

Abstract
It has been suggested in many studies that combined treatment with chemotherapeutic agents and apoptosis-inducing ligands belonging to TNFR family is a more effective strategy for cancer treatment. However, the role of androgen regulation of TNFR family-induced apoptosis in prostate cancer is poorly understood. In this study, we investigated the dose-dependent effects of androgen on TNF-α and TRAIL-mediated apoptosis in LNCaP. To investigate the interaction between the androgen receptor (AR) and the caspase-2 gene, chromatin immunoprecipitation analysis was used, and we are the first to identify that AR interacts in vivo with an androgen-responsive elements in intron 8 of caspase-2 gene. We have found that DHT inhibited apoptosis in dose-dependent manner. There is a direct, androgen-dependent correlation between the levels of activated Akt and caspase activation after treatment with TNF-α and TRAIL. We have also found that there are at least two different regulatory mechanisms of p53 expression by androgen: at the gene and protein levels. At the same time, the level of AR was found to be higher in LNCaP-si-p53 compared to LNCaP-mock cells. These data indicate that there is a mutual regulation of expression between p53 and AR. Our study suggests that androgen-dependent outcome of apoptotic treatment can occur, at least in part, via the caspase-2, Akt and p53-mediated pathways.
Keywords: androgen, androgen receptor, Akt, p53, caspase-2, apoptosis
Introduction
Many different factors contribute to the development of prostate cancer including somatic mutations of the androgen receptor (AR) or AR amplification (Nelson et al., 2003). Some mutations in the AR result in altered ligand binding specificity permitting activation by nonandrogenic steroid hormones or even by antiandrogens. AR amplification with concomitant overexpression of AR can increase the sensitivity of prostate cancer cells to low levels of androgens that eventually result in the development of androgen-independent prostate cancer (Linja et al., 2001). It has recently been reported that the AR is the main factor that determines the molecular changes required for driving prostate cancer cells from androgen-responsive to an androgen-refractory state (Chen et al., 2004). These authors performed microarray-based gene expression profiling analysis on seven pairs of hormone-sensitive (HS) and hormone-refractory (HR) prostate cancer xenografts. In an analysis of 12 000 probe sets, only AR mRNA was differentially expressed in all seven pairs of tumors. These data indicate that AR overexpression alone may be sufficient for a switch from HS to HR. Moreover, these authors introduced wild-type AR cDNA into HS LNCaP that resulted in a threefold increase in AR levels, and showed that these transfectants could grow in at least 80% lower concentrations of androgen. In addition, these cells became resistant to the antiandrogen bicalutamide. The authors suggested that even a modest change in the level of AR can upset the balance of cofactors that regulate AR activity. There are also other mechanisms for developing of an HR state including crosstalk between the AR and other signal transduction pathways, alterations in the expression of steroid coactivators and corepressors, and androgen-independent mechanisms (Isaacs and Isaacs, 2004).
The treatment of most cancers is based on therapeutic agents directed against rapidly proliferating cells. However, in the case of prostate cancer, the proliferative growth fraction, even of metastatic cells, is usually less than 10% (Pinski et al., 2001). The development of new approaches to induce apoptosis in a proliferation-independent manner is critical for prostate cancer treatment.
Defects in apoptotic signaling pathways are common in cancer cells (Zornig et al., 2001). Inhibition of apoptosis is important for tumor initiation since apoptosis is involved in the process of eliminating cells with different anomalies that lead to malignant transformation. Impaired apoptosis may also enhance tumor progression and promote metastasis, and this results in an increase of cancer cell resistance to various forms of therapy (Johnstone et al., 2002). Therefore, an understanding of how to induce apoptosis in apoptosis-resistant tumor cells represents the principal therapeutic goal of this type of apoptotic research.
It has been suggested in many studies that combined treatment with chemotherapeutic agents and apoptosis-inducing ligands belonging to TNFR family is a more effective strategy for cancer treatment. However, the role of androgen regulation of TNFR family-induced apoptosis is poorly understood. We have previously shown that androgen withdrawal decreased the sensitivity of the androgen-dependent cell line LNCaP to treatment with TNF-α (Guseva et al., 2004) and TRAIL even in the presence of the phosphotidylinositol-3 kinase (PI3K)/Akt inhibitor wortmannin (Rokhlin et al., 2002a, b). Thus, androgen may play a critical role in regulating sensitivity of LNCaP to apoptosis induced by TNFR family ligands.
