

Abstract
Abnormalities in lipid metabolism have been proposed as contributing factors to both defective insulin secretion from the pancreatic beta cell and peripheral insulin resistance in type 2 diabetes. Previously, we have shown that prolonged exposure of isolated rat islets of Langerhans to excessive fatty acid levels impairs insulin gene transcription. This study was designed to assess whether palmitate alters the expression and binding activity of the key regulatory factors pancreas-duodenum homeobox-1 (PDX-1), MafA, and Beta2, which respectively bind to the A3, C1, and E1 elements in the proximal region of the insulin promoter. Nuclear extracts of isolated rat islets cultured with 0.5 mM palmitate exhibited reduced binding activity to the A3 and C1 elements, but not the E1 element. Palmitate did not affect the overall expression of PDX-1, but reduced its nuclear localization. In contrast, palmitate blocked the stimulation of MafA mRNA and protein expression by glucose. Combined, adenovirus-mediated, over-expression of PDX-1 and MafA in islets completely prevented the inhibition of insulin gene expression by palmitate. These results demonstrate that prolonged exposure of islets to palmitate inhibits insulin gene transcription by impairing nuclear localization of PDX-1 and cellular expression of MafA.
The prevalence of diabetes mellitus is increasing dramatically in Western countries, in part due to the increase in obesity. Type 2 diabetes mellitus, the most frequent form of the disease, is characterized by defective insulin secretion from the pancreatic beta cells and peripheral insulin resistance. According to the lipotoxicity hypothesis, abnormalities in lipid metabolism contribute to both defects (1), and in particular to the inexorable decline of beta-cell function observed during the course of the disease (2). However, the mechanisms of lipotoxicity in the beta cell remain largely unknown.
In vitro, prolonged exposure to excessive concentrations of fatty acids inhibits glucose-stimulated insulin secretion (3–7) and insulin gene expression (8–11). Previous studies in our laboratory have shown that deleterious effects of fatty acids appear mediated by distinct mechanisms: whereas inhibition of insulin secretion is observed after culture with palmitate, oleate, and other fatty-acids (7), insulin gene expression is only affected by palmitate and is mediated via de novo synthesis of ceramide (11). In isolated rat islets, we have shown that palmitate markedly blunts the activation by glucose of an insulin promoter reporter construct, indicating a transcriptional mode of action (11). However, the mechanisms by which palmitate affects the insulin promoter are unknown.
Both beta-cell specific expression and metabolic regulation of the insulin gene are conferred by a highly conserved region lying approximately 340 base pairs (bp) upstream of the transcription initiation site that constitutes the promoter/enhancer region (12–14). The main glucose-responsive elements on the insulin promoter are the highly conserved A3 (15), C1 (16), and E1 (16) sites, which respectively bind the homeodomain protein pancreas-duodenum homeobox-1 (PDX-1) (17), the basic region leucine zipper MafA (18–20), and a heterodimeric complex of basic helix-loop-helix (bHLH) proteins consisting of the ubiquitous class A (E12/E47 and E2/5) and beta-cell restricted class B (Beta2/NeuroD) proteins (21).
The aims of this study were 1) to examine whether prolonged exposure of isolated rat islets to palmitate alters the binding activity of MafA, PDX-1, and Beta2 to their cognate sequences on the insulin promoter and, if so; 2) to ascertain whether this effect is due to reduced expression or post-translational modifications of the transcription factors; and 3) whether over-expression of the proteins prevents palmitate inhibition of insulin gene expression.
EXPERIMENTAL PROCEDURES
Reagents
RPMI-1640 was obtained from Invitrogen (Carlsbad, CA). [α-32P]UTP and [α-32P]dCTP were from Perkin-Elmer Lifesciences (Boston, MA). Fatty-acid-free bovine serum albumin (BSA) was from Serologicals Corporation (Norcross, GA). Palmitic acid (sodium salt) and all other reagents (analytical grade) were from Sigma.
Animals
Normal 225–275 g male Wistar rats were purchased from Charles River Laboratories (Wilmington, MA). Animals were housed on a 12-h light/dark cycle with free access to standard laboratory chow and water. All procedures using animals were approved by the Pacific Northwest Research Institute Animal Care and Use Committee.
Rat Islet Isolation and Culture
Rat islets were isolated by collagenase digestion and dextran density gradient centrifugation as described (11), then allowed to recover overnight in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, and 11.1 mM glucose. Experimental culture conditions for each experiment were as described under “Results.” The preparation of culture media containing palmitate was as described (11), with a final molar ratio of palmitate/BSA of 5:1. All control conditions contained the same amount of BSA and vehicle (EtOH/H2O, 1:1, v/v) as those with palmitate.
Extraction of Total, Cytosolic, and Nuclear Proteins
Total cellular proteins were extracted from islet lysates as described (22). To separate nuclear from cytosolic proteins, batches of 350–500 islets were washed in phosphate buffered saline (PBS), resuspended in 400 μl of cold hypotonic buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 0.1 mM EDTA, 0.1 mM EGTA, 0.5 mM phenylmethanesulfonyl fluoride [PMSF], 0.5 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μg/ml pepstatin), and allowed to swell on ice for 15 min before adding 25 μl of 10% (w/v) Nonidet P-40. After vortexing vigorously, the nuclei were pelleted by centrifugation (14,000 rpm; 1 min; 4°C) and resuspended in 50 μl of high-salt buffer (20 mM HEPES [pH 7.9], 0.4 M NaCl, 0.2 mM DTT, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM PMSF, 0.5 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 μg/ml pepstatin). All protein isolates were stored at −80°C. Protein content was quantified using the BCA Kit (Pierce, Rockford, IL).
