

Abstract
Developmental abnormalities in endocardial cushions frequently contribute to congenital heart malformations including septal and valvular defects. While compelling evidence has been presented to demonstrate that members of the TGF-β superfamily are capable of inducing endothelial-to-mesenchymal transdifferentiation in the atrioventricular canal, and thus play a key role in formation of endocardial cushions, the detailed signaling mechanisms of this important developmental process, especially in vivo, are still poorly known. Several type I receptors (ALKs) for members of the TGF-β superfamily are expressed in the myocardium and endocardium of the developing heart, including the atrioventricular canal. However, analysis of their functional role during mammalian development has been significantly complicated by the fact that deletion of the type I receptors in mouse embryos often leads to early embryonal lethality. Here, we used the Cre/loxP system for endothelial-specific deletion of the type I receptor Alk2 in mouse embryos. The endothelial-specific Alk2 mutant mice display defects in atrioventricular septa and valves, which result from a failure of endocardial cells to appropriately transdifferentiate into the mesenchyme in the AV canal. Endocardial cells deficient in Alk2 demonstrate decreased expression of Msx1 and Snail, and reduced phosphorylation of BMP and TGF-β Smads. Moreover, we show that endocardial cells lacking Alk2 fail to delaminate from AV canal explants. Collectively, these results indicate that the BMP type I receptor ALK2 in endothelial cells plays a critical non-redundant role in early phases of endocardial cushion formation during cardiac morphogenesis.
Keywords: Atrioventricular cushion, ALK2, BMP, TGF-β, Transformation, Cardiac development
Introduction
Cardiovascular malformations are the most common life-threatening birth defects in humans, affecting approximately one in one hundred live births (Srivastava, 2001; Hoffman and Kaplan, 2002). The etiology of these conditions is still very poorly known, but it often appears to be multi-factorial, involving both environmental and genetic causes. Among cardiac deformities, atrioventricular (AV) septal defects are very frequent (Schroeder et al., 2003). They arise from a failure of endocardial cushions to fuse, which leads to a range of secondary abnormalities, including incomplete formation of the septum between atria and ventricles and malformation of the AV valves. AV cushion formation is initiated by factors that induce endothelial-to-mesenchymal transdifferentiation (EMT) (Eisenberg and Markwald, 1995), and indeed, it has been shown that the vast majority of cushion mesenchymal cells are derived from the endothelium (Kisanuki et al., 2001; Anderson et al., 2003; de Lange et al., 2004). Subsequent morphogenetic events lead to elongation, outgrowth and remodeling of cushions to form mature septa and delicate thin valve leaflets. During EMT, endothelial cells delaminate from the endocardium into the underlying acellular hyaluronate-rich substance called cardiac jelly that separates endocardial and myocardial layers, and transform into cushion mesenchymal cells. Co-culture experiments have shown that signals from both the adjacent myocardium and from the extracellular matrix of the cardiac jelly play a critical role in the endocardial cell EMT (Eisenberg and Markwald, 1995).
Many soluble growth factors, transmembrane receptors, intracellular signaling molecules, nuclear factors and glycosaminoglycans are known to induce EMT. Initially, it was shown that members of the TGF-β superfamily are able to induce EMT in the chick during cardiac morphogenesis (Potts and Runyan, 1989; Eisenberg and Markwald, 1995). The Wnt/β-catenin pathway was shown to be required for EMT, first in zebrafish (Hurlstone et al., 2003) and subsequently also in mice (Liebner et al., 2004), while recent studies suggest that Notch signaling plays a role upstream of TGF-βs during murine endocardial cushion transformation (Timmerman et al., 2004). Normal cushion tissue formation has also been shown to be controlled by regulators of the Ras signaling pathway (Lakkis and Epstein, 1998; Camenisch et al., 2002b; Gitler et al., 2003b). Recently, dynamic temporal and spatial changes in calcineurin signaling and NFATc1/VEGF expression were demonstrated to play key roles in endocardial EMT and in valve development (Chang et al., 2004). However, detailed signaling mechanisms as well as crosstalk between different signaling pathways remain largely unknown.
Several studies using a three-dimensional explant culture model in conjunction with antisense oligonucleotides and neutralizing antibodies have shown that TGF-βs, particularly TGF-β2 and -3, as well as BMPs are essential growth factors for initiation and regulation of EMT (Potts and Runyan, 1989; Potts et al., 1991; Runyan et al., 1992; Markwald et al., 1996; Nakajima et al., 2000). In addition, studies using neutralizing antibodies in the chick explant culture model have suggested that the prototypical TGF-β type I receptor, ChALK5, is not involved in the cushion EMT, while inhibition of a related type I receptor, ChALK2, prevents the transdifferentiation (Lai et al., 2000). Subsequently, it was shown that constitutively active ALK2 alone is sufficient to stimulate EMT (Desgrosellier et al., 2005). Mice deficient both in BMP5 and BMP7 display defective cushion development (Solloway and Robertson, 1999), and it was recently reported that a tissue-specific deletion of the BMP type I receptor Alk3 in myocardial cells leads to hypoplastic endocardial cushions and decreased Tgf-β2 expression (Gaussin et al., 2002). In fact, mice deficient in SMAD6, an inhibitory intracellular signal mediator of TGF-βs, display endocardial cushion hyperplasia (Galvin et al., 2000). However, single TGF-β null mutants do not demonstrate impaired endocardial EMT (Shull et al., 1992; Kulkarni et al., 1993; Kaartinen et al., 1995; Proetzel et al., 1995; Sanford et al., 1997), while mice deficient in TGF-β2 display defects in later stages of valve remodeling (Sanford et al., 1997; Bartram et al., 2001).
TGF-β superfamily members signal via heteromeric receptor complexes composed of two type II and two type I receptors. Different receptor complexes have been shown to bind TGF-β ligands with different affinities, which determine downstream signaling responses (Derynck and Feng, 1997). For instance, TGF-βs typically bind to a receptor complex composed of the TGF-β type II receptor and ALK5, while BMPs regularly signal via BMP type II receptor and ALK3 (Massague and Chen, 2000). The type I receptor ALK2 has been shown to display a more restricted signaling specificity for BMPs 5, -6 and -7 (Macias-Silva et al., 1998). However, it has been suggested that ALK2 might mediate TGF-β signals in specific cell types in vitro, such as mouse mammary epithelial cells (Miettinen et al., 1994).
