in AKT expression was observed in cells overexpressing PKCɛ, suggesting the down-regulation of PKCɛ expression induced by TRAIL mediated the decrease in AKT expression.
Discussion
In this study, we explored the role of PKCɛ in the apoptosis and survival of glioma cells using the apoptotic stimulus TRAIL and siRNAs directed against PKCɛ mRNA. We found that TRAIL induced activation of PKCɛ within 15 to 30 minutes of TRAIL treatment which was further increased after 3 hours. The early and late activation of PKCɛ are probably mediated by two distinct mechanisms; a cleavage-independent activation at the early time points and a cleavage-dependent activation at the later time point which could be attributed to the generation of a constitutively active catalytic fragment. Indeed, similar results of cleavagedependent activation of PKCɛ were recently reported in TNF-α treated cells (9).
TRAIL also induced translocation of PKCɛ to the plasma membrane. The translocation of PKC is associated with the activation of this enzyme and it is considered as an important molecular event in the function of this kinase family (29). Translocation of PKC is mediated by binding to selective anchoring proteins or selective receptors for activated C-kinases (RACK; ref. 30) and several domains of PKCɛ have implicated its translocation and anchoring to the membrane (31). The mechanisms by which TRAIL induces translocation of PKCɛ and the role of this translocation in PKCɛ effects are currently not understood. However, membranal translocation of PKCɛ has been associated with the apoptotic effect of UV radiation (32).
TRAIL induced cleavage and down-regulation of PKCɛ and generation of a 43 kDa fragment in all the cells that were sensitive to TRAIL. In contrast, no cleavage of PKCɛ was observed in the TRAIL-resistant glioma cells, suggesting that the cleavage and loss of PKCɛ were involved in the apoptotic response of TRAIL. Various studies have shown that PKC isoforms are proteolytically cleaved in response to apoptotic stimuli and that the apoptotic effect of some of these isoforms is associated with the accumulation of the cleaved constitutive active catalytic fragment (6, 33). Indeed, cleavage of PKCδ (26, 34), PKCθ (35), PKCμ (7), and PKCζ (36) have been reported in response to various apoptotic stimuli such as radiation, chemotherapeutic drugs and ligation of the FAS and TNF-α receptors, and caspase 3 has been implicated in the cleavage of these PKC isoforms (6, 7, 26, 34).
PKCɛ undergoes cleavage in response to serum deprivation (37), chemotherapeutic agents (38) and TNF-α treatment (9). Koriyama et al. (29) and Hoppe et al. (37) showed both in vitro and in vivo, that caspase 3 mediated the cleavage of PKCɛ in their cellular systems. In contrast, Basu et al. (9) reported that in the MCF-7 cells that lack functional caspase 3, the cleavage of PKCɛ is mediated by caspase 7. We found that TRAIL induced the generation of a 43 kDa fragment in all the glioma cells that were examined in this study, and no other catalytic fragments were detected. Using different caspase inhibitors, we found that the caspase 3 and caspase 8 inhibitors completely inhibited the cleavage of PKCɛ and the apoptosis induced by TRAIL, whereas partial inhibition was observed with the caspase 9 inhibitor. Thus, our data suggest that in glioma cells, TRAIL exerts apoptosis via activation of caspases 8 and 9 and that caspase 3 cleaves PKCɛ at the atypical cleavage site, SSPD in the hinge region.
We found that TRAIL induced a large decrease in the expression of the full-length PKCɛ in parallel to the increased generation of its cleaved catalytic fragment. PKCɛ has been associated with antiapoptotic functions in various cellular systems including lung cancer cells (10), T lymphocytes (39), and prostate cancer cells (40). We therefore hypothesized that the down-regulation of PKCɛ mediated the apoptotic effect of TRAIL. We found that overexpression of PKCɛ in the A172 cells inhibited the apoptosis induced by TRAIL, acting downstream from caspase 8 activation and upstream of Bid cleavage and activation of the mitochondrial pathway. Overexpression of PKCɛ inhibited the apoptosis induced by TRAIL by 50% to 60%, suggesting that the down-regulation of PKCɛ may not be the only factor involved in the apoptotic effect of TRAIL. A partial protective effect of PKCɛ on the apoptosis of glioma cell lines treated with TRAIL was also observed by Shinohara et al. (41).