Immunoblotting
Proteins (5–30 μg) were separated by 10% SDS-PAGE and transferred to nitrocellulose. Membranes were blocked in 5% non-fat milk in TBS-T [10 mM Tris (pH 7.5), 150 mM NaCl, and 0.1% Tween 20] for 1 h at room temperature and probed with primary antibodies against MafA (1:1000; Bethyl Laboratories, Montgomery, TX), Beta2 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA), PDX-1 (1:5000; kindly provided by C. Wright, Vanderbilt University, Nashville, TN) or TFIID (1:200; Santa Cruz Biotechnology) overnight at 4°C. Detection was performed using a horse radish peroxidase (HRP)-labeled anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) and enhanced chemiluminescence (Perkin-Elmer Lifesciences) on Kodak X-Mat film. Bands were quantified using Optiquant System software (Packard Instrument Co.).
Immunohistochemistry and Confocal Microscopy
Cultured islets were washed with 1X PBS, then fixed with 4% formaldehyde for 30 min on ice, then washed twice with 1X PBS and embedded in 80 μl of liquefied HistoGel (Richard-Allan Scientific, Kalamazoo, MI). After hardening, the islet-HistoGel pellet was further fixed in 4% formaldehyde for 3 h on ice. Following standard ethanol dehydration and paraffin embedding, 0.4 μm sections were mounted on slides and rehydrated. Antigen retrieval was performed using 10 mM sodium citrate, heated to 90ºC for 9 min then allowed to cool to room temperature. The sections were washed with PBS twice, blocked with 5% donkey serum in PBS+1% BSA for 1 h at room temperature, then incubated overnight at 4ºC with an anti-PDX-1 antibody diluted 1:10,000 in PBS+1% BSA. An anti-rabbit secondary antibody conjugated to Cy5 (Jackson Immuno Research Laboratories) was used at 1:1000 in PBS+1% BSA for 1 h at room temperature for detection. Nuclei of the sectioned islets were detected by the addition of 1 μg/ml propidium iodide just prior to mounting with Aqua Poly/Mount (Polysciences, Warrington, PA). Images were acquired using a Fluoview 500 Olympus confocal microscope.
Electrophoretic Mobility Shift Assay (EMSA)
Double-stranded oligodeoxynucleotide probes against human insulin A3 (5’-CCC CTG GTT AAG ACT CTA ATG ACC CG-3’), Rat2 C1 (5’-AGC TTG GAA ACT GCA GCT TCA GCC CCT CTG-3’), and Rat2 E1 (5’-TCT GGC CAT CTG CTG ATC CA-3’), elements were labeled by end filling with the DNA Polymerase Large (Klenow) Fragment Kit (Promega) and [α-32P]dCTP (Perkin Elmer). In addition, we used an oligodeoxynucleotide probe (Zd: 5’-TTT GCT CTC CTG GAG ACA TTT GCC CCC AGC TGT GAG C-3’) to the Z mini-enhancer of the human insulin promoter (23). Nuclear extracts (5–10 μg) were incubated with 30,000 cpm of labeled probe, with or without cold competitors, in a final volume adjusted to 30 μl with binding buffer (15 mM HEPES [pH 7.5], 60 mM KCl, 5 mM MgCl2, 2 mM EDTA, 12% glycerol, 3.3 mM dithiothreitol, and 100 ng poly[dI-dC]) at room temperature for 30 min. Binding reactions were resolved on 4.5% acrylamide gels run in 0.5% TBE (44.5 mM Tris, 44.5 mM boric acid, 1 mM EDTA) for 2 h at 4°C and visualized by autoradiography. The identity of the protein in the binding complexes was determined by super-shift using antibodies against PDX-1 or MafA. The bands were quantified using Optiquant System software (Packard Instrument Co.).
RNA Isolation and Real-Time Fluorescence-Based Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from aliquots of 100 islets as described (24). PCR primers and probes for the rat MafA, PDX-1 and beta-actin genes were designed using the Primer Express software program (Applied Biosystems): MafA (5’-3’); forward primer, CTT CAG CAA GGA GGA GGT CAT C; reverse primer, GCG TAG CCG CGG TTC TT; probe, 6FAM-CTG AAA CAG AAG CGG CGC ACG C-TAMRA; PDX-1 (5’-3’); forward primer, CCG CGT TCA TCT CCC TTT C; reverse primer, TGC CCA CTG GC-TTTTCCA; probe, 6FAM-TGG ATG AAA TCC ACC AAA GCT CAC GC-TAMRA; and beta-actin (5’-3’); forward primer, ACG AGG CCC AGA GCA AGA; reverse primer, TTG GTT ACA ATG CCG TGT TCA; probe, 6FAM-TGG GTC CTC CAC TTC ACG GCG-TAMRA. Onestep RT-PCR was carried out using the Gold RT-PCR kit (Perkin Elmer) and an ABI Prism 7700 Sequence detector as previously described (25). Results are expressed as a ratio of target gene to beta-actin.
Ribonuclease Protection Assay (RPA)
RPAs were carried out using the Direct Protect Lysate RPA kit (Ambion, Austin, TX) and [α-32P]UTP labeled probes generated from a 360-bp sequence of the rat II preproinsulin gene and a conserved 245-bp sequence of the mouse beta-actin gene. Protected fragments were resolved on 5% denaturing gels as described (11). The probes were determined to be in excess for each experiment. The bands were quantified using Optiquant System software (Packard Instrument Co.).