Studies to unravel the in vivo roles of several BMP ligands and their receptors in mammals have often been hampered by the fact that mice deficient in Bmps 2 or -4, as well as mice deficient in type 1 receptors (Alk2 and Alk3) die at or immediately after gastrulation, before cardiac morphogenesis (Winnier et al., 1995; Mishina et al., 1995; Zhang and Bradley, 1996; Gu et al., 1999; Mishina et al., 1999). To define the role of Alk2 in endocardial cell transformation in vivo, we used the Tie2-Cre transgenic mouse line to target Cre recombinase into endothelial cells (Kisanuki et al., 2001; Koni et al., 2001). Tie2-Cre transgenic mice heterozygous for the Alk2 knockout allele were subsequently crossed with homozygous Alk2Fx/Fx mice (Kaartinen and Nagy, 2001; Kaartinen et al., 2004; Dudas et al., 2004). The resulting Alk2/Tie2-Cre mutants display AV and ventricular septal defects, and show that ALK2 is required for successful endothelial cell to mesenchyme transdifferentiation in AV cushions.
Materials and methods
Mice and genotyping
Mice homozygous for the Alk2FX allele and mice heterozygous for the Alk2KO allele were generated and genotyped as described previously (Kaartinen and Nagy, 2001; Kaartinen et al., 2004; Dudas et al., 2004). Tie2-Cre and Rosa26R Cre (R26R) reporter mice were obtained from the Jackson Labs and genotyped by PCR (for detailed protocols see: http://www.jax.org). All mice were maintained on mixed genetic backgrounds. All studies were carried out at the Animal Care Facility of the Saban Research Institute of Childrens Hospital Los Angeles in accordance with institutional guidelines.
Histological analyses
Embryonic tissues were fixed with 4% buffered formaldehyde for 2–12 h, dehydrated and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin. For cryostat sectioning, tissues were fixed in 4% buffered formaldehyde and equilibrated in 30% sucrose prior to freezing in HistoPrep (Fisher). Frozen sections were cut at 8 μm. Embryos or sections were stained for β-galactosidase activity as described (Hogan et al., 1994). Briefly, the specimens were fixed in 4% buffered formaldehyde for 30 min at room temperature, washed 3 times for 10 min in the detergent wash and developed for 2 to 6 h in the X-gal staining solution.
Expression studies
Wholemount and section in situ hybridization on embryos was carried out as described (Hogan et al., 1994; Moorman et al., 2001). Probes specific for Msx1 (Furuta et al., 1997), Snail (Cano et al., 2000), Hey2 (Donovan et al., 2002), Pdgfrα (Gitler et al., 2003a) and Tgf-β2 (Blavier et al., 2001) were used. To analyze phosphorylation of Smads, de-paraffinized sections were boiled for 10 min in 10 mM citrate buffer, pH 6.0 in a pressure cooker and immunostained using antibodies specific for phospho-Smad-1/5/8 or phopho-Smad-2 (Cell signaling). For immunohistochemistry, frozen sections were stained with monoclonal antibodies to CD31 (Sigma) or to NFATc1 (Developmental Studies Hybridoma Bank at the University of Iowa) according to standard procedures and counterstained with DAPI (Harlow and Lane, 1988).
Apoptosis and cell proliferation
Apoptotic cells were detected using the DeadEnd Fluorometric TUNEL system (Promega). Cell proliferation was analyzed using the BrdU incorporation assay (Zymed) or by immunostaining for phosphohistone H3 (Cell signaling).
Explant cultures
Collagen gels (1 mg/ml, type I rat tail collagen from BD) were prepared in OptiMEM supplemented with 1% fetal calf serum, 1× ITS (insulin, transferrin and selenium) and penicillin/streptomysin (1× ) all from Invitrogen (Sugi et al., 2004). AV regions of the hearts were dissected from E10 embryos, cut longitudally to expose the lumen and placed on the collagen gels. Additional media were added to the cultures 2 h later and incubation was continued under standard tissue culture conditions (37°C, 100% humidity, 8% CO2). For some cultures, TGF-β3 (10 ng/ml, Sigma) or BMP2 (50 ng/ml, Sigma) was added to the culture media.
Results
Alk2 is expressed in endothelial cells in the mouse AV canal
It has previously been shown that the Alk1 gene (encoding the TGF-β/BMP type I receptor) is expressed in endothelial cells and that ALK1 mediates TGF-β signaling in concert with the prototypical TGF-β type I receptor ALK5 during the proliferative phase of angiogenesis (Goumans et al., 2003). Moreover, it has been suggested that in chick the type I receptor ALK2, which is closely related to ALK1, is involved in formation of AV-cushions during cardiac development by functioning in endothelial-to-mesenchymal transformation (Lai et al., 2000; Desgrosellier et al., 2005). Therefore, we were interested in whether ALK2 would also be involved in endothelial development in mammals, particularly during cardiac morphogenesis. As a first step, we analyzed expression of Alk2 in the AV-canal at embryonic day 10 (E10), when the cushions are forming (Fig. 1A). Alk2 mRNA was detected in endocardial cells, as well as in some underlying mesenchymal cells, as previously described by Gu et al. (1999). To analyze the function of Alk2 in mammalian endothelial cells of the AV canal, we used the tissue-specific Cre/loxP gene targeting strategy. It has previously been shown that the Tie2 promoter/enhancer will efficiently target transgene expression to the endothelium, and that mice expressing Cre recombinase under the control of the Tie2 promoter can be used to abrogate a gene of interest efficiently and specifically in the endothelium (Kisanuki et al., 2001; Koni et al., 2001). To verify that this is indeed the case in the transgenic line that we opted to use (Koni et al., 2001), we followed the fate of cells recombined by the Tie2-Cre transgene by using the ROSA26 Cre reporter (R26R) assay as previously reported (Soriano, 1999; Chai et al., 2000; Jiang et al., 2000). The double transgenic embryos (Tie2-Cre+/−;R26R+/−) were harvested at E12, and stained for β-galactosidase activity. Positive staining was seen specifically in endothelial cells (Fig. 1B) and in mesenchymal cells derived from the endocardium, e.g., in the mesenchyme of endocardial cushions (Fig. 1C) confirming that this Tie2-Cre transgenic line could efficiently be used to induce recombination in the endothelial cell lineage. Therefore, we crossed the Tie2-Cre mice that also were heterozygous for the Alk2 knockout allele with mice homozygous for the floxed Alk2 (Alk2FX/FX) allele (Kaartinen and Nagy, 2001; Kaartinen et al., 2004; Dudas et al., 2004). Efficiency of the Cre-induced recombination in the AV canal at E10 was verified by using the RT-PCR strategy as previously described (Dudas et al., 2004). This analysis showed that the Alk2 gene was effectively recombined by the Tie2-Cre transgene (Fig. 1D). To compare the overall vascular phenotype between controls and Alk2/Tie2-Cre mutants early in development, we crossed Alk2KO/WT;Tie2-Cre males with females that were homozygous both for the AlkFX and R26R alleles (Alk2FX/FX;R26R+/+), harvested embryos at E9.5 and stained them for β-galactosidase activity (Figs. 1E–H). Blood vessels were strongly stained both in controls and mutants demonstrating that vascular development was not significantly affected by the loss of Alk2 activity in endothelial cells.