The overexpressed PKCɛ underwent cleavage in TRAIL-treated cells, similar to the endogenous PKCɛ, suggesting that the partial protective effect of PKCɛ may be due to an apoptotic effect of the cleaved fragment. We found that a PKCɛ mutant in which aspartic acid 383 was mutated to alanine (D383A) and which did not undergo cleavage in response to TRAIL, was significantly more effective than the wild-type PKCɛ in protecting A172 cells from apoptosis induced by TRAIL. Thus, our results suggest that the cleavage of PKCɛ contributed to the apoptotic effect of TRAIL in glioma cells. The cleaved PKCɛ has been associated with both pro- and antiapoptotic effects in various cellular systems. Thus, apoptotic effects of the cleaved PKCɛ catalytic fragment were observed in the GH3B6 cells (38), whereas Basu et al. (9) reported that the catalytic domain of PKCɛ exerted an antiapoptotic effect in TNF-α-treated cells.
The down-regulation of PKCɛ in TRAIL-treated glioma cells raised the possibility that the expression of PKCɛ is essential for the survival of these cells. Using siRNAs directed against PKCɛ mRNA, we reduced PKCɛ expression in the cells by 90%. Silencing of PKCɛ expression induced cell apoptosis in all the glioma cell lines and primary cultures that were examined, further suggesting an important role of PKCɛ in the survival of glioma cells.
We found that the decrease in PKCɛ expression by either siRNAs or TRAIL induced a selective decrease in the expression of AKT, whereas the expression of other apoptosis-related proteins was not significantly affected. AKT (PKB) is a family of serine-threonine kinases that regulates cell survival in a variety of cellular systems including gliomas (42, 43). The survival effects of AKTare exerted by phosphorylating proteins such as BAD, caspase 9, and the forkhead transcription factors or by activating antiapoptotic pathways such as NF-κB (43). The activity of AKT is regulated by phosphorylation on Thr308 by PDK-1 and on Ser473 by an unidentified kinase referred to as PDK-2 (44). In addition, the activity of AKT is also regulated by its degradation via diverse mechanisms. Indeed, proteasomedependent degradation of AKT has been reported in response to treatment of tumor cells with Hsp90-specific inhibitors (45), whereas caspase-dependent and independent degradation of AKT occurs in response to p53 inhibition of the a6β4 integrin survival signaling (46), UV radiation (47), and inhibition of the vascular endothelial growth factor receptor pathway (48). The mechanisms by which loss of PKCɛ induced a decrease in the expression of AKT are currently not understood. One possibility is that down-regulation of PKCɛ induced activation of caspase 3 that results in the cleavage and degradation of AKT. Indeed, silencing of PKCɛ induced activation of caspase 3 and overexpression of PKCɛ decreased the activation of caspase 3 induced by TRAIL. Alternatively, down-regulation of the Hsp90 protein, which is required for the stability of AKT, is another possible mechanism because PKC has been associated with the regulation of Hsp90 under various conditions (49). Finally, the direct regulation of AKT expression by PKCɛ may be also considered because interaction between AKT and PKCɛ has been shown in various cellular systems (50).
In summary, the results of both TRAIL-induced apoptosis and PKCɛ silencing indicate that the expression of AKT is regulated by PKCɛ and that PKCɛ is essential for the survival of glioma cells. Our results also suggest that in TRAIL-treated cells, the cleaved PKCɛ contributes to the apoptotic effect of TRAIL, in addition to the loss of this isoform from the cells. Thus, in addition to delineating the role of PKCɛ in TRAIL-induced apoptosis, the results of this study have broader implications for the role of PKCɛ signaling in the regulation of AKT expression and for glioma cell function. We (13) and others (14) have recently reported that PKCɛ is highly expressed in glioblastomas. Thus, our results that PKCɛ is essential for the survival of glioma cells identify an important role of PKCɛ in these tumors.