Generation of Recombinant Adenoviruses and Islet Transduction
Recombinant adenoviruses (Ad) encoding PDX-1 and MafA under the cytomegalovirus (CMV) promoter were generated by the Pacific Northwest Research Institute Adenoviral Core Facility, as described (11), using a mouse PDX-1 cDNA provided by M. Montminy (Salk Institute for Biological Studies, La Jolla, CA) and a mouse MafA cDNA provided by R. Stein (Vanderbilt University). A recombinant Ad expressing firefly luciferase under the control of the CMV promoter was used as a control. The appropriate titer for each recombinant Ad was assessed in preliminary experiments by infecting 100 islets with increasing viral concentrations (from 105–1010 plaque-forming units (PFU)/islet) overnight. The islets were then washed in PBS prior to initiating experimental conditions.
Statistics
Data are expressed as mean ± S.E. Intergroup comparisons were performed by Student’s paired t test or analysis of variance with post-hoc Dunnett t test, where appropriate. p<0.05 was considered significant.
RESULTS
Palmitate inhibits MafA and PDX-1 binding to their cognate sequences on the insulin promoter in isolated islets
Isolated rat islets were cultured for 24, 48, or 72 h with 2.8 or 16.7 mM glucose in the absence or presence of 0.5 mM palmitate. Binding of islet nuclear extracts to the A3, C1, E1 and Zd probes was assessed by EMSA. Figure 1A shows a representative gel after a 72-h culture. Discrete complexes were observed with each probe, and disappeared in excess of unlabeled probes.
Fig. 1.
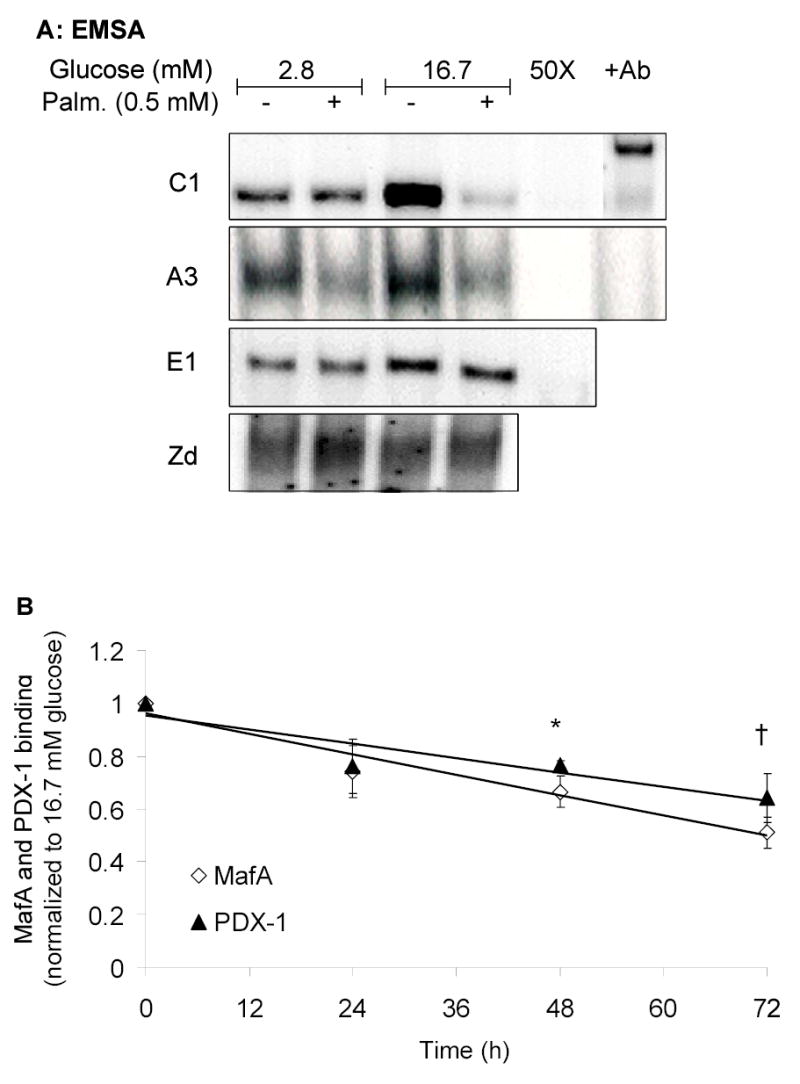
Palmitate inhibits MafA and PDX-1 binding to the insulin promoter. A, Representative EMSA using nuclear extracts of islets cultured for 72 h at 2.8 or 16.7 mM glucose in the presence or absence of 0.5 mM palmitate. Binding complexes were displaced by 50-fold excess (50X) of unlabeled probe and supershifted by specific antibodies: (+Ab) to MafA (C1 site) and PDX-1 (A3 site). B, Average results obtained after quantification of the binding complexes. Each time point represents the mean ± S.E. of 3–5 separate experiments and is normalized to the control values at 16.7 mM glucose. *, p<0.05; †, p<0.001 (ANOVA by time point).