Fig. 1.

Targeted deletion of Alk2 in the endothelium. (A) Alk2 is expressed in endocardial cushions at E10 (arrow points to the positive in situ hybridization signal [blue] in endothelial cells). (B–C) The Tie2-Cre transgene specifically induces recombination in endothelial cells (B) and in cells derived from them, i.e., in the mesenchyme of endocardial cushions (arrows in panel C) as demonstrated using R26R reporter assay and staining for β-galactosidase activity (Blue stain), counter stained with eosin. (D) The RT-PCR analysis of mRNA isolated from the AV-canal tissues at E10.0 in wild-type (WT = Tie2-Cre+/−;Alk2WT/WT), heterozygote (He = Tie2-Cre+/−;Alk2KO/WT) and homozygote (Ho = Tie2-Cre+/−;Alk2FX/KO) mutant embryos. Shorter amplification product (284 bp) was detected in heterozygote and homozygote embryos, confirming a successful recombination event in targeted tissues resulting in a truncated allele. (E–H) Wholemount staining of control (Tie2-Cre+/−;Alk2FX/WT;R26R+/−) and Alk2/Tie2-Cre mutant (Tie2-Cre+/−;Alk2FX/KO;R26R+/−) embryos for β-galactosidase activity (blue color); R26R lineage tracing assay. Comparable reporter activity can be seen in mutants (F, H) and controls (E, G), including the AV canal (arrows, G and H). Numbers (3, 4, 6) in panels G and H depict the aortic arch arteries. (I–L) Endocardial cushions (arrowheads in panels I–L) form in Alk2/Tie2-Cre mutants (J, L), but appear considerably smaller than in controls at E10.5 (I, K). (M) Quantification of mesenchymal cells in AV cushions at E10.5; mean ± SD; *P < 0.01 (n = 8, total number of mesenchymal cells were counted per parasagittal section; 3 sections analyzed per sample). A, Atrium; V, ventricle. Magnification: A, ×20; B, ×5; C, ×10; D–F, ×3; G–H, ×7; I–J, ×10; K–L, ×40.
Alk2/Tie2-Cre mutants display AV-canal defects
Genotype analysis of embryos from Alk2KO/WT/Tie2-Cre+/− ×Alk2FX/FX crosses at E14.5 revealed that only 10% instead of the expected 25% of Alk2/Tie2-Cre mutants could be recovered at that time point (Table 1). In histological sections at E10, the endocardial cushions appeared smaller in mutants than in controls (Figs. 1I–L). A dramatic reduction in mesenchymal cell number was particularly evident in the superior cushion and differed significantly from that of controls (P < 0.01) (Fig. 1M).
Table 1.
Genotype distribution of embryos from Alk2KO;Tie2-Cre+/− ×Alk2FX/FX crosses
| Genotype | E10–E11 (n = 307) | E14–E15 (n = 97) |
|---|---|---|
| Alk2KO/Tie2-Cre+ | 89 (22.5%) | 11 (10.2%) |
| Alk2WT/Tie2-Cre+ | 108 (27.3%) | 28 (28.8%) |
| Alk2KO/Tie2-Cre− and Alk2WT/Tie2-Cre− | 199 (50.2%) | 58 (59.8%) |
At E14.5, the surviving Alk2/Tie2-Cre embryos (n = 17) showed a range of cardiac defects (Fig. 2). Most of them (15 out of 17 studied) had a ventricular septal defect, of varying severity, where the secondary ventricular foramen has not closed. In addition, 11 had defective AV septation and valve development. The AV cushions were of variable size, shape and degree of fusion with one another, or with other septal structures. For example, the primary atrial foramen was still patent, allowing blood to shunt between right and left atria in 5 embryos. Outgrowth and formation of AV leaflets were also variable. In some, cushion tissue was sparse or absent altogether from most of the AV junction, especially on the right, although prominent trabecular structures, perhaps those that would normally support the developing leaflet cushions, were still evident. In contrast, outflow tract (OFT) septation and OFT leaflet formation appeared normal (Fig. 2 and data not shown).
Fig. 2.

Tissue-specific abrogation of Alk2 in endothelial cells leads to septal and valvular defects. Transverse sections on 3 different levels from rostral to caudal in a control (A–C) and mutant (D –F) at E14.5. Defects can be seen both in ventricular (arrow in panel E) and atrioventricular (arrows in panel F) septation, as well as in AV valves (arrowhead in panel F). In contrast, tissue-specific abrogation of Alk2 in cardiac myocytes does not impair cardiac development (G–I) at E14. Sections on comparable levels are shown. Magnification: ×5.
Alk2 is not required in cardiac myocytes during heart organogenesis
Although our expression studies suggest that Alk2 is not strongly expressed in the myocardium, we could not exclude the possibility that low level Alk2 expression would still play a critical role in this cell type. Therefore, we deleted Alk2 in the myocardium by using the cardiac myocyte-specific Cre mouse line (αMHC-Cre) as previously described (Gaussin et al., 2002). Alk2/αMHC-Cre mice are viable and failed to display any detectable altered cardiac phenotype (Fig. 2) demonstrating, that unlike Alk3 (Gaussin et al., 2002), expression of Alk2 in cardiac myocytes is not required during this developmental stage.
