

Abstract
Progressive kidney failure is a genetically and clinically heterogeneous group of disorders. Podocyte foot processes and the interposed glomerular slit diaphragm are critical components of the permeability barrier in the kidney. Mutations in genes encoding for structural proteins of the podocyte lead to the development of proteinuria resulting in progressive kidney failure and focal segmental glomerulosclerosis (FSGS). Here, we show that the canonical transient receptor potential 6 (TRPC6) ion channel is expressed in podocytes and represents a component of the glomerular slit diaphragm. We identified five families with autosomal dominant FSGS in which disease segregated with mutations in the TRPC6 gene on chromosome 11q. Two of the TRPC6 mutants displayed increased current amplitudes. Together, this data demonstrates that TRPC6 channel activity at the slit diaphragm is essential for proper regulation of podocyte structure and function.
Proteinuria is a common feature of kidney dysfunction of glomerular origin and is itself a risk factor for both renal and extra-renal disease1. A variety of recent evidence supports the notion of the glomerular podocyte as a central component of the renal filtration barrier 2. Podocytes (glomerular visceral epithelial cells) are located within the glomerulus. Their complex cytoarchitecture includes cellular extensions (foot processes) that connect to the glomerular basement membrane on the outer aspect of the glomerular capillaries. Together with the interposed slit diaphragm, a specialized multi-protein junction, they form a critical component of the ultrafiltration barrier 3. This explains why structural damage of the podocyte may lead to the development of proteinuria. Mutations in ACTN4 (α-actinin-4), NPHS1 (nephrin) and NPHS2 (podocin) have all been shown to lead to the development of proteinuric kidney disease 4. Winn and colleagues identified a family with autosomal dominant FSGS in which disease co-segregated with a point mutation in the TRPC6 gene on chromosome 11q 5,6. This prompted us to examine the renal expression and interactions of TRPC6 as well as the spectrum and function of TRPC6 genetic variants in FSGS families.
TRPC6 is a member of the transient receptor potential (TRP) superfamily of cation-selective ion channels. The TRPC subfamily (TRPC1-7) is a group of calcium-permeable cation channels that are important for the increase in intracellular [Ca2+] following engagement of G-protein-coupled receptors and receptor tyrosine kinases 7. TRPCs form homo-and heterotetramers that can interact with a variety of other proteins 8. Because all previously described genes mutated in FSGS and nephrotic syndrome are highly expressed in the glomerular podocyte, we examined the expression of TRPC6 in the kidney to define its localization. Confocal microscopy of adult rat kidney sections revealed wide expression of TRPC6 throughout the kidney in tubules and glomeruli (Fig. 1a). This observation is consistent with recent reports detecting TRPC6 mRNA in glomeruli 6,9. Most of TRPC6 expression within the glomerulus was confined to podocytes (Fig.1a, left panel) as shown by immunofluorescent double labeling with the podocyte marker synaptopodin (Fig.1a, middle panel) resulting in a yellow staining pattern (Fig. 1a, right panel) 10. In addition, we found a signal in glomerular endothelial cells. Whereas TRPC6 labeling with the anti-TRPC6 antibody produced a strong glomerular staining and staining within tubules (Fig. 1b left panel), the pre-incubation of TRPC6 antibody with a TRPC6 control peptide resulted in a negative signal (Fig. 1b right panel). Next, we studied the expression of TRPC1-6 mRNAs in isolated glomeruli and cultured murine podocytes by RT-PCR (Fig 1c). Whereas TRPC1-6 are all expressed in the glomerulus, only TRPC1, 2, 5 and 6 were found to be expressed in cultured podocytes. We analyzed TRPC6 expression in a cultured mouse podocyte cell line 11. Labelling was detected at the cell membrane (Fig. 1d). To determine the precise subcellular localization of TRPC6, we carried out immunogold labeling of ultrathin frozen sections from adult kidney cortex (Fig. 1e). Gold particles were found in the cell body of podocytes and in primary processes (Fig. 1e, white arrows). In podocyte foot processes gold particles labeled areas in close vicinity to the slit diaphragm region (Fig. 1e, black arrows). We also detected TRPC6 expression in glomerular endothelial cells (Fig. 1e) and on a few mesangial cells (data not shown). In order for TRPC6 to reach the slit diaphragm in podocyte foot processes, TRPC6 protein has to be transported from the cell body through the major processes into the foot processes. Membrane proteins with high protein turnover can be found in various subcellular localizations. Similarly, podocalyxin, a major sialoprotein in podocytes is detected intracellularly throughout the entire exocytotic pathway consistent with a high rate synthesis 12. High power view of a section through the slit diaphragm area shows the close association of TRPC6 to the slit diaphragm (Fig. 1f, black arrows).