The presence of MafA and PDX-1, respectively, in the complexes formed with the C1 and A3 probes was confirmed by supershifting the bands with specific antibodies. The presence of 16.7 mM glucose visibly enhanced the intensity of the binding complex to the C1 probe and, to a lesser extent, to the A3 and E1 probes. The addition of 0.5 mM palmitate markedly reduced binding to the C1 and A3 elements in the presence of 16.7 mM glucose (Fig. 1A). Binding to the E1 and Zd sites was not affected by palmitate. The loss of binding to the A3 and C1 elements in the presence of 0.5 mM palmitate at elevated glucose (16.7 mM), was detectable after 24 h of culture and statistically significant after 48 h (p<0.05) and 72 h (p<0.001) (Fig. 1B). These results demonstrate that palmitate specifically impairs the binding activity of MafA and PDX-1 to their cognate sequences on the insulin promoter in the presence of high glucose levels in beta cells, consistent with our previous observation that palmitate blunts glucose-activation of an insulin promoter-reporter (11).
Palmitate alters PDX-1 nuclear localization and MafA gene expression
To determine whether the loss of binding activity observed in Figure 1 was due to diminished protein expression or altered localization, expression of PDX-1, MafA, and Beta2 in whole-cell, nuclear, or cytosolic protein extracts of rat islets cultured for 24 h in 2.8 or 16.7 mM glucose in the absence or presence of 0.5 mM palmitate was assessed by immunoblotting (Fig. 2A).
Fig. 2.


Palmitate affects PDX-1 localization and MafA expression. Islets were cultured for 24 h at 2.8 or 16.7 mM glucose in the presence or absence of 0.5 mM palmitate. A, Representative immunoblots with total (lanes 1–4), cytosolic (lanes 5–6), or nuclear (lanes 7–8) protein fractions. TFIID was used as a control for loading variations. B, PDX-1 localization (green) was visualized by immunostaining and laser-scanning confocal microscopy in islets cultured in 16.7 mM glucose in the absence (“control”) or presence of palmitate (0.5 mM). Propidium iodide (red) was used for nuclear staining. C, Quantification of PDX-1 expression in total, cytosolic and nuclear extracts after 24 h of culture at 16.7 mM glucose in the presence or absence of 0.5 mM palmitate. Results are mean ± S.E. of 6-8 separate experiments. *, p<0.05. D, Quantification of MafA expression in nuclear and total-cell lysates after 24h of culture at 16.7 mM glucose in the presence or absence of 0.5 mM palmitate. Results are presented as mean ± S.E. of 5–7 different experiments. *, p<0.001.
Expression of PDX-1 in total-cell lysates was not significantly affected by either glucose or palmitate (Fig. 2A). At 16.7 mM glucose, PDX-1 was predominantly localized in the nuclear fraction as shown by Western blotting and immunohistochemistry (Fig. 2A&B). In the presence of palmitate, however, the expression of PDX-1 in the nucleus was reduced (Fig 2A–C). These results suggest that palmitate impairs the translocation of PDX-1 from the cytosol to the nucleus that normally occurs upon glucose stimulation (26,27).
At 2.8 mM glucose, MafA expression was undetectable in whole-cell lysates by immunoblotting (Fig. 2A). In contrast, at 16.7 mM glucose MafA protein was readily detected, demonstrating a strong stimulation of MafA protein expression by glucose. In the presence of palmitate, glucose-induced MafA protein expression was reduced, on average, by 44±3% (n=7; p<0.001) in total-cell lysates (Fig. 2A&D). MafA protein was undetectable in the cytosolic fraction, and its levels in the nuclear fractions were reduced by 47±7% by palmitate (n=5; p<0.01; Fig. 2A&D), in parallel with its reduced expression in whole-cell lysates. These results indicate that palmitate inhibits glucose-induced MafA protein expression. Neither Beta2 nor the control protein TFIID was affected by palmitate.
To further investigate the mechanisms by which palmitate affects protein expression, MafA and PDX-1 mRNA levels were measured in islets after 24-h culture in 2.8 or 16.7 mM glucose, with or without 0.5 mM palmitate (Fig. 3A). Glucose significantly increased MafA mRNA expression (n=7; p<0.01). In the presence of palmitate, the stimulatory effect of glucose was no longer significant (Fig. 3A). The time course of this inhibitory effect is shown in Figure 3B. In contrast, neither glucose nor palmitate had a significant effect on the expression of PDX-1 mRNA (Fig. 3A), consistent with the unaltered total protein expression observed (Fig. 2A&C). These results indicate that palmitate affects PDX-1 and MafA binding activities by two distinct mechanisms: PDX-1 is altered at the post-translational level in its nuclear localization, whereas palmitate inhibits glucose-stimulation of MafA mRNA levels.
Fig. 3.

Palmitate inhibits the expression of MafA mRNA, but not PDX-1 mRNA. A, Islets were cultured for 24 h at 2.8 or 16.7 mM glucose in the presence or absence of 0.5 mM palmitate. MafA, PDX-1 and beta-actin mRNA were measured by real-time, fluorescence-based RT-PCR. Results are expressed as fold increase of the ratio of MafA or PDX-1 /beta-actin mRNA over the control value (at 2.8 mM glucose), and are mean ± SE of 7-8 replicate experiments. *, p<0.01. B, Islets were cultured for up to 24 h at 2.8 mM glucose or 16.7 mM glucose with or without 0.5 mM palmitate and collected at the indicated time points. Results are expressed as fold increase of the ratio of MafA /beta-actin mRNA over the control value (at 2.8 mM glucose), and are mean ± SE of 4 replicate experiments. *, p<0.05.