Endothelial cells deficient in Alk2 fail to populate the mesenchyme of the AV cushions in vivo
To follow the fate of Alk2-deficient endothelial cells during AV canal development, we applied the R26R reporter assay described above. Analysis of parasagittal sections of control embryos (at E10–E12) revealed that both the endocardium as well as the underlying mesenchyme of AV cushions stained positive for the β-galactosidase activity (Figs. 3A, E, G). In contrast, the mesenchyme of mutant AV cushions displayed only a few positively staining cells, while AV canal endocardial cells deficient in Alk2 demonstrated strong positive β-galactosidase staining (Figs. 3B, F, H). Comparison of the cardiac phenotype between Alk2/Tie2-Cre and Alk2/Tie2-Cre;R26R mutants did not reveal any histological differences between these two backgrounds (data not shown). Consistent with the histological studies (Fig. 2), the cushions of Alk2 mutants were overall smaller and less cellular than those of controls (Figs. 3A,B). In addition to AV canal cushions, we also examined the contribution of the endocardially derived cells in the OFT mesenchyme. While mesenchymal cells of the proximal outflow tract from control embryos stained strongly positive for β-galactosidase at E11.0 (Fig. 3C) as previously reported (Kisanuki et al., 2001), no corresponding positively staining cells could be seen in the mesenchyme of Alk2/Tie2-Cre mutants (Fig. 3D). At E14, the mesenchyme of both the AV septum and AV valves was populated by positively staining cells derived from the endothelium in controls (Fig. 4). In contrast, the corresponding mesenchymal structures in Alk2 mutants showed only a few positively staining cells. In addition, many mutants showed abnormal endocardial thickenings (Fig. 4F). Consistent with the histological analyses, Alk2/Tie2-Cre mutants displayed ventricular septal defects (Fig. 4).
Fig. 3.

Fate mapping of endothelial cells in Alk2/Tie2-Cre mutants. While endothelial cells covering endocardial AV-cushions stain positive both in controls (A, E, G) and Alk2/Tie2-Cre mutants (B, F, H) in the R26R reporter assay, only the wild-type mesenchyme demonstrates a large number of positively staining cells (parasagittal sections; arrowheads in panel B point to a few positively staining cells seen in the mesenchyme of Alk2 mutants). In controls, the proximal outflow tract mesenchyme displayed a large number of positively staining cells at E11 (arrow in panel C), while in mutants, no positively staining cells could be seen (arrow in panel D) (C–D, transverse sections). In older mutant embryos, the AV canal mesenchyme displayed a larger number of cells, but still only a few of them stained positive in the lineage tracing assay (F and H), while about 90% of the mutant mesenchyme demonstrated positive reporter activity (E and G). (A–D) Samples (A–B, E10; C–D, E11) were harvested and stained as wholemounts and sectioned after post-fixation. (E–H) Samples (E –F, E11; G –H; E12) were cryostat sectioned, and subsequently frozen sections were stained for β-galactosidase activity. SC, superior endocardial cushion; IC, inferior endocardial cushion. A, atrium; V, ventricle. Magnification: A –B: × 40; E –F: ×20; C–D, G–H: ×10.
Fig. 4.

Endothelial cells deficient in ALK2 fail to populate the valvular and septal mesenchyme. In controls (A –C), both the membraneous portion of the interventricular septum and the valve mesenchyme are composed of cells derived from the endothelium (blue, denoted with white asterisks), while Alk2/Tie2-Cre mutants (D–I) do not demonstrate similar mesen-chymal staining in corresponding locations (black asterisks). Arrows in panels D and G point to the ventricular septal defect. Arrowhead in panel F points to the abnormal epithelial thickening in a mutant. VS, ventricular septum; RV, right ventricle; LV, left ventricle. Magnification: A, D –G: ×10; B–C, E–I: ×20.
Altered gene expression and Smad activation in the AV canal of Alk2/Tie2-Cre mutants
The homeodomain protein Msx1 is a well-known effector of BMP signaling (Suzuki et al., 1997), and its expression has been shown to be induced by BMPs (Bei and Maas, 1998). Moreover, it has been demonstrated that the Msx1 gene is expressed in AV canal endocardial cells during EMT (Gitler et al., 2003a). Therefore, we investigated whether abrogation of Alk2 in endothelial cells could influence Msx1 expression. First, we isolated total RNA from the AV canal tissues harvested at E10 and analyzed them by RT-PCR. Msx1 expression was clearly attenuated in Alk2/Tie2-Cre mutants when compared to controls (Fig. 5AA). Next, we compared the localization of Msx1 mRNA by section in situ hybridization. In controls, Msx1 was strongly expressed in endocardial cells in the AV canal as well as in the cushion mesenchyme at E10 (Fig. 5A). In contrast, in Alk2/Tie2-Cre mutants, the endothelium of the superior AV cushion did not display any detectable staining, and there were only a few positively staining cells in the AV-cushion mesenchyme (Fig. 5B). Similarly, a transcriptional repressor Snail has been shown to play a key role in EMT (Savagner et al., 1997; Batlle et al., 2000; Cano et al., 2000; Timmerman et al., 2004). In the control AV-canal, the pattern of Snail expression was very similar to that of Msx1: strong expression both in the AV-canal endocardium and in the underlying mesenchyme (Fig. 5C). Moreover, Snail mRNA was detected in atrial endocardial cells, but not in those of the ventricles. Similar to controls, in Alk2/Tie2-Cre mutants, Snail was expressed in the atrial, but not in the ventricular endocardium. However, in the AV canal endothelium, expression of Snail was notably reduced when compared to controls or to the level of atrial expression in the same section (Fig. 5D). Recent studies have indicated that Notch signaling promotes endocardial cell transformation (Timmerman et al., 2004). Because a basic helix–loop–helix transcription factor Hey2 is an important mediator of Notch signaling (Donovan et al., 2002; Sakata et al., 2002), we compared the Hey2 expression in Alk2 Tie2-Cre mutants and controls. We found that in both control and mutant samples Hey2 was strongly expressed in the AV canal endocardium (Figs. 5E, F). In concordance with the earlier published studies, positive expression was also found throughout the ventricular myocardium and the OFT endothelium (data not shown) (Donovan et al., 2002). In addition, we analyzed expression of Pdgfrα, Msx2, Slug and ErbB2, all genes with a presumed role in the AV canal development; none of these genes displayed detectable differences in expression between controls and Alk2/Tie2-Cre mutants (Figs. 5G, H and data not shown).