FIGURE 1.
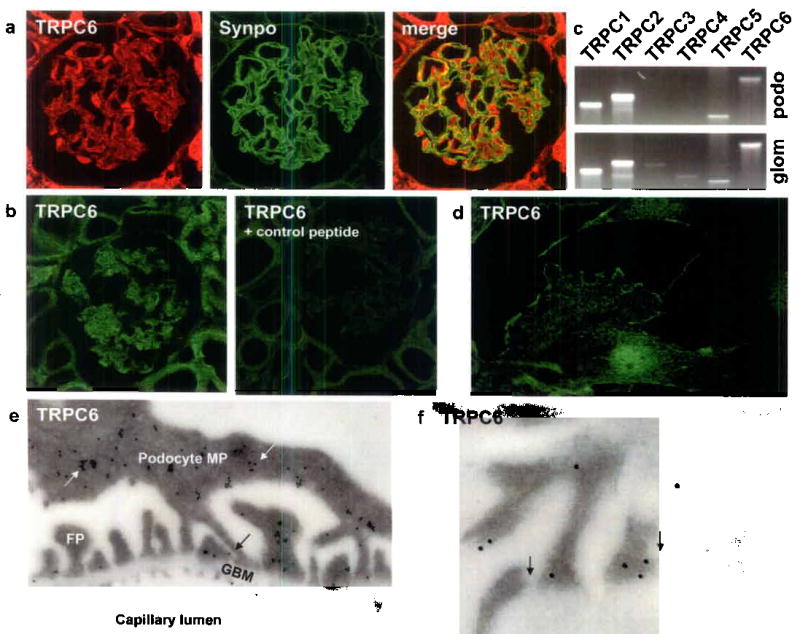
TRPC6 expression in the kidney glomerulus. (a) Confocal microscopy shows TRPC6 (red) expression in the glomerulus. TRPC6 co-localizes with the podocyte marker Synaptopodin (green) resulting in a yellow overlap. (b) Compared to the TRPC6 antibody labeling, preabsorption of TRPC6 antibody with the control peptide results in a negative staining. (c) Analysis of TRPC1-6 mRNA expression in cultured podocytes by RT-PCR. In addition to TRPC6 mRNA, also TRPC1, TRPC2 and TRPC5 mRNA is detected in total podocyte RNA (podo). TRPC3 and TRPC4 mRNA is detected in total glomerular RNA (glom) but not in total podocyte RNA. (d) TRPC6 localizes in cultured podocytes to the cell membrane as shown by immunostaining. (e) Immunogold labeling displays TRPC6 localization in podocyte major processes (MP, white arrows) and foot processes (FP). Of note, TRPC6 in podocyte FP is located in close vicinity to the slit diaphragm (black arrows). (f) High-power view of slit diaphragm areas (arrows) display heavy TRPC6 immunogold labeling.
Next, we tested whether TRPC6 co-localizes with the human disease-associated slit diaphragm proteins nephrin, podocin and CD2AP. Since available antibodies against TRPC6 and slit diaphragm proteins are all rabbit-polyclonal, we transfected cultured podocytes with GFP-tagged TRPC6. Confocal microscopy of GFP-TRPC6 transfected podocytes stained with antibodies against nephrin, podocin and CD2AP (Fig. 2a) revealed expression of GFP-TRPC6 at the podocyte cell membrane partially co-localizing with nephrin, podocin and CD2AP 13–15. These findings suggest that within podocytes, TRPC6 is at least in part associated with the slit diaphragm.
FIGURE 2.