Adenoviral co-expression of PDX-1 and MafA prevents palmitate inhibition of insulin gene expression
To further explore the roles of PDX-1 and MafA in mediating palmitate inhibition of insulin gene expression, we next sought to determine whether over-expression of these transcription factors could restore insulin mRNA expression in the presence of palmitate. First, isolated islets were infected with adenoviruses encoding for PDX-1 or MafA at a titer of 105 PFU/islet, and expression of the proteins was examined by immunoblotting after 24, 48, or 72 h. As shown in Figure 4A, protein expression was sustained for up to 72 h. Next, to determine whether the over-expressed proteins were functional, islets were infected with increasing titers of each virus, and binding activity of islet nuclear extracts to the A3 and C1 elements was examined by EMSA. As shown in Figure 4B, binding of over-expressed PDX-1 and MafA to their respective sequences exceeded endogenous levels and increased with increasing viral titers. For subsequent experiments, the lowest titer of virus (105 PFU/islet) was used. Islets infected with PDX-1, MafA, or both were then cultured for 72 h in the presence of 16.7 mM glucose with or without 0.5 mM palmitate. Insulin gene expression was measured by RPA (Fig. 4C) and quantified (Fig. 4D). As previously shown (10,11), palmitate significantly decreased insulin gene expression after 72 h in control, luciferase-infected islets (p<0.01). Over-expression of MafA alone increased insulin gene expression relative to that of the luciferase infected control islets at 16.7 mM glucose. Paradoxically, over-expression of PDX-1 reduced insulin mRNA levels. In the presence of palmitate, neither MafA nor PDX-1 alone was able to prevent the decrease in insulin gene expression (Fig. 4C). However, combined over-expression of MafA and PDX-1 completely prevented the inhibitory effect of palmitate on insulin gene expression (Fig. 4C&D), confirming the key role of these two transcription factors in the mechanisms of lipotoxicity on the insulin gene.
Fig. 4.
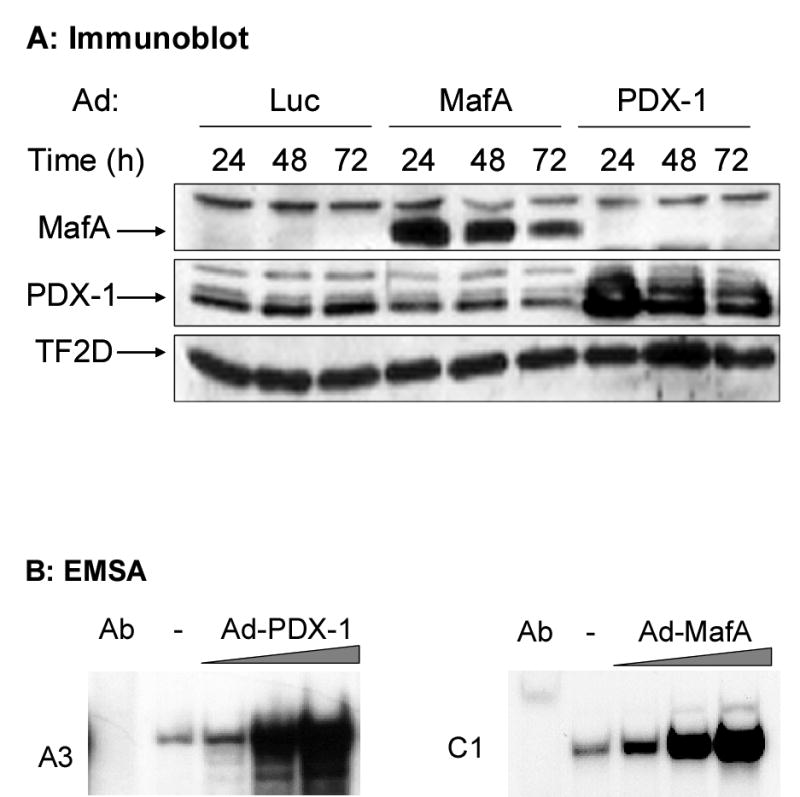

Adenoviral over-expression of PDX-1 and MafA restores insulin gene expression in islets exposed to palmitate. A, Immunoblot of islet proteins after infection with Ad expressing luciferase, MafA or PDX-1 and cultured for 24, 48 or 72 h at 16.7 mM glucose. B, Islets were infected with 105–107 PFU/islet of either Ad-MafA or Ad-PDX-1 and cultured at 16.7 mM glucose for 16 h. Binding to the C1 and A3 elements was assessed in islets infected with increasing concentrations of Ad-MafA and Ad-PDX-1, respectively. C, Islets were infected with Ad-Luc, Ad-MafA, Ad-PDX-1 or a combination of Ad-MafA and Ad-PDX-1 (at 105 PFU/islet). Preproinsulin and beta-actin mRNA levels were measured by RPA. D, Quantification of 4 replicate experiments presented as mean ± S.E of the ratio of preproinsulin/beta-actin mRNA, normalized to the control value (luciferase-infected islets cultured in 16.7 mM glucose). *, p<0.01.
DISCUSSION
The aims of this study were to determine whether prolonged exposure of isolated rat islets to palmitate alters the binding activity of MafA, PDX-1, and Beta2 to the insulin promoter, to ascertain whether this effect was due to reduced expression or post-translational modifications of the transcription factors, and to determine whether over-expression of these proteins could prevent palmitate inhibition of insulin gene expression. Our results show that exposure of rat islets to elevated levels of palmitate inhibits binding of MafA and PDX-1, but not Beta2, to the insulin promoter. Interestingly, the mechanisms of this inhibition differ in that palmitate alters the cellular localization of PDX-1 by preventing its translocation to the nucleus, whereas glucose-stimulated MafA mRNA expression is blunted, resulting in reduced overall MafA protein levels. Importantly, palmitate inhibition of insulin gene expression is prevented by combined over-expression of MafA and PDX-1.