Fig. 5.

Msx1 and Snail are differentially expressed in Alk2/Tie2-Cre mutants. (AA) Semi-quantitative RT-PCR analysis of Msx1 and Snail expression in the AV canal at E10; C, control, M1 and M2, two different mutant samples. β-actin was used as a quality and loading control (β-Act). Msx1 is strongly expressed both in the endothelium (arrow) and in the underlying mesenchyme in controls (A). In Alk2/Tie2-Cre mutants (B), the AV canal endothelium shows no positive signal (arrow). Only a few positively staining cells can be seen in the mesenchyme (arrowhead). In controls, the transcriptional repressor Snail is expressed in both the AV canal endothelium (arrow), mesenchyme and in the atrial endothelium (arrowhead in panel C). Similar to controls, Snail is expressed in the mutant atrial endocardium (arrowheads in panel D), while the expression in the AV canal endothelium is notably reduced (arrow in panel D). Insets in panels A–D show the high magnification images (×63); red arrows depict the endocardial cells. Endocardial expression of Hey2 (E–F) and Pdgfrα (G –H) is comparable between controls (E, G) and mutants (F, H). Both control (I, K) and Alk2/Tie2-Cre mutant (J, L) samples display similar expression patterns of endothelial cell-specific proteins, CD31 and NFATc1. Parasagittal sections of either E10 hearts (A–D and G–K) or E11 hearts (E –F). A, atrium; V, ventricle. A –H, section in situ hybridization; I–L, immunohistochemistry. Magnification: ×20.
To exclude the possibility that the loss of Alk2 in the AV canal endocardium would cause a developmental delay in endocardial cell maturation, which would subsequently disturb cushion formation, we compared expression of CD31 (a well established endothelial cell marker) and NFATc1 (previously shown to be strongly and specifically expressed in endothelial cells of AV cushions at the time when the cushion mesenchyme is formed) (Ranger et al., 1998; de la Pompa et al., 1998), in the AV canal at E10 between controls and mutants. As can be seen in Figs. 5I–L, the expression of both of these markers was comparable between the genotypes suggesting that delayed endothelial maturation of the AV canal was not responsible for the observed phenotypes.
Endothelial cells deficient in Alk2 fail to transdifferentiate in vitro
AV endocardial explants from both mouse and chick embryos have been shown to transdifferentiate in vitro, when placed on three-dimensional collagen gels (Bernanke and Markwald, 1984; Sugi and Markwald, 1996; Yamamura et al., 1997). We used this culture system and isolated AV canal tissues from controls and Alk2/Tie2-Cre mutants at E10.0. After dissection, explants were placed, endocardium down, onto collagen gels, and incubated under standard culture conditions for up to 50 h. The explants attached tightly on the gel and started to beat. After 24 h in culture, a large number of cells had migrated into the collagen gel from control explants (Fig. 6). About 90% of these cells showed a typical elongated fibroblastoid phenotype, while less than 10% of cells were of an intermediate rounded phenotype. These round cells presumably represent a population that is activated, but does not transdifferentiate, as seen in explants treated with the γ-secretase inhibitor, DAPT (Timmerman et al., 2004). In contrast, the number of migrating fibroblastoid cells in Alk2 mutants was reduced to about 10% of that of controls. Exogenous addition of BMP2 (50 ng/ml) or TGF-β3 (10 ng/ml) could not rescue the migration defect of Alk2/Tie2-Cre mutants (Fig. 6G).
Fig. 6.
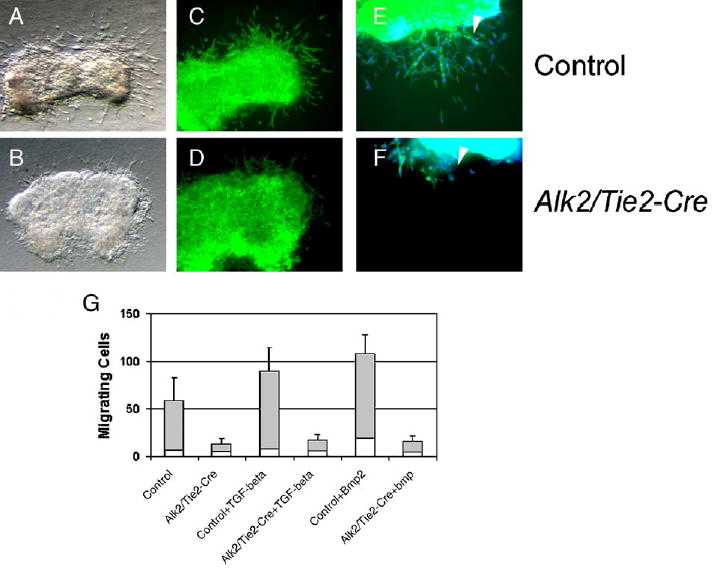
Endothelial cells deficient in Alk2 fail to transdifferentiate in vitro. (A–F) AV-canal explants were harvested at E10 and cultured for 24 h on 3-D collagen gels. Phase contrast images of control (A) and Alk2/Tie2-Cre (B) explants. (C–D) FITC-phalloidin staining (×10) and (E –F) FITC-Phalloidin staining (×20); counterstaining with DAPI. Transformed cells invading the collagen gel are indicated by arrowheads. The number of transformed cells is significantly reduced in Alk2/Tie2-Cre mutants when compared to controls. (G) Quantitative analysis of explants. Treatment of mutant explants with TGF-β3 (10 ng/ml) or BMP2 (50 ng/ml) stimulated transdifferentiation of endothelial cells in control explants, but was not able to rescue the Alk2-mutant phenotype.