Acknowledgments
Grant support: James S. McDonnell Foundation, 21st Century Scientist Award/Brain Cancer Research, and by NIH grant RO1 CA109196.
We thank Michelle Johnston and Donghong Ju for their excellent technical assistance and Sandra Rempel for critical review of the manuscript.
References
- 1.Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988;334:661–5. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 2.Hug H, Sarre TF. Protein kinase C isoenzymes: divergence in signal transduction? Biochem J. 1993;291:329–43. doi: 10.1042/bj2910329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutcher I, Webb PR, Anderson NG. The isoform-specific regulation of apoptosis by protein kinase C. Cell Mol Life Sci. 2003;60:1061–70. doi: 10.1007/s00018-003-2281-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruvolo PP, Deng X, Carr BK, May WS. A functional role for mitochondrial protein kinase Cα in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem. 1998;273:25436–42. doi: 10.1074/jbc.273.39.25436. [DOI] [PubMed] [Google Scholar]
- 5.Jamieson L, Carpenter L, Biden TJ, Fields AP. Protein kinase Cι activity is necessary for Bcr-Abl-mediated resistance to drug-induced apoptosis. J Biol Chem. 1999;274:3927–30. doi: 10.1074/jbc.274.7.3927. [DOI] [PubMed] [Google Scholar]
- 6.Ghayur T, Hugunin M, Talanian RV, et al. Proteolytic activation of protein kinase Cδ by an ICE/CED 3-like protease induces characteristics of apoptosis. J Exp Med. 1996;184:2399–404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Endo K, Oki E, Biedermann V, et al. Proteolytic cleavage and activation of protein kinase C [μ] by caspase-3 in the apoptotic response of cells to 1-β-D-arabinofuranosylcytosine and other genotoxic agents. J Biol Chem. 2000;275:18476–81. doi: 10.1074/jbc.M002266200. [DOI] [PubMed] [Google Scholar]
- 8.Sitailo LA, Tibudan SS, Denning MF. Bax activation and induction of apoptosis in human keratinocytes by the protein kinase Cδ catalytic domain. J Invest Dermatol. 2004;123:434–43. doi: 10.1111/j.0022-202X.2004.23403.x. [DOI] [PubMed] [Google Scholar]
- 9.Basu A, Lu D, Sun B, Moor AN, Akkaraju GR, Huang J. Proteolytic activation of protein kinase C-ɛ by caspase-mediated processing and transduction of antiapoptotic signals. J Biol Chem. 2002;277:41850–6. doi: 10.1074/jbc.M205997200. [DOI] [PubMed] [Google Scholar]
- 10.Ding L, Wang H, Lang W, Xiao L. Protein kinase C-ɛ promotes survival of lung cancer cells by suppressing apoptosis through dysregulation of the mitochondrial caspase pathway. J Biol Chem. 2002;277:35305–13. doi: 10.1074/jbc.M201460200. [DOI] [PubMed] [Google Scholar]
- 11.Jung YS, Ryu BR, Lee BK, et al. Role for PKC-ɛ in neuronal death induced by oxidative stress. Biochem Biophys Res Commun. 2004;320:789–94. doi: 10.1016/j.bbrc.2004.05.217. [DOI] [PubMed] [Google Scholar]
- 12.Comalada M, Xaus J, Valledor AF, Lopez-Lopez C, Pennington DJ, Celada A. PKC ɛ is involved in JNK activation that mediates LPS-induced TNF-α, which induces apoptosis in macrophages. Am J Physiol Cell Physiol. 2003;285:C1235–45. doi: 10.1152/ajpcell.00228.2003. [DOI] [PubMed] [Google Scholar]
- 13.Mandil R, Ashkenazi E, Blass M, et al. Protein kinase Cα and protein kinase Cδ play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 2001;61:4612–9. [PubMed] [Google Scholar]
- 14.Sharif TR, Sharif M. Overexpression of protein kinase Cɛ in astroglial brain tumor derived cell lines and primary tumor samples. Int J Oncol. 1999;15:237–43. [PubMed] [Google Scholar]