In this study, we investigated the dose-dependent effects of androgen on TNF-α and TRAIL-mediated apoptosis in LNCaP. We found that DHT inhibited (in a dose-dependent manner) caspase activity after treatment with TNF-α or wortmannin plus TRAIL. We also found that Akt and p53 pathways play an important role in androgen-dependent TNFR family-induced apoptosis, and that caspase-2 expression is androgen regulated by direct binding of AR with an androgen-responsive element (ARE) in caspase-2 gene.
Results
Androgen regulates TNFR family-induced apoptosis
It has been reported that LNCaP displays a bell-shaped growth curve in response to increasing doses of DHT. Low doses DHT (in the pm range) result in an increase in cell proliferation, whereas higher doses (in nm and μm ranges) result in growth arrest (Sonnenschein et al., 1989). However, how androgens affect the apoptotic response of LNCaP to TNFR family death ligands has not yet been investigated. We have previously described that LNCaP is sensitive to TNF-α treatment but resistant to TRAIL (Rokhlin et al., 2002a, b; Guseva et al., 2004). However, in the presence of an inhibitor of the PI3K/Akt pathway, wortmannin, LNCaP was converted from TRAIL resistant to sensitive. To investigate the role of androgens in TNF-α- and TRAIL-mediated caspase activity and cell death, LNCaP was treated with these death receptor ligands in the presence of different concentrations of DHT. Caspase activity was estimated in live cells at different time points of treatment and cell death was measured after 72 h of treatment. As can be seen from Figure 1, even 0.1 pm of DHT decreased caspase activity (1A) and cell death (1B) after treatment with TNF-α in the absence or in the presence of wortmannin. In the case of simultaneous treatment with wortmannin and TRAIL, caspase inhibition was observed starting at 1 nm of DHT, whereas lower concentrations of DHT (0.1 and 10 pm) that inhibited TNF-α-mediated caspase activity did not affect TRAIL-dependent caspase activity, although cell death was diminished. These data indicate that TNF-α-mediated apoptosis is much more sensitive to the inhibitory effects of androgen compared to TRAIL-induced apoptosis. We also found that DHT inhibited Fas-mediated apoptosis in LNCaP after treatment with anti-Fas agonistic Ab IPO-4 in the presence of wortmannin (data not shown).
Figure 1.
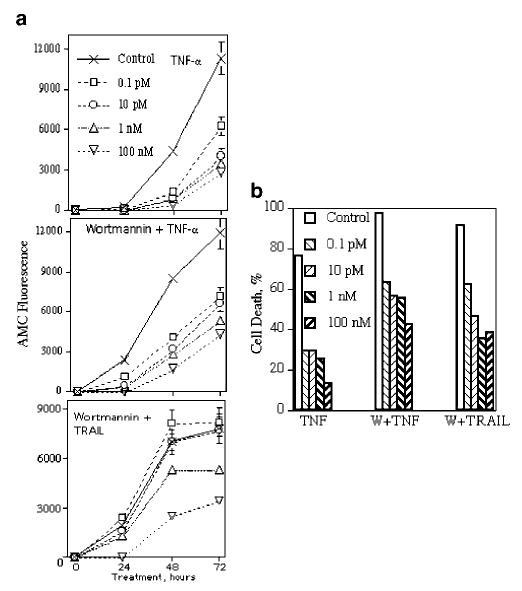
Effects of DHT on caspase activity (a) and cell death (b) in LNCaP after treatment with TNF-α and TRAIL. Caspase activity was measured in living cells with the fluorogenic substrate Ac-DEVD-AMC. The substrate (20 μm final concentration) was mixed with cells in growth medium, cells (7000/well/100 μl) were plated on 96-well plates and incubated with the substrate for 24 h to make cells adherent. Cells were then treated with DHT at indicated concentrations, and 1 h later, cells were treated with TNF-α (20 ng/ml), wortmannin (1 μm) plus TNF-α or wortmannin plus TRAIL (100 ng/ml). Substrate hydrolysis was monitored using a fluorescence reading system set to 360 nm for excitation and 460 nm for emission. Cell death was estimated by the calcein AM assay after 72 h of treatment. Each point or column represents mean values of four replicates in one of two experiments, which all gave similar results
We subsequently examined caspase activity by Western blot analysis using antibodies against caspase-2, -7 and -8. As shown in Figure 2a, the levels of caspase-2 proenzyme were decreased in a dose-dependent manner under DHT treatment, but caspase-2 activation was inhibited by all doses of DHT. In the case of caspase-7, DHT did not change the levels of proenzyme but the effect of DHT on activated band formation resembled the effect on caspase-2. All these effects are apparent only in the case of wortmannin + TRAIL treatment; caspase activation under TNF-α treatment is weaker compared to TRAIL treatment (data not shown) and therefore is not detectable by Western blot analysis.