Activation of BMP and TGF-β Smads is reduced in AV canal endocardial cells of Alk2/Tie2-Cre mutant mice
To establish how loss of Alk2 affects downstream signaling, we examined phosphorylation of both BMP and TGF-β Smads using antibodies that specifically recognize their phosphorylated, active forms. While the myocardium displayed a large number of the nuclei staining positive for phospho-Smad 1/5/8 both in controls and in mutants, the AV canal endocardium demonstrated positive staining only in controls (Figs. 7A–B). Surprisingly, the staining pattern of phospho-Smad2 was also affected in Alk2 mutants when compared to controls (Figs. 7C–D). These findings suggest that loss of Alk2 in AV canal endocardial cells affects activation not only of BMP Smads, which are directly phosphorylated by ALK2, but also that of related TGF-β Smads.
Fig. 7.

Smad activation is affected in Alk2/Tie2-Cre mutants. Both Smad1/5/8 phosphorylation (A–B) and Smad2 (C–D) phosphorylation are notably reduced in the AV-canal endocardium of Alk2/Tie2-Cre mutants (*** in panels B and D) at E10 when compared to those of controls (arrowheads in panels A and C). A, atrium; V, ventricle. Magnification: ×20.
Discussion
Formation of endocardial cushions during cardiac morphogenesis is required for normal septal and valvular morphogenesis. A critical step in this process is transformation of endocardial cells into invasive mesenchymal cells (EMT). As outlined above, the key role of TGF-β superfamily member signaling in induction of EMT is well established, particularly in chick models. However, substantial interspecies differences have been detected in cell signaling between avian and mouse AV canal explants (Camenisch et al., 2002a), and very little is known about differential roles of type I receptors in mammalian endocardial cells. To better elucidate individual functions of the type I receptors during mouse AV canal morphogenesis, we deleted the Alk2 gene specifically in endothe-lial cells. These Alk2/Tie2-Cre mice demonstrate severe cardiac defects in structures derived from endocardial cushions.
Our in vivo lineage tracing experiments demonstrated that in Alk2/Tie2-Cre mutants endothelial cells fail to transdifferentiate to the mesenchyme. While at E10 the mutant cushions were noticeably smaller and less cellular than those of controls, at later time points, mutant cushions contained a surprising number of mesenchymal cells that did not stain positive for the β-galactosidase activity in a lineage tracing assay. This could indicate that, in Alk2/Tie2-Cre mutants, other cell types, e.g., epicardial cells, which have been implicated in formation of the endocar-dial cushion mesenchyme (Gittenberger-de Groot et al., 1998), can substitute for endothelial cells formation of the mesenchyme. We also considered the possibility that the Tie2-Cre transgene is transcriptionally regulated by ALK2-mediated signaling, since TGF-β superfamily members have been shown to be able to upregulate Tie2 expression in cultured endothelial cells (Mandriota and Pepper, 1998). However, this scenario is unlikely, since the lineage tracing experiments demonstrated comparable staining intensity at E9.5 between control and mutant AV-canal endothelial cells. Conversely, it is possible that there were still some endothelial cells that failed to express Cre in sufficient quantities for efficient gene recombination, and that, in Alk2 mutants, these cells could partially compensate the phenotype resulting from a total lack of endocardial cell transformation. This is supported by our findings that at E10 some isolated endothelial cells (both in controls and mutants) failed to stain positive for the β-galactosidase activity in a lineage-tracing assay. Moreover, the resulting morphologies are consistent with a depletion of size and cell number in the AV cushions. The mutant embryos recovered at E14.5 might have survived because incomplete removal of the Alk2 gene allowed some endocardial cells to undergo EMT: the resulting structures, although dysmorphic, would be otherwise normal and hence able to fuse where size and alignment allowed, extend along trabecular myocardium and become infiltrated by myocardium, all features observed among the surviving Alk2/Tie2-Cre embryos.
Previous studies have shown that mesenchymal cells in the proximal or conal OFT cushions are derived from the endothelium (Kisanuki et al., 2001), and that proximal OFT septation is mediated via the BMP type II receptor (Delot et al., 2003). Therefore, we were interested in determining whether ALK2 would also be involved in this process. While our analysis demonstrated that ALK2 function indeed is necessary for EMT in the proximal OFT, as well as in the AVC, it also pointed out that despite the lack of ALK2-mediated EMT, normal OFT septation or semilunar valve development can still occur. However, based on these experiments, we cannot exclude the possibility that defective EMT in conal cushions together with defective AV cushions may contribute to the formation of VSDs observed in Alk2/Tie2-Cre mutants. We have previously shown that mice lacking Alk2 in neural crest cells display persistent truncus arteriosus as a result of defective outflow tract septation (Kaartinen et al., 2004). Moreover, mice lacking Alk3 in the neural crest display similar, albeit not identical outflow tract defects (Stottmann et al., 2004), indicating that these two related type I receptors play distinct non-redundant functions. As the function of Alk3 in endothelial cells, if any, is currently unknown, we cannot determine whether Alk2 and Alk3 also display non-redundant functions in the endothelium during vascular or cardiac development.
Although ligands signaling via ALK2 in vivo are currently unknown, it is likely that they include BMPs, such as BMP2, -5, -6 and -7 (Macias-Silva et al., 1998). In the AV canal, a spectrum of BMPs is expressed by the myocardium, and BMP signaling via ALK3 was recently shown to upregulate mesenchymal expression of TGF-β2 (Gaussin et al., 2002). Subsequently, it was suggested that BMP2 plays also a critical role in induction of autocrine TGF-β pathways in endocardial cells (Sugi et al., 2004). However, our observations that in Alk2/Tie2-Cre mutants the defect in EMT cannot be rescued by exogenous addition of TGF-βs, and that activation of both BMP Smads and TGF-β Smads is affected in Alk2/Tie2-Cre mutants, suggests that the role of BMP signaling in the endocardium is not solely the induction of TGF-β expression. Our current results are more consistent with a model in which BMP signaling via ALK2 is required to render endothelial cells permissive to TGF-β signaling (see a schematic model in Fig. 8). Alternatively, it can be argued that ALK2 mediates TGF-β signaling to induce EMT, as suggested by Miettinen et al. (1994) using a mouse mammary epithelial cell line model. However, this paradigm has been challenged by recent studies demonstrating that activation of TGF-β Smads is absolutely required, both in human and mouse epithelial cell lines, for TGF-β-induced EMT (Piek et al., 1999; Valcourt et al., 2005), whereas signaling leading to activation of BMP Smads (i.e., action mediated via ALK2) alone is neither necessary nor sufficient for EMT induction. A third possibility is that heteromeric TGF-β receptor complexes may be formed between different type I and type II receptors. Indeed, this type of signaling mechanism involving TGFβRII, ALK5 and ALK1 has been demonstrated to play an important regulatory role in endothelial cell proliferation (Goumans et al., 2003).