TRPC6 co-localizes and directly interacts with slit diaphragm proteins. (a) GFP-TRPC6 co-localizes with CD2AP, nephrin and podocin at the cell membrane of cultured podocytes as shown by confocal microscopy (arrows). (b) GFPTRPC6 associates with FLAG-tagged nephrin and podocin but not with CD2AP in co-transfected HEK293 cells. GFP-tagged TRPC6 was detected in total cell lysate by immunoblotting using an anti-GFP antibody (upper panel). FLAG-tagged fusion proteins were immunoprecipitated, eluted and visualized with an anti-FLAG antibody (middle panel). Co-immunoprecipitated GFP-TRPC6 was detected in eluate fractions (lower panel). FLAG-myopodin served as a negative control for TRPC6-binding. (c) Endogenous co-immunoprecipitation of TRPC6 with slit diaphragm proteins from cultured podocytes. TRPC6 interacts with nephrin and podocin but not with CD2AP. Mr, relative molecular mass; WB, primary antibody used for Western Blotting.
To test whether TRPC6 interacts with nephrin, podocin or CD2AP, we performed co-immunoprecipitation studies (Fig. 2b). Therefore, we co-expressed recombinant mouse GFP-TRPC6 with FLAG-tagged mouse nephrin, podocin, and CD2AP, respectively, in human embryonic kidney (HEK293) cells. FLAG-fusion proteins from cell lysates were immunoprecipitated using anti-FLAG-M2 beads and eluates were analyzed by immunoblotting using anti-FLAG and anti-GFP antibodies. To detect the interaction of GFP-TRPC6 with FLAG-tagged nephrin, podocin or CD2AP, we analyzed eluates with anti-GFP antibody. Immunoblotting showed that GFP-TRPC6 was absent from the eluates derived from cells co-transfected with FLAG-CD2AP (Fig. 2b). In contrast, GFP-TRPC6 was present in eluates derived from cells co-transfected with FLAG-nephrin and FLAG-podocin, indicating a direct biochemical interaction of TRPC6 with nephrin and podocin but not with CD2AP. We also performed the reverse co-immunoprecipitation where we immunoprecipitated the GFP-fusion proteins first (data not shown). This set of experiment yielded identical results. We also performed endogenous co-immunoprecipitation using whole cellular extracts from cultured differentiated podocytes known to express the vital slit diaphragm components nephrin, podocin and CD2AP 16. The immunoprecipitation with anti-TRPC6 antibody revealed an interaction of TRPC6 with nephrin and podocin but not with CD2AP (Fig. 2c).
Nephrin is a central component of the slit diaphragm and mice deficient in the nephrin gene suffer from proteinuria and partial foot process effacement 17,18. We therefore examined the effect of nephrin deficiency on the localization of TRPC6 in the glomerulus (Fig. 3). Neonatal wild type mice displayed a low expression of glomerular TRPC6 (Fig. 3, left upper panel). Some of the expression is found in podocytes as revealed by immunofluorescence double labeling with the podocyte marker synaptopodin (Fig. 3, middle upper panel), resulting in a partial yellow overlap (Fig. 3, right upper panel). The targeted deletion of nephrin leads to an induction of podocyte TRPC6 expression and to focal accumulation of the TRPC6 protein (Fig. 3a). This data suggest that the lack of nephrin induces podocyte TRPC6 expression and leads to altered cellular localization of TRPC6.
FIGURE 3.
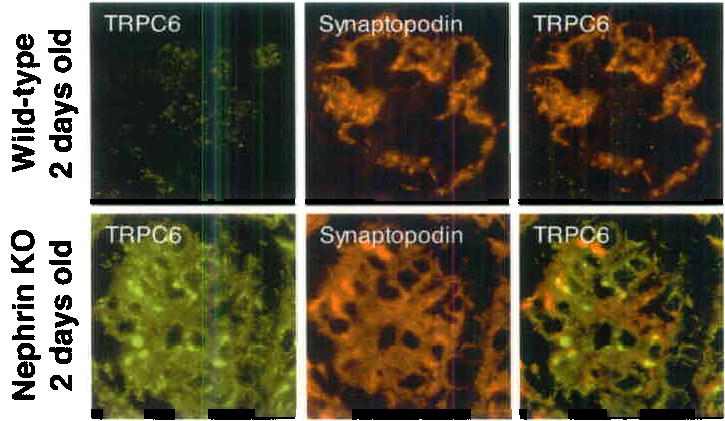
TRPC6 is upregulated in 2 days old nephrin-KO mouse glomeruli as shown by fluorescence microscopy and immunogold labeling. Weak TRPC6 expression was detected in glomeruli of 2 days old wt-mice (upper panel), TRPC6 was found to be upregulated in glomeruli of 2 days old nephrin-KO mice (lower panel). Of note, TRPC6 forms aggregates within the glomerulus. Double labeling of TRPC6 with the podocyte marker synaptopodin demonstrates the localization of TRPC6 in kidney podocytes resulting in a yellow staining pattern.