We (9–11) and others (8,28,29) have shown previously that concurrent exposure to chronically elevated levels of glucose and palmitate inhibits insulin gene expression. More importantly, this inhibitory effect of palmitate was observed to occur at the transcriptional level (11). We now demonstrate that the mechanisms of this transcriptional inhibition involve two key transcription factors of the insulin gene, PDX-1 and MafA. Although reduced binding of PDX-1 and MafA to the A3 and C1 elements, respectively, was demonstrated indirectly by EMSA (Fig. 1), the observation that palmitate decreases nuclear localization of PDX-1 and mRNA expression of MafA indicates that the major site of action of palmitate lies upstream of the binding activities of the transcription factors.
Insulin gene expression is highly regulated by glucose, which coordinately enhances both the rate of transcription and mRNA stability (14). Several studies have shown that binding to the A3 and C1 elements is modulated by glucose (reviewed in (14)). A major site of glucose regulation of PDX-1 function is at the post-translational level (14). Although the precise nature of the post-translational modifications is still debated, glucose promotes nuclear translocation of PDX-1 (26,27,30–34) and its binding to the A3 element (27,30). We observed a glucose-induced increase in binding to the A3 element, as previously reported in adult islets (14,17,27,35), which was abolished in the presence of palmitate (Fig. 1). Also in line with prior studies (26,36,37), we observed no glucose-induced increase in total PDX-1 protein expression (Fig. 2A&C). Palmitate did not inhibit PDX-1 expression, but significantly reduced its nuclear localization (Fig. 2A–C). Interestingly, at 48 h both Gremlich et al. (28) and Yoshikawa et al. (29) observed that PDX-1 mRNA expression was substantially reduced in the presence of palmitate. In our experiments, we did not observe either a glucose induced increase, nor a palmitate mediated decrease in PDX-1 mRNA expression (Fig. 3A). Rather, the most compelling effect of palmitate after 24 h of exposure was at the level of PDX-1 nuclear localization (Fig. 2A–C). Since PDX-1 is thought to regulate its own transcription (38,39), we postulate that the early defect seen in the presence of palmitate is a sequestration in the cytosol, which, at later time points, might also result in decreased overall cellular expression of PDX-1, as observed by Gremlich et al. (28) and Yoshikawa et al. (29). Consistent with this hypothesis, Kharroubi et al. (40) failed to observe a decrease in PDX-1 expression in both INS-1 cells and rat islets after 24 h treatment with palmitate.
Binding to the C1 element was markedly increased in response to glucose (Fig. 1). MafA has only recently been identified as the activator of the C1 element (18,20), and is localized exclusively in the nucleus of pancreatic beta cells (Fig. 2A; and (20)). At low glucose concentrations, MafA protein levels are essentially undetectable (Fig. 2A; and (19)). As shown here with rat islets (Fig. 2&3) and previously in MIN6 cells (19), glucose significantly enhances mRNA and protein expression of MafA. Importantly, our results demonstrate for the first time that palmitate inhibits glucose stimulation of MafA mRNA (Fig. 3) and, as a consequence, its protein expression (Fig. 2), providing an additional mechanism whereby this fatty acid impairs insulin gene transcription.
As a trans-activator of the insulin gene, MafA alone has a much greater effect than either PDX-1 or Beta2 (19). In this study, adenoviral over-expression of MafA enhanced insulin gene expression, suggesting that MafA might be a factor limiting transcription of the insulin gene in rat islets. This is further supported by the recent observation that MafA binds to and activates elements of the PDX-1 promoter (41). In contrast, over-expression of PDX-1 actually reduced insulin mRNA levels in islets (Fig. 4C). This phenomenon has been described previously (17,42) and was suggested to result from the titration of co-activators by the over-expressed protein (17). Importantly, combined over-expression of MafA and PDX-1 completely prevented palmitate inhibition of insulin gene expression. This observation identifies MafA and PDX-1 as critical targets of lipotoxicity at the level of the insulin gene.
We previously demonstrated that palmitate inhibition of insulin gene expression requires de novo synthesis of ceramide (11). The results of the present study raise the question as to how ceramide might affect PDX-1 and MafA. Although the signaling mechanisms of PDX-1 regulation by glucose are still debated the involvement of the phosphatidylinositol-3 kinase (PI3K) pathway has been suggested (27,31–33,43). Since ceramide has been shown to interfere with this pathway, particularly at the level of protein kinase B in all cell types thus far tested (44), it is tempting to speculate that ceramide might dampen a stimulatory signal from glucose to PDX-1 via the PI3K pathway. Alternatively, ceramide may activate c-Jun NH2-terminal Kinase (JNK) in beta cells, as it does in other cells (45,46). In beta cells, JNK activation has been shown to inhibit PDX-1 binding (47) and nuclear localization (48). These hypotheses are currently under investigation in our laboratory. The mode of regulation of MafA by glucose is essentially unknown. Our results suggest that palmitate inhibition of MafA expression is at the transcriptional level. Elucidating the mechanisms of these effects awaits characterization of the promoter/enhancer region of the MafA gene.