Figure 2.
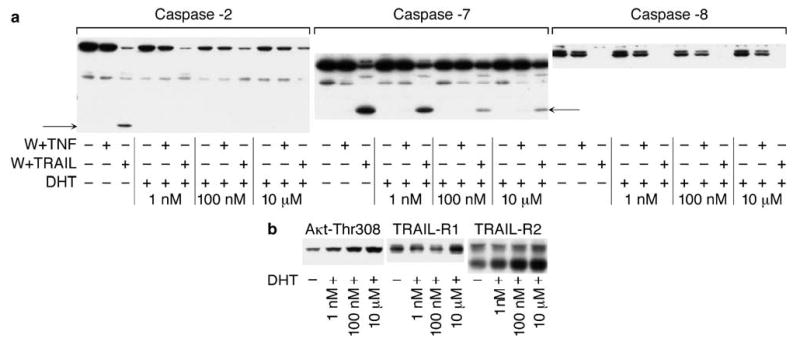
DHT effects on caspase activation (a) and expression of activated form Akt (Thr308), TRAIL-R1 and TRAIL-R2 (b). LNCaP was cultured for 48 h in the presence of the indicated concentrations of DHT. Cells were then treated for 3 h with wortmannin (1 μm) plus TNF-α (20 ng/ml) or TRAIL (100 ng/ml) (a). Cells were lysed in 1% Triton buffer, 20 μg of proteins were separated on 4–20% SDS–PAGE and expression of proteins was examined by Western blot analysis. Activated bands are indicated by arrows
DHT modulates Akt activity and regulates caspase activation downstream from TRAIL-DISC
In contrast to caspases-2 and -7, we observed that TRAIL-mediated caspase-8 activation was not inhibited by DHT treatment (Figure 2a). Thus, DHT cannot inhibit caspase-8 activity and these data indicate that modulation of caspase activity by DHT occurs downstream of the DISC. This idea was confirmed by examination of TRAIL-R1 and -R2 levels under treatment with different doses of DHT. DHT treatment increased the levels of TRAIL receptors, including treatment with 10 μm of DHT. Thus, on the one hand, DHT increased TRAIL receptors levels, but on the other hand, DHT decreased TRAIL-dependent caspase activity. Therefore, DHT accomplishes its inhibitory effects downstream of the DISC and the PI3K-Akt pathway is apparently involved in DHT-dependent modulation of apoptosis (Figure 2b).
In agreement with DHT-mediated inhibition of TNF-α and TRAIL-induced caspase activity in LNCaP, 1 nm of DHT sharply increased the level of the activated form of Akt (Akt-Thr308) (Figure 2b), and 10 μm DHT further increased the level of Akt-Thr308. Thus, there is a direct correlation between the DHT-mediated levels of Akt-Thr308 and caspase inhibition under treatment with TNF-α and TRAIL even in the presence of an inhibitor of the PI3K/Akt pathway. Obviously, DHT can over-ride the apoptosis-inducing effect of wortmannin since the inhibitory effects of DHT were observed under treatment with ligands in the presence of wortmannin.