To explore the role of TRPC6 gene variation in kidney disease, we screened probands of 71 pedigrees with familial FSGS for alterations in the TRPC6 gene by DNA sequence analysis. Of these 71 families screened, 43 showed evidence of disease in members of multiple generations, and 28 showed evidence of disease in 2 or more members of a single generation. Probands of 49% of the families were of western European ancestry; 5% of African ancestry, and 27% described themselves as Hispanic. The phenotype in affected members of largest of these families (FS-Z) showed cosegregation only with chromosome 11q markers (with 2 point lod score examining linkage of affected individuals with the disease-associated haplotype equal to 1.8 at θ=0). We identified different heterozygous sequence variants in five unrelated families with adult onset disease, all of which predicted changes in the encoded gene product: N143S, S270T, K874Stop, R895C and E897K (Table 1). We genotyped other available family members for the relevant variants. In all families, patterns of TRPC6 variant and disease inheritance followed a pattern of cosegregation (with less than complete penetrance). In each family, inheritance was consistent with an autosomal dominant pattern (Figure 4; see Supplementary Note for clinical details). Each of the observed amino acid substitutions occurred in evolutionarily conserved residues (Figure 5a). Two mutations predict amino-acid substitutions in the N-terminal intracellular domain of TRPC6, two predict amino-acid substitutions in the C-terminal intracellular domain, and one encodes a premature stop codon near the C terminus (Fig 5b). We genotyped 180 control individuals for the disease-associated variants. None of these disease associated variants were identified in the controls. In addition, none of these substitutions were found in the public SNP databases.
TABLE 1.
Characteristics of TRPC6 mutations
| Family | Ethnicity | Mutation | Exon | Age range at disease presentation (yrs) | Number of family members with ESRD | Change in current amplitude |
|---|---|---|---|---|---|---|
| FG-EA | African-American | N143S | 2 | 27–39 | 5 of 36 | No |
| FG-BN | Colombian | S270T | 2 | 17–52 | 3 of 12 | No |
| FS-Z | Polish | K874Stop | 12 | 27–57 | 9 of 53 | No |
| FG-FQ | Mexican | R895C | 13 | 18–46 | 6 of 25 | Yes |
| FS-XR | Irish/German | E897K | 13 | 24–35 | 2 of 12 | Yes |
Summary of families and TRPC6 variants identified, with age range of disease presentation, number of family members known to have end-stage renal disease (ESRD), and whether the variant protein produced an altered current amplitude when expressed in HEK cells. See Supplementary Note for additional clinical details.
FIGURE 4.
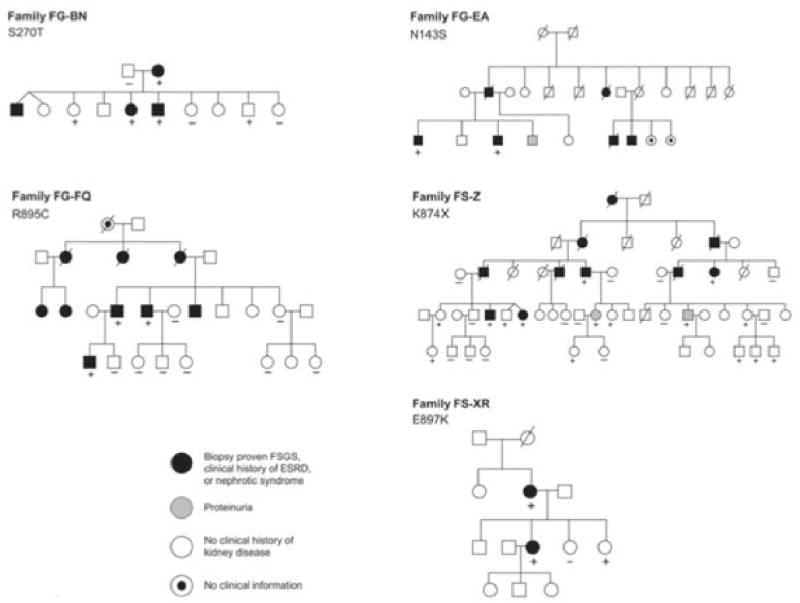
Familial FSGS pedigrees. TRPC6 variants segregating within each family are indicated. Genotyped individuals are indicated as carrying (+) or not carrying (–) the variant identified in each family.