In conclusion, this study uniquely demonstrates that the mechanisms by which palmitate impairs insulin gene transcription involve reduced binding of PDX-1 and MafA to the insulin promoter. Interestingly, both factors are affected at different levels by palmitate: PDX-1 is affected post-translationally in its nuclear localization, whereas MafA is affected at the transcriptional level. The importance of these observations for the mechanisms of lipotoxicity is illustrated by the complete prevention of the decrease in insulin gene expression in the presence of palmitate observed after combined over-expression of PDX-1 and MafA. Although the contribution of fatty-acid inhibition of insulin gene transcription to beta-cell dysfunction in human type 2 diabetes remains to be established, it has been shown in rodents that intact regulation of insulin gene expression by glucose is required to maintain adequate intracellular insulin stores in the face of increased demand (49). Identification of the precise molecular targets of fatty acids should facilitate the design of new therapeutic strategies to prevent the deterioration of beta-cell function during the course of type 2 diabetes.
Footnotes
We thank Drs. Christopher Wright (Vanderbilt University) for the PDX-1 antibody, Roland Stein (Vanderbilt University) for the MafA cDNA, and Marc Montminy (Salk Institute for Biological Studies) for the PDX-1 cDNA. We also thank Drs. Paul Robertson and Christopher Rhodes for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health (R01DK58096, V.P.), and the 2003 Thomas R. Lee Career Development Award from the American Diabetes Association (V.P.).
The abbreviations used are: bp, base pair; PDX-1, pancreas-duodenum homeobox-1; bHLH, basic helix-loop-helix; BSA, bovine serum albumin; FBS, fetal bovine serum; PBS, phosphate buffered saline; PMSF, phenylmethanesulfonyl fluoride; HRP, horse radish peroxidase; EMSA, electrophoretic mobility shift assay; RT-PCR, reverse transcriptase polymerase chain reaction; RPA, ribonuclease protection assay; Ad, adenovirus; CMV, cytomegalovirus; PFU, plaque-forming units; ANOVA, analysis of variance; PI3K, phosphatidylinositol-3 kinase; JNK, c-Jun NH2-terminal kinase.
References
- 1.Unger RH. Endocrinology. 2003;144:5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 2.Poitout V, Robertson RP. Endocrinology. 2002;143:339–342. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 3.Sako Y, Grill VE. Endocrinology. 1990;127:1580–1589. doi: 10.1210/endo-127-4-1580. [DOI] [PubMed] [Google Scholar]
- 4.Elks ML. Endocrinology. 1993;133:208–214. doi: 10.1210/endo.133.1.8319569. [DOI] [PubMed] [Google Scholar]
- 5.Zhou YP, Grill VE. J Clin Invest. 1994;93:870–876. doi: 10.1172/JCI117042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou YP, Grill V. J Clin Endocrinol Metab. 1995;80:1584–1590. doi: 10.1210/jcem.80.5.7745004. [DOI] [PubMed] [Google Scholar]
- 7.Moore PC, Ugas MA, Hagman DK, Parazzoli SD, Poitout V. Diabetes. 2004;53:2610–2616. doi: 10.2337/diabetes.53.10.2610. [DOI] [PubMed] [Google Scholar]
- 8.Ritz-Laser B, Meda P, Constant I, Klages N, Charollais A, Morales A, Magnan C, Ktorza A, Philippe J. Endocrinology. 1999;140:4005–4014. doi: 10.1210/endo.140.9.6953. [DOI] [PubMed] [Google Scholar]
- 9.Jacqueminet S, Briaud I, Rouault C, Reach G, Poitout V. Metabolism. 2000;49:532–536. doi: 10.1016/s0026-0495(00)80021-9. [DOI] [PubMed] [Google Scholar]
- 10.Briaud I, Harmon JS, Kelpe CL, Segu VB, Poitout V. Diabetes. 2001;50:315–321. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. J Biol Chem. 2003;278:30015–30021. doi: 10.1074/jbc.M302548200. [DOI] [PubMed] [Google Scholar]
- 12.Stein, R. (2001) in Handbook of Physiology. Section 7: The Endocrine System (Cherrington, A., and Jefferson, J., eds) Vol. II, pp. 25–78, American Physiology Society, Washington, DC
- 13.Ohneda K, Ee H, German M. Semin Cell Dev Biol. 2000;11:227–233. doi: 10.1006/scdb.2000.0171. [DOI] [PubMed] [Google Scholar]
- 14.Melloul D, Marshak S, Cerasi E. Diabetologia. 2002;45:309–326. doi: 10.1007/s00125-001-0728-y. [DOI] [PubMed] [Google Scholar]
- 15.Melloul D, Ben-Neriah Y, Cerasi E. Proc Natl Acad Sci USA. 1993;90:3865–3869. doi: 10.1073/pnas.90.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma A, Fusco-DeMane D, Henderson E, Efrat S, Stein R. Molecular Endo. 1995;9:1468–1476. doi: 10.1210/mend.9.11.8584024. [DOI] [PubMed] [Google Scholar]