AR inhibits caspase activity in PC3
To investigate whether the apoptotic response is androgen dependent in other cells, we transfected PC3 with plasmids containing wild-type AR and estimated caspase activity after treatment with TNF-α, anti-Fas Ab and TRAIL. Two transfectants were selected and one PC3-AR cell line was obtained from Dr T Brown (Toronto) and labeled here as PC3-AR-1. All three transfectants expressed comparable levels of AR to LNCaP (data not shown). As has been described previously (Heisler et al., 1997), androgens are toxic to PC3-AR cells and cell death was observed after culturing for 2–3 weeks in medium supplemented with FCS. Therefore, PC3-AR cells as well as PC3-Hygro control cells were selected and cultured in steroid-free condition (SFC). Surprisingly, PC3-AR cell lines were found to be relatively resistant to treatment with the three different ligands compared to PC3-Hygro (Figure 3). Importantly, this resistance occurs in the absence of androgens. The addition of DHT could further inhibit caspase activity in some of PC3-AR cell lines, but the major effects were observed after treatment for 48 h in SFC without DHT. Of note, AR was localized in cytosol, whereas in nuclei fraction of cells AR was not detected (data not shown). Therefore, at least in short term treatment, AR plays a protective role in TNFR family-induced apoptosis even in the absence of androgens.
Figure 3.
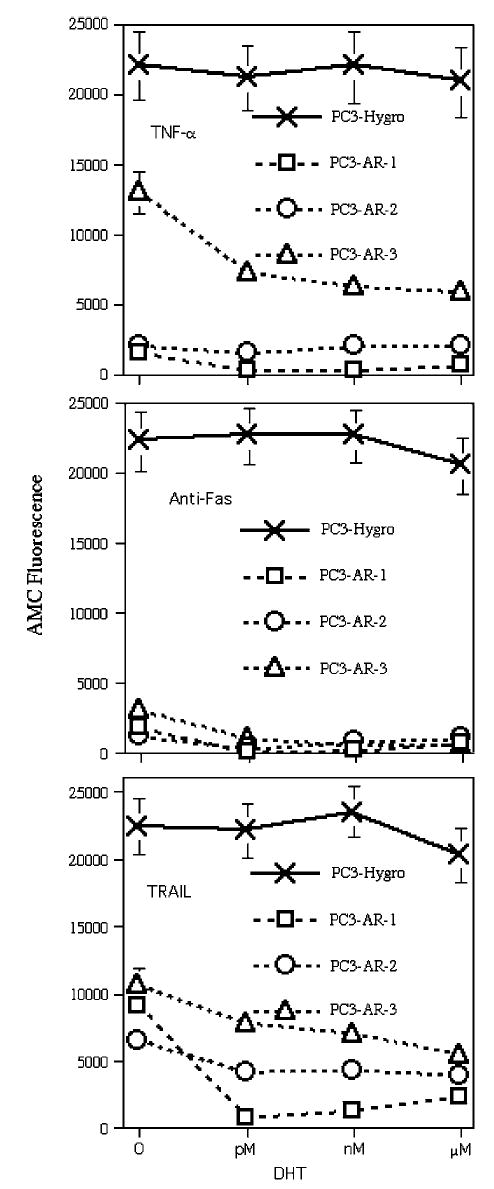
Effects of DHT on caspase activity in PC3-AR transfectants. Cells were plated on 96-well plates and cultured in SFC. Caspase activity was measured in living cells as described in the legend to Figure 1. Cells were treated with 10 pm, 10 nm and 10 μm of DHT together with TNF-α (20 ng/ml), anti-Fas mAb (100 ng/ml) or TRAIL (100 ng/ml). Substrate hydrolysis was measured after 48 h of treatment. PC-3(AR)2 cell line was obtained from Dr TJ Brown and marked here as PC3-AR-1
Role of p53 in androgen-dependent TNFR family-induced apoptosis
We have previously shown that p53 plays an important role in TNF-α-mediated apoptosis in LNCaP (Rokhlin et al., 2000). To further investigate the role of p53 in apoptosis, we switched off the expression of p53 using a specific si-p53 lentivirus construct. Western blot analysis confirmed that p53 expression was very low in LNCaP-si-p53 cells (data not shown). Caspase acivation in LNCaP-mock and LNCaP-si-p53 were compared to treatment with TNF-α and TRAIL in the presence of wortmannin. As can be seen from Figure 4, caspase activation in LNCaP-si-p53 after wortmannin + TNF-α treatment was diminished more than fivefold compared to LNCaP-mock. Moreover, while DHT treatment decreased caspase activity in LNCaP-mock, DHT did not change caspase activity in LNCaP-si-p53. In the case of wortmannin + TRAIL treatment, caspase activity was decreased twofold in LNCaP-si-p53 and DHT treatment resulted in further inhibition of caspase activity. Apparently, p53 plays a more important role in androgen-regulated TNF-α-induced apoptosis than in TRAIL-induced apoptosis.