FIGURE 5.

Sequence alterations. TRPC channel sequence alignments (using T-COFFEE). Mutated residues all occur in highly conserved amino acid residues.
To study whether mutations in TRPC6 affect calcium channel function we expressed either wild type or each of the five mutant TRPC6 channels in HEK293-M1 cells stably transfected with the Gαq-coupled M1 muscarinic receptor. TRPC6 currents were recorded before and after activation of M1 receptors by carbachol. Although we were able to measure currents from the N143S, S270T and K874Stop TRPC6 channels, the currents did not differ noticeably from wild type TRPC6 currents (data not shown). In contrast, currents from R895C and E897K mutant channels were significantly larger than wild type TRPC6 currents (Figure 6). Similarly, Winn and coworkers identified increased current amplitude with the TRPC6 P112Q channel 5,6. We also noted subtle differences in the rectification of the current-voltage relationship of these two mutants. However, we believe these changes in rectification result from the increased current density rather than directly from structurally related changes in channel gating or permeation. If the currents through R895C and E897K TRPC6 channels are similarly increased in vivo, these mutations could lead to a gain-of-function alteration in activity and thus increased calcium influx.
FIGURE 6.
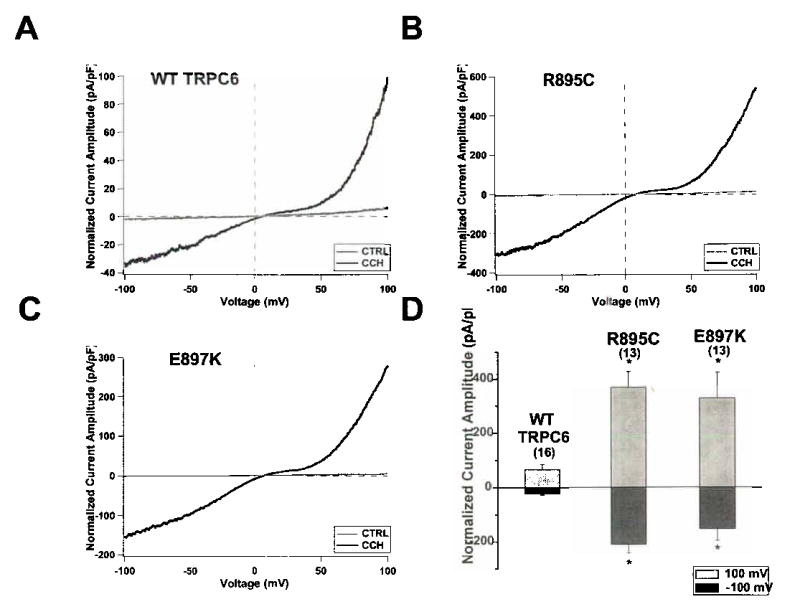
Representative whole-cell currents measured from HEK293-M1 cells transiently transfected with WT TRPC6 (a), R895C (b) or E897K (c) cDNA. Current traces were recorded as cells were perfused with control bath solution (CTRL, gray traces) or 100 μM carbachol (CCh, black traces). Voltage ramps from –100 mV to 100 mV over 150 ms were applied every 3.45 seconds from a holding potential of 0 mV. Current amplitude was normalized for cell capacitance. (d) Average normalized current amplitude measured at –100 mV (dark bars) and 100 mV (light bars) from cells expressing WT TRPC6, R895C or E897K. R895C and E897K current amplitudes were significantly increased at both –100 mV and 100 mV compared to WT current amplitudes (*, p < 0.01). The number of experiments for each is shown in parentheses, and the error bars show the S.E.M. for each measurement.
Three of the TRPC6 mutations identified did not produce apparent changes in current amplitude. Nevertheless, we believe these to be disease causing because of the nature of the changes (substitutions in very highly conserved residues or premature stop codon), cosegregation with the disease phenotype, and the absence of these changes from control individuals. This suggests that an abnormality other than increased current amplitude is the cause of disease in the individuals with these mutations. The several possibilities include altered channel regulation (despite normal amplitude), altered interaction with other slit-diaphragm proteins, and altered protein turnover.