- 17.Marshak S, Totary H, Cerasi E, Melloul D. Proc Natl Acad Sci USA. 1996;93:15057–15062. doi: 10.1073/pnas.93.26.15057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olbrot M, Rud J, Moss LG, Sharma A. Proc Natl Acad Sci U S A. 2002;99:6737–6742. doi: 10.1073/pnas.102168499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. J Biol Chem. 2002;277:49903–49910. doi: 10.1074/jbc.M206796200. [DOI] [PubMed] [Google Scholar]
- 20.Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Mol Cell Biol. 2003;23:6049–6062. doi: 10.1128/MCB.23.17.6049-6062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shieh SY, Tsai MJ. J Biol Chem. 1991;266:16708–16714. [PubMed] [Google Scholar]
- 22.Wrede, C. E., Dickson, L. M., Lingohr, M. K., Briaud, I., and Rhodes, C. J. (2002) J Biol Chem
- 23.Sander M, Griffen SC, Huang J, German MS. Proc Natl Acad Sci U S A. 1998;95:11572–11577. doi: 10.1073/pnas.95.20.11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 25.Tran PO, Gleason CE, Robertson RP. Diabetes. 2002;51:1772–1778. doi: 10.2337/diabetes.51.6.1772. [DOI] [PubMed] [Google Scholar]
- 26.Rafiq I, Kennedy HJ, Rutter GA. J Biol Chem. 1998;273:23241–23247. doi: 10.1074/jbc.273.36.23241. [DOI] [PubMed] [Google Scholar]
- 27.Macfarlane WM, McKinnon CM, Felton-Edkins ZA, Cragg H, James RF, Docherty K. J Biol Chem. 1999;274:1011–1016. doi: 10.1074/jbc.274.2.1011. [DOI] [PubMed] [Google Scholar]
- 28.Gremlich S, Bonny C, Waeber G, Thorens B. J Biol Chem. 1997;272:30261–30269. doi: 10.1074/jbc.272.48.30261. [DOI] [PubMed] [Google Scholar]
- 29.Yoshikawa H, Tajiri Y, Sako Y, Hashimoto T, Umeda F, Nawata H. Metabolism. 2001;50:613–618. doi: 10.1053/meta.2001.22565. [DOI] [PubMed] [Google Scholar]
- 30.Petersen HV, Peshavaria M, Pedersen AA, Philippe J, Stein R, Madsen OD, Serup P. FEBS Lett. 1998;431:362–366. doi: 10.1016/s0014-5793(98)00776-5. [DOI] [PubMed] [Google Scholar]
- 31.Rafiq I, da Silva Xavier G, Hooper S, Rutter GA. J Biol Chem. 2000;275:15977–15984. doi: 10.1074/jbc.275.21.15977. [DOI] [PubMed] [Google Scholar]
- 32.Macfarlane W, Shepherd R, Cosgrove K, James R, Dunne M, Docherty K. Diabetes. 2000;49:418–423. doi: 10.2337/diabetes.49.3.418. [DOI] [PubMed] [Google Scholar]
- 33.Elrick LJ, Docherty K. Diabetes. 2001;50:2244–2252. doi: 10.2337/diabetes.50.10.2244. [DOI] [PubMed] [Google Scholar]
- 34.Campbell SC, Macfarlane WM. Biochem Biophys Res Commun. 2002;299:277–284. doi: 10.1016/s0006-291x(02)02633-5. [DOI] [PubMed] [Google Scholar]
- 35.Wu H, MacFarlane WM, Tadayyon M, Arch JR, James RF, Docherty K. Biochem J 344 Pt. 1999;3:813–818. [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Cahill CM, Pineyro MA, Zhou J, Doyle ME, Egan JM. Endocrinology. 1999;140:4904–4907. doi: 10.1210/endo.140.10.7158. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Zhou J, Doyle ME, Egan JM. Endocrinology. 2001;142:1820–1827. doi: 10.1210/endo.142.5.8128. [DOI] [PubMed] [Google Scholar]
- 38.Marshak S, Benshushan E, Shoshkes M, Havin L, Cerasi E, Melloul D. Mol Cell Biol. 2000;20:7583–7590. doi: 10.1128/mcb.20.20.7583-7590.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerrish K, Van Velkinburgh JC, Stein R. Mol Endocrinol. 2004;18:533–548. doi: 10.1210/me.2003-0371. [DOI] [PubMed] [Google Scholar]
- 40.Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Endocrinology. 2004;145:5087–5096. doi: 10.1210/en.2004-0478. [DOI] [PubMed] [Google Scholar]
- 41.Samaras SE, Zhao L, Means A, Henderson E, Matsuoka TA, Stein R. J Biol Chem. 2003;278:12263–12270. doi: 10.1074/jbc.M210801200. [DOI] [PubMed] [Google Scholar]
- 42.Seijffers R, Ben-David O, Cohen Y, Karasik A, Berezin M, Newgard CB, Ferber S. Endocrinology. 1999;140:3311–3317. doi: 10.1210/endo.140.7.6796. [DOI] [PubMed] [Google Scholar]
- 43.Macfarlane WM, Smith SB, James RFL, Clifton AD, Doza YN, Cohen P, Docherty K. J Biol Chem. 1997;272:20936–20944. doi: 10.1074/jbc.272.33.20936. [DOI] [PubMed] [Google Scholar]
- 44.Summers SA, Nelson DH. Diabetes. 2005;54:591–602. doi: 10.2337/diabetes.54.3.591. [DOI] [PubMed] [Google Scholar]
- 45.Mathias S, Pena LA, Kolesnick RN. Biochem J. 1998;335 ( Pt 3):465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruvolo PP. Pharmacol Res. 2003;47:383–392. doi: 10.1016/s1043-6618(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 47.Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC. J Biol Chem. 2002;277:30010–30018. doi: 10.1074/jbc.M202066200. [DOI] [PubMed] [Google Scholar]
- 48.Kawamori D, Kajimoto Y, Kaneto H, Umayahara Y, Fujitani Y, Miyatsuka T, Watada H, Leibiger IB, Yamasaki Y, Hori M. Diabetes. 2003;52:2896–2904. doi: 10.2337/diabetes.52.12.2896. [DOI] [PubMed] [Google Scholar]
- 49.Leibowitz G, Uckaya G, Oprescu AI, Cerasi E, Gross DJ, Kaiser N. Endocrinology. 2002;143:3214–3220. doi: 10.1210/en.2002-220174. [DOI] [PubMed] [Google Scholar]