- 15.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–90. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 16.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–8. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 17.Pan G, O’Rourke K, Chinnaiyan AM, et al. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–3. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 18.Kischkel FC, Hellbardt S, Behrmann I, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–88. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, El Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–33. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 20.Lin Y, Devin A, Cook A, et al. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IκB kinase and c-Jun N-terminal kinase. Mol Cell Biol. 2000;20:6638–45. doi: 10.1128/mcb.20.18.6638-6645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-κB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–20. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 22.Chen X, Thakkar H, Tyan F, et al. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene. 2001;20:6073–83. doi: 10.1038/sj.onc.1204736. [DOI] [PubMed] [Google Scholar]
- 23.Harper N, Hughes MA, Farrow SN, Cohen GM, MacFarlane M. Protein kinase C modulates tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by targeting the apical events of death receptor signaling. J Biol Chem. 2003;278:44338–47. doi: 10.1074/jbc.M307376200. [DOI] [PubMed] [Google Scholar]
- 24.Meng XW, Heldebrant MP, Kaufmann SH. Phorbol 12- myristate 13-acetate inhibits death receptor-mediated apoptosis in Jurkat cells by disrupting recruitment of Fas-associated polypeptide with death domain. J Biol Chem. 2002;277:3776–83. doi: 10.1074/jbc.M107218200. [DOI] [PubMed] [Google Scholar]
- 25.Blass M, Kronfeld I, Kazimirsky G, Blumberg PM, Brodie C. Tyrosine phosphorylation of protein kinase Cδ is essential for its apoptotic effect in response to etoposide. Mol Cell Biol. 2002;22:182–95. doi: 10.1128/MCB.22.1.182-195.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deutsch E, Cohen A, Kazimirsky G, et al. Role of protein kinase Cδ in reactivation of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2004;78:10187–92. doi: 10.1128/JVI.78.18.10187-10192.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brodie C, Steinhart R, Kazimirsky G, et al. PKCδ associates with and is involved in the phosphorylation of RasGRP3 in response to phorbol esters. Mol Pharmacol. 2004;66:76–84. doi: 10.1124/mol.66.1.76. [DOI] [PubMed] [Google Scholar]
- 29.Koriyama H, Kouchi Z, Umeda T, et al. Proteolytic activation of protein kinase Cδ and ɛ by caspase-3 in U937 cells during chemotherapeutic agent-induced apoptosis. Cell Signal. 1999;11:831–8. doi: 10.1016/s0898-6568(99)00055-8. [DOI] [PubMed] [Google Scholar]
- 30.Ron D, Kazanietz MG. New insights into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J. 1999;13:1658–76. [PubMed] [Google Scholar]
- 31.Wang QJ, Lu G, Schlapkohl WA, et al. The V5 domain of protein kinase C plays a critical role in determining the isoform-specific localization, translocation, and biological function of protein kinase C-δ and - ɛ. Mol Cancer Res. 2004;2:129–40. [PubMed] [Google Scholar]
- 32.Chen N, Ma W, Huang C, Dong Z. Translocation of protein kinase Cɛ and protein kinase Cδ to membrane is required for ultraviolet B-induced activation of mitogen-activated protein kinases and apoptosis. J Biol Chem. 1999;274:15389–94. doi: 10.1074/jbc.274.22.15389. [DOI] [PubMed] [Google Scholar]