Mutations in several podocyte genes have been identified as culprits for progressive renal failure or increased glomerular disease susceptibility 4. The presence of TRPC6 mutations co-segregating with kidney disease, the evolutionary conservation of the altered amino acids, slit-diaphragm localization and interactions, and the gain-of-function changes observed in two mutants suggest that TRPC6 channel function is essential for normal renal ultrafiltration. The question remains why the onset of kidney disease in patients with TRPC6 mutations occurs at a relatively advanced age. Similarly to the adult onset and dominantly inherited form of FSGS due to mutations in the widely expressed protein α-actinin-4, gain-of-function mutations in TRPC6 may produce subtle changes in intracellular function that lead to irreversibly altered cell behavior only after time and in the presence of other renal insults19. In addition, podocytes express several other TRPC channels including TRPC1,2,5 and 6 (Fig.1b). Partial functional redundancy may also help account for the late onset of glomerular disease. The ability of TRPC6 to form heteromers with other TRPC channels suggests a complex cellular regulation of calcium homeostasis.
Podocyte electrophysiology has been under investigation for many years20. However, published work has been limited to measurements of the membrane potential and whole-cell conductance in rodent podocytes and their response to vasoactive agonists 20. The location of podocytes within the glomerulus, surrounding the glomerular capillaries, exposes these cells to the transmural hydrostatic pressure driving ultrafiltration. Podocyte foot processes contain a contractile apparatus that may be regulated by slit-diaphragm-derived calcium signaling 21. Mature podocytes express several types of receptors and second messenger systems 3. These include receptors for muscarine, angiotensin, prostaglandin E2, and atrial natriuretic peptide, which all activate intracellular Ca2+, phospholipase C, inositol 1,4,5-triphosphate, cAMP and cGMP signaling cascades. Thus, podocytes may employ foot process and slit diaphragm-derived intracellular signals to respond to their cellular environment. Nephrin by itself has been shown to be a signalling molecule generating podocyte survival signals 22. Fyn kinase is associated with nephrin 23 and known to regulate TRPC6 channel opening by tyrosine phosphorylation 24. The disruption of nephrin from the slit diaphragm as has been reported in secondary forms of FSGS25 may mediate its effects on podocytes by modulation of TRPC6 originated calcium flux. Changes in TRPC6 calcium currents in podocyte foot processes appear to be central to the ability of the podocyte to regulate its intracellular and cytoskeletal behavior.
METHODS
Immunohistochemistry and immunoelectron microscopy
Female adult Sprague-Dawley rats (BW 200g) were perfused via the abdominal aorta with 2% paraformaldehyde in PBS for 3 min at 220 mm Hg followed by cryoprotectant sucrose-PBS solution (800 mOsmol) for 5 min at 220 mm Hg. Mouse kidneys (wild type and nephrin−/−) were harvested and snap-frozen according to standard protocols and fixed with 4% formaldehyde after sectioning in ice-cold acetone for 10 min. For immunofluorescent labeling, sections were washed once with PBS and incubated with blocking solution (2% FCS, 2% BSA, 0.2% fish gelatin) for 30 min at room temperature before further incubation with the primary antibody for 1 h at room temperature. For double labeling, a second primary antibody was applied for 1 h. Antigen-antibody complexes were visualized with secondary antibodies conjugated with fluorochromes. Specimens were analyzed using a confocal microscope (Zeiss). Immunogold labeling of ultrathin frozen sections of perfusion-fixed mouse kidney were incubated with the rabbit anti-TRPC6 antibody (Chemicon), followed by gold-labeled anti-rabbit secondary antibody. Images were obtained using a Philips CM10 electron microscope.
We used the following antibodies: mouse mab G1 to Synaptopodin and rabbit polyclonal antisera to TRPC6 (Chemicon, Sigma, Alomone 1:50 dilution) and polyclonal antibodies against CD2AP (gift from Dr. Andrew Shaw, Washington University), nephrin and podocin (all in 1:200 dilution). As a negative control, the primary antibodies were either omitted or, in the case of TRPC6 staining, the primary antibody was pre-incubated with a TRPC6 control peptide for 1 hour before use in the immunolabeling. Specimens were analyzed using a confocal microscope (Zeiss). For immunofluorescent labeling of cultured podocytes, cells were processed as decribed before 11. Immunofluorescent labelling of nephrin-deficient kidneys was performed as described previously 18.