Fig. 8.

Hypothetical model for induction of EMT by ALK2-mediated signaling. BMP binds to a type II receptor, which recruits ALK2. ALK2 becomes phosphorylated, which leads to subsequent phosphorylation of BMP Smads (1/5/8). Expression of genes encoding the putative downstream effectors, Msx1 and Snail, is induced, and endothelial cells become competent to respond to TGF-βs. This in turn leads to activation of TGF-β Smads, which is prerequisite for EMT.
In summary, the role of TGF-βs and BMPs in endocardial cushion formation has been extensively studied in chick, using in vitro techniques. In contrast, much less is known about signaling mechanisms of these growth factors during mammalian development in vivo. Our present results demonstrate that signaling via the type-I receptor ALK2 is required for appropriate activation of BMP Smads and induction of EMT during formation of mouse endocardiac cushions in vivo, and for subsequent morphogenetic steps that lead to development and maturation of AV-valve leaflets and septa. Our present results suggest that Alk2-mediated signaling maybe part of a critical pathway involved in pathogenesis of AV septal and valve malformations, which are among the most common congenital cardiac birth defects in humans.
Acknowledgments
We thank Saverio Bellusci, and Anil Bhushan for probes, and Ivan Vesely for support during the study. This work was supported by grants from the NIH (HL074862 and DE013085) to VK and by the AHA and NIH to JE.
References
- Anderson RH, Webb S, Brown NA, Lamers W, Moorman A. Development of the heart: (2) Septation of the atriums and ventricles. Heart. 2003;89:949–958. doi: 10.1136/heart.89.8.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, Speer CP, Poelmann RE, Gittenberger-de Groot AC. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation. 2001;103:2745–2752. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia DH. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Bei M, Maas R. FGFs and BMP4 induce both Msx1-independent and Msx1-dependent signaling pathways in early tooth development. Development. 1998;125:4325–4333. doi: 10.1242/dev.125.21.4325. [DOI] [PubMed] [Google Scholar]
- Bernanke DH, Markwald RR. Effects of two glycosaminoglycans on seeding of cardiac cushion tissue cells into a collagen-lattice culture system. Anat Rec. 1984;210:25–31. doi: 10.1002/ar.1092100105. [DOI] [PubMed] [Google Scholar]
- Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell. 2001;12:1457–1466. doi: 10.1091/mbc.12.5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA, Klewer SE. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol. 2002a;248:170–181. doi: 10.1006/dbio.2002.0731. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Schroeder JA, Bradley J, Klewer SE, McDonald JA. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2–ErbB3 receptors. Nat Med. 2002b;8:850–855. doi: 10.1038/nm742. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P, Jr, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR. A field of myocardial–endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- de Lange FJ, Moorman AF, Anderson RH, Manner J, Soufan AT, Gier-de Vries C, Schneider MD, Webb S, van den Hoff MJ, Christoffels VM. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- Delot EC, Bahamonde ME, Zhao M, Lyons KM. BMP signaling is required for septation of the outflow tract of the mammalian heart. Development. 2003;130:209–220. doi: 10.1242/dev.00181. [DOI] [PubMed] [Google Scholar]
- Derynck R, Feng XH. TGF-beta receptor signaling. Biochim Biophys Acta. 1997;1333:F105–F150. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- Desgrosellier JS, Mundell NA, McDonnell MA, Moses HL, Barnett JV. Activin receptor-like kinase 2 and Smad6 regulate epithelial–mesenchymal transformation during cardiac valve formation. Dev Biol. 2005;280:201–210. doi: 10.1016/j.ydbio.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Donovan J, Kordylewska A, Jan YN, Utset MF. Tetralogy of fallot and other congenital heart defects in Hey2 mutant mice. Curr Biol. 2002;12:1605–1610. doi: 10.1016/s0960-9822(02)01149-1. [DOI] [PubMed] [Google Scholar]
- Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech Dev. 2004;121:173–182. doi: 10.1016/j.mod.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW, Hogan BL. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 1997;124:2203–2212. doi: 10.1242/dev.124.11.2203. [DOI] [PubMed] [Google Scholar]
- Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone MA, Jr, Falb D, Huszar D. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. 2000;24:171–174. doi: 10.1038/72835. [DOI] [PubMed] [Google Scholar]
- Gaussin V, Van de PT, Mishina Y, Hanks MC, Zwijsen A, Huylebroeck D, Behringer RR, Schneider MD. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci U S A. 2002;99:2878–2883. doi: 10.1073/pnas.042390499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Lu MM, Jiang YQ, Epstein JA, Gruber PJ. Molecular markers of cardiac endocardial cushion development. Dev Dyn. 2003a;228:643–650. doi: 10.1002/dvdy.10418. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, Parada LF, Epstein JA. Nf1 has an essential role in endothelial cells. Nat Genet. 2003b;33:75–79. doi: 10.1038/ng1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Vrancken Peeters MP, Mentink MM, Gourdie RG, Poelmann RE. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ Res. 1998;82:1043–1052. doi: 10.1161/01.res.82.10.1043. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- Gu Z, Reynolds EM, Song J, Lei H, Feijen A, Yu L, He W, MacLaughlin DT, van den Eijnden-van Raaij, Donahoe PK, Li E. The type I serine/threonine kinase receptor ActRIA (ALK2) is required for gastrulation of the mouse embryo. Development. 1999;126:2551–2561. doi: 10.1242/dev.126.11.2551. [DOI] [PubMed] [Google Scholar]
- Harlow, E., Lane, D., 1988. Antibodies. A Laboratory Manual. CSH Press, New York.
- Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- Hogan, B., Beddington, R., Costantini, F., Lacy, E., 1994. Manipulating the Mouse Embryo. A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York.
- Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, Cuppen E, Zivkovic D, Plasterk RH, Clevers H. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003;425:633–637. doi: 10.1038/nature02028. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Nagy A. Removal of the floxed neo gene from a conditional knockout allele by the adenoviral Cre recombinase in vivo. Genesis. 2001;31:126–129. doi: 10.1002/gene.10015. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial–mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YT, Beason KB, Brames GP, Desgrosellier JS, Cleggett MC, Shaw MV, Brown CB, Barnett JV. Activin receptor-like kinase 2 can mediate atrioventricular cushion transformation. Dev Biol. 2000;222:1–11. doi: 10.1006/dbio.2000.9698. [DOI] [PubMed] [Google Scholar]
- Lakkis MM, Epstein JA. Neurofibromin modulation of ras activity is required for normal endocardial–mesenchymal transformation in the developing heart. Development. 1998;125:4359–4367. doi: 10.1242/dev.125.22.4359. [DOI] [PubMed] [Google Scholar]
- Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, Dejana E. Beta-catenin is required for endothelial–mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. 2004;166:359–367. doi: 10.1083/jcb.200403050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias-Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. J Biol Chem. 1998;273:25628–25636. doi: 10.1074/jbc.273.40.25628. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Pepper MS. Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ Res. 1998;83:852–859. doi: 10.1161/01.res.83.8.852. [DOI] [PubMed] [Google Scholar]
- Markwald R, Eisenberg C, Eisenberg L, Trusk T, Sugi Y. Epithelial–mesenchymal transformations in early avian heart development. Acta Anat (Basel) 1996;156:173–186. doi: 10.1159/000147845. [DOI] [PubMed] [Google Scholar]
- Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995;9:3027–3037. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Crombie R, Bradley A, Behringer RR. Multiple roles for activin-like kinase-2 signaling during mouse embryogenesis. Dev Biol. 1999;213:314–326. doi: 10.1006/dbio.1999.9378. [DOI] [PubMed] [Google Scholar]
- Moorman AF, Houweling AC, de Boer PA, Christoffels VM. Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J Histochem Cytochem. 2001;49:1–8. doi: 10.1177/002215540104900101. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Yamagishi T, Hokari S, Nakamura H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP) Anat Rec. 2000;258:119–127. doi: 10.1002/(SICI)1097-0185(20000201)258:2<119::AID-AR1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Piek E, Westermark U, Kastemar M, Heldin CH, van Zoelen EJ, Nister M, ten Dijke P. Expression of transforming-growth-factor (TGF)-beta receptors and Smad proteins in glioblastoma cell lines with distinct responses to TGF- beta1. Int J Cancer. 1999;80:756–763. doi: 10.1002/(sici)1097-0215(19990301)80:5<756::aid-ijc21>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Potts JD, Runyan RB. Epithelial–mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor beta. Dev Biol. 1989;134:392–401. doi: 10.1016/0012-1606(89)90111-5. [DOI] [PubMed] [Google Scholar]
- Potts JD, Dagle JM, Walder JA, Weeks DL, Runyan RB. Epithelial–mesenchymal transformation of embryonic cardiac endothelial cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth factor beta 3. Proc Natl Acad Sci U S A. 1991;88:1516–1520. doi: 10.1073/pnas.88.4.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- Runyan RB, Potts JD, Weeks DL. TGF-beta 3-mediated tissue interaction during embryonic heart development. Mol Reprod Dev. 1992;32:152–159. doi: 10.1002/mrd.1080320211. [DOI] [PubMed] [Google Scholar]
- Sakata Y, Kamei CN, Nakagami H, Bronson R, Liao JK, Chin MT. Ventricular septal defect and cardiomyopathy in mice lacking the transcription factor CHF1/Hey2. Proc Natl Acad Sci U S A. 2002;99:16197–16202. doi: 10.1073/pnas.252648999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non- overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial–mesenchymal transition. J Cell Biol. 1997;137:1403–1419. doi: 10.1083/jcb.137.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JA, Jackson LF, Lee DC, Camenisch TD. Form and function of developing heart valves: coordination by extracellular matrix and growth factor signaling. J Mol Med. 2003;81:392–403. doi: 10.1007/s00109-003-0456-5. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Doetschman T. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solloway MJ, Robertson EJ. Early embryonic lethality in Bmp5;Bmp7 double mutant mice suggests functional redundancy within the 60A subgroup. Development. 1999;126:1753–1768. doi: 10.1242/dev.126.8.1753. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srivastava D. Genetic assembly of the heart: implications for congenital heart disease. Annu Rev Physiol. 2001;63:451–469. doi: 10.1146/annurev.physiol.63.1.451. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–2218. doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugi Y, Markwald RR. Formation and early morphogenesis of endocardial endothelial precursor cells and the role of endoderm. Dev Biol. 1996;175:66–83. doi: 10.1006/dbio.1996.0096. [DOI] [PubMed] [Google Scholar]
- Sugi Y, Yamamura H, Okagawa H, Markwald RR. Bone morphogenetic protein-2 can mediate myocardial regulation of atrio-ventricular cushion mesenchymal cell formation in mice. Dev Biol. 2004;269:505–518. doi: 10.1016/j.ydbio.2004.01.045. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Ueno N, Hemmati-Brivanlou A. Xenopus msx1 mediates epidermal induction and neural inhibition by BMP4. Development. 1997;124:3037–3044. doi: 10.1242/dev.124.16.3037. [DOI] [PubMed] [Google Scholar]
- Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL. Notch promotes epithelial–mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. TGF-{beta} and the Smad signaling pathway support transcriptomic reprogramming during epithelial–mesenchymal cell transition. Mol Biol Cell. 2005;16:1987–2002. doi: 10.1091/mbc.E04-08-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- Yamamura H, Zhang M, Markwald RR, Mjaatvedt CH. A heart segmental defect in the anterior–posterior axis of a transgenic mutant mouse. Dev Biol. 1997;186:58–72. doi: 10.1006/dbio.1997.8559. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bradley A. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development. 1996;122:2977–2986. doi: 10.1242/dev.122.10.2977. [DOI] [PubMed] [Google Scholar]