- 33.Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase Cδ. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- 34.Reyland ME, Anderson SM, Matassa AA, Barzen KA, Quissell DO. Protein kinase Cδ is essential for etoposide-induced apoptosis in salivary gland acinar cells. J Biol Chem. 1999;274:19115–23. doi: 10.1074/jbc.274.27.19115. [DOI] [PubMed] [Google Scholar]
- 35.Datta R, Kojima H, Yoshida K, Kufe D. Caspase- 3-mediated cleavage of protein kinase Cτ in induction of apoptosis. J Biol Chem. 1997;272:20317–20. doi: 10.1074/jbc.272.33.20317. [DOI] [PubMed] [Google Scholar]
- 36.Frutos S, Moscat J, Diaz-Meco MT. Cleavage of ζPKC but not λ/ιPKC by caspase-3 during UV-induced apoptosis. J Biol Chem. 1999;274:10765–70. doi: 10.1074/jbc.274.16.10765. [DOI] [PubMed] [Google Scholar]
- 37.Hoppe J, Hoppe V, Schafer R. Selective degradation of the PKC-ɛ isoform during cell death in AKR-2B fibroblasts. Exp Cell Res. 2001;266:64–73. doi: 10.1006/excr.2001.5211. [DOI] [PubMed] [Google Scholar]
- 38.Leverrier S, Vallentin A, Joubert D. Positive feedback of protein kinase C proteolytic activation during apoptosis. Biochem J. 2002;368:905–13. doi: 10.1042/BJ20021253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bertolotto C, Maulon L, Filippa N, Baier G, Auberger P. Protein kinase Cτ and ɛ promote T-cell survival by a rsk-dependent phosphorylation and inactivation of BAD. J Biol Chem. 2000;275:37246–50. doi: 10.1074/jbc.M007732200. [DOI] [PubMed] [Google Scholar]
- 40.McJilton MA, Van Sikes C, Wescott GG, et al. Protein kinase Cɛ interacts with Bax and promotes survival of human prostate cancer cells. Oncogene. 2003;22:7958–68. doi: 10.1038/sj.onc.1206795. [DOI] [PubMed] [Google Scholar]
- 41.Shinohara H, Kayagaki N, Yagita H, et al. A protective role of PKCɛ against TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in glioma cells. Biochem Biophys Res Commun. 2001;284:1162–7. doi: 10.1006/bbrc.2001.5104. [DOI] [PubMed] [Google Scholar]
- 42.Marte BM, Downward J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–8. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 43.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–98. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 44.Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:561–76. [PMC free article] [PubMed] [Google Scholar]
- 45.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858–66. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 46.Bachelder RE, Ribick MJ, Marchetti A, et al. p53 inhibits α6β4 integrin survival signaling by promoting the caspase 3-dependent cleavage of AKT/PKB. J Cell Biol. 1999;147:1063–72. doi: 10.1083/jcb.147.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Widmann C, Gibson S, Johnson GL. Caspasedependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti-apoptotic signals. J Biol Chem. 1998;273:7141–7. doi: 10.1074/jbc.273.12.7141. [DOI] [PubMed] [Google Scholar]
- 48.Riesterer O, Zingg D, Hummerjohann J, Bodis S, Pruschy M. Degradation of PKB/Akt protein by inhibition of the VEGF receptor/mTOR pathway in endothelial cells. Oncogene. 2004;23:4624–35. doi: 10.1038/sj.onc.1207596. [DOI] [PubMed] [Google Scholar]
- 49.Wu JM, Xiao L, Cheng XK, Cui LX, Wu NH, Shen YF. PKCɛ is a unique regulator for hsp90 β gene in heat shock response. J Biol Chem. 2003;278:51143–9. doi: 10.1074/jbc.M305537200. [DOI] [PubMed] [Google Scholar]
- 50.Wu D, Thakore CU, Wescott GG, McCubrey JA, Terrian DM. Integrin signaling links protein kinase Cɛ to the protein kinase B/Akt survival pathway in recurrent prostate cancer cells. Oncogene. 2004;23:8659–72. doi: 10.1038/sj.onc.1207900. [DOI] [PubMed] [Google Scholar]