Co-immunoprecipitation studies
Recombinant mouse GFP-TRPC6 was co-expressed with FLAG-tagged mouse CD2AP, nephrin and podocin, respectively, in HEK293 cells. FLAG-fusion proteins from cell lysates were immunoprecipitated using anti-FLAG-M2 beads and eluates were analyzed by immunoblotting using anti-FLAG and anti-GFP antibodies (Sigma).
For endogenous co-immunoprecipitation studies, whole cell extracts from cultured differentiated podocytes were prepared and incubated with anti-TRPC6 antibody overnight. The reaction was then incubated with protein G-coupled beads (Sigma) for 2 hours. Co-immunoprecipitated protein complexes were visualized by Western Blotting using antibodies against TRPC6 (1:300 dilution), CD2AP (1:2000), nephrin (1:300) and podocin (1:1,500). As a negative control, we used protein G coupled beads without antibody.
Cell culture and transfection
Wild type murine podocytes were cultured as described26. A GFP-TRPC6 construct, which included the complete mouse TRPC6 cDNA, was kindly provided by Mike Zhu (Ohio State University, Columbus, OH, United States). Cultured podocytes were transfected using standard protocols 10.
RT-PCR
We isolated total RNA from murine cultured podocytes using the Trizol reagent (Invitrogen) according to the manufacturer’s instructions. cDNA synthesis was performed with SuperScript II Reverse Transcriptase (Invitrogen) using Oligo(dT)12-18 oligonucleotide primers according to the manufacturer's instructions.
Clinical recruitment
Blood was obtained from members of families with familial FSGS after informed consent was given in accordance with a protocol approved by the Institutional Review Board at Brigham and Women’s Hospital. In these families, clinical history and pedigree information was obtained. Urine measurements for albumin excretion were performed using a DCA 2000 microalbumin/creatinine analyzer (Bayer). Genomic DNA was isolated from peripheral blood leukocytes using Qiagen columns.
Genotyping
DNA from FSGS affected probands from 71 families was analyzed for mutations in the TRPC6 gene using bidirectional sequencing. Sequence analysis was performed using PCR amplified genomic DNA. High-throughput capillary sequencing instrumentation and Sanger dideoxy DNA sequencing was used for mutation detection. Verification of sequence alterations in probands, family members, and control subjects was done using MALDI-TOF mass spectrometry (Sequenom) based SNP genotyping at the Harvard-Partners Core Genotyping Facility (primer sequences are available as Supplementary Table 1).
Sequence Alignment
Sequence alignments were performed using the T-Coffee program comparing human TRPC6 protein sequences to other human TRPC channels (1, 3–7) and TRPC6 protein sequences in Fly (Drosophila), Rat (Rattus norvegicus), Mouse (Mus musculus), Guinea Pig (Cavia porcellus), and Chimpanzee (Pan troglodyte) 27. Protein sequence data was obtained from the NCBI database (http://www.ncbi.nlm.nih.gov).
Site directed mutagenesis
Site-directed mutagenesis was performed to insert the variants identified into a full-length human TRPC6 cDNA clone (pcDNA3.1-TRPC6). Two mutagenic oligonucleotide primers were designed containing the desired mutation, flanked by unmodified nucleotide sequence. Mutagenesis was performed with an amplification reaction using the human TRPC6 cDNA template, mutagenic primers, and Pfu DNA polymerase. Clones were then selected and sequenced to identify and verify mutants.
Electrophysiological analysis of mutant and wild type TRPC6
35 mm dishes of HEK293-M1 cells, human embryonic kidney cells stably transfected with the M1 muscarinic receptor, were transiently transfected with 2 μg of WT or mutant TRPC6 cDNA and 0.25 μg of eGFP cDNA using Lipofectamine 2000 (Invitrogen). After transfection cells were plated on glass coverslips at low density. We recorded from cells 24–72 hours after transfection. Cells were maintained at 37° C in DMEM/Ham’s F-12 (1:1), 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin and 100 μg/mL G-418 in 5% CO2.
We visualized eGFP-positive cells with a fluorescence microscope (Olympus) and recorded currents using an Axopatch 200B amplifier and pClamp8 software (Axon Instruments). During voltage ramps, currents were sampled at 10 kHz and the recordings were filtered at 2kHz. In each experiment the membrane potential was held either at −60 mV or 0 mV; no differences were noted between experiments performed at a holding potential of −60 mV and 0 mV. Borosilicate glass pipettes with resistances of 2–4 MΩ were used for recording. The bath solution contained 135 mM NaCl, 5 mM CsCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 10 mM Glucose, pH 7.4. The pipette solution contained 135 mM CsMES, 10 CsCl, 3 mM MgATP, 0.2 mM NaGTP, 0.2 mM EGTA, 0.13 mM CaCl2, 10 HEPES, pH 7.3. Free calcium concentration in the pipette solution was approximately 100 nM as calculated by MaxChelator. Average current amplitudes at –100 mV and 100 mV were compared with the Student’s t test.
Acknowledgments
We thank the family members for their participation in these studies and Amin Arnaout for helpful discussions on the manuscript. This work was supported by NIH grants DK54931 (to M.R.P.), DK057683 (to P.M) and DK55001 (to R.K.) as well as the Howard Hughes Medical Institute (P.L.S., D.C.). J.R. was supported by the KMD Foundation and the KUFA-ASN Research Grant.
References
- 1.Zandi-Nejad K, Eddy AA, Glassock RJ, Brenner BM. Why is proteinuria an ominous biomarker of progressive kidney disease? Kidney Int Suppl. 2004:S76–89. doi: 10.1111/j.1523-1755.2004.09220.x. [DOI] [PubMed] [Google Scholar]
- 2.Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24:333–5. doi: 10.1038/74139. [DOI] [PubMed] [Google Scholar]
- 3.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 4.Pollak MR. Inherited podocytopathies: FSGS and nephrotic syndrome from a genetic viewpoint. J Am Soc Nephrol. 2002;13:3016–23. doi: 10.1097/01.asn.0000039569.34360.5e. [DOI] [PubMed] [Google Scholar]
- 5.Winn MP, et al. Mutation in TRPC6 causes familial focal segmental glomeruloslcerosis. J Am Soc Nephrol. 2004;15:33A. [Google Scholar]
- 6.Winn, M.P. et al. A Mutation in the TRPC6 Cation Channel Causes Familial Focal Segmental Glomerulosclerosis. Science (2005). [DOI] [PubMed]
- 7.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 8.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–24. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 9.Facemire CS, Mohler PJ, Arendshorst WJ. Expression and relative abundance of short transient receptor potential channels in the rat renal microcirculation. Am J Physiol Renal Physiol. 2004;286:F546–51. doi: 10.1152/ajprenal.00338.2003. [DOI] [PubMed] [Google Scholar]
- 10.Mundel P, et al. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139:193–204. doi: 10.1083/jcb.139.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mundel P, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–58. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 12.Schnabel E, Dekan G, Miettinen A, Farquhar MG. Biogenesis of podocalyxin--the major glomerular sialoglycoprotein--in the newborn rat kidney. Eur J Cell Biol. 1989;48:313–26. [PubMed] [Google Scholar]
- 13.Boute N, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–54. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 14.Kestila M, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–82. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 15.Kim JM, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300:1298–300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 16.Reiser J, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–7. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Putaala H, Soininen R, Kilpelainen P, Wartiovaara J, Tryggvason K. The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet. 2001;10:1–8. doi: 10.1093/hmg/10.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Hamano Y, et al. Determinants of vascular permeability in the kidney glomerulus. J Biol Chem. 2002;277:31154–62. doi: 10.1074/jbc.M204806200. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan JM, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–6. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 20.Pavenstadt H, Bek M. Podocyte electrophysiology, in vivo and in vitro. Microsc Res Tech. 2002;57:224–7. doi: 10.1002/jemt.10078. [DOI] [PubMed] [Google Scholar]
- 21.Drenckhahn D, Franke RP. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab Invest. 1988;59:673–82. [PubMed] [Google Scholar]
- 22.Huber TB, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol. 2003;23:4917–28. doi: 10.1128/MCB.23.14.4917-4928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verma R, et al. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem. 2003;278:20716–23. doi: 10.1074/jbc.M301689200. [DOI] [PubMed] [Google Scholar]
- 24.Hisatsune C, et al. Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem. 2004;279:18887–94. doi: 10.1074/jbc.M311274200. [DOI] [PubMed] [Google Scholar]
- 25.Schmid H, et al. Gene expression profiles of podocyte-associated molecules as diagnostic markers in acquired proteinuric diseases. J Am Soc Nephrol. 2003;14(11):2958–66. doi: 10.1097/01.asn.0000090745.85482.06. [DOI] [PubMed] [Google Scholar]
- 26.Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 27.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]