Figure 4.
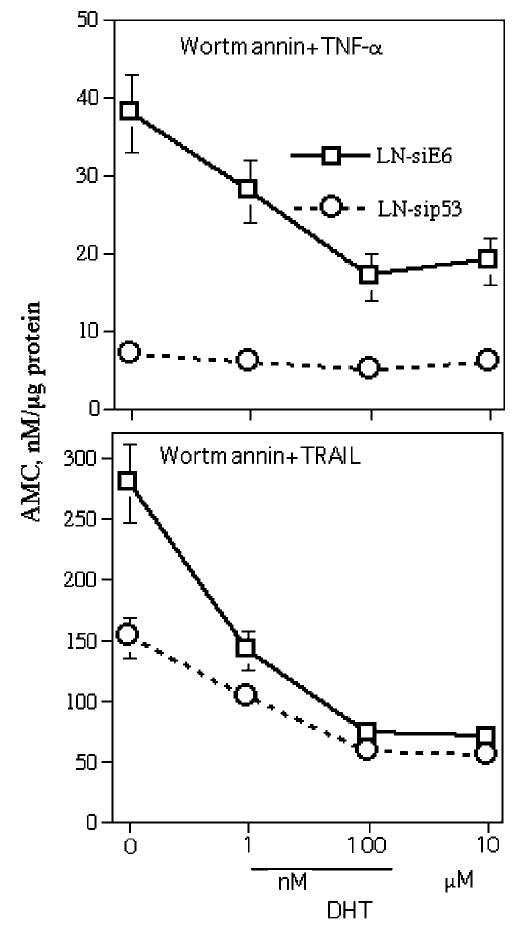
Effects of DHT on caspase activity in LNCaP-si-E6 (mock transfectant) and LNCaP-si-p53. Cells were pretreated for 48 h with indicated concentrations of DHT and then treated for 3 h with wortmannin (1 μm) plus TNF-α (20 ng/ml) or with wortmannin plus TRAIL (100 ng/ml). Caspase activity was estimated in cell lysates with the substrate Ac-DEVD-AMC
We next examined the levels of p53 under DHT treatment by Northern and Western blot analysis. As can be seen from Figure 5a, the levels of p53 mRNA decreased after treatment with 1 nm of DHT and then further decreased after treatment with 10 μm DHT. A time course of DHT treatment showed that 4 h did not have any effect on mRNA p53 levels, 8 h resulted in a slight decrease and 24 h decreased the mRNA p53 to the same level as 3 days of treatment. However, culturing of LNCaP for 3 days in SFC did not decrease the level of p53 mRNA. Figure 5b shows that DHT decreased the level of p53 protein in dose- and time-dependent manner correspondingly to the decreased mRNA levels. At the same time, culturing LNCaP in SFC decreased the level of p53 protein to the same extent as treatment with high concentrations of DHT. Thus, the interaction of AR with high concentrations of DHT as well as the absence of androgens in SFC resulted in decreased levels of p53 protein. The fact that culturing in SFC did not decrease the level of mRNA but decreased the protein level of p53 indicate that there are at least two different regulatory mechanisms of p53 expression by androgens: high concentrations of DHT regulate p53 expression at both the gene and protein levels, whereas androgen deprivation resulted only in decreased protein levels of p53.
Figure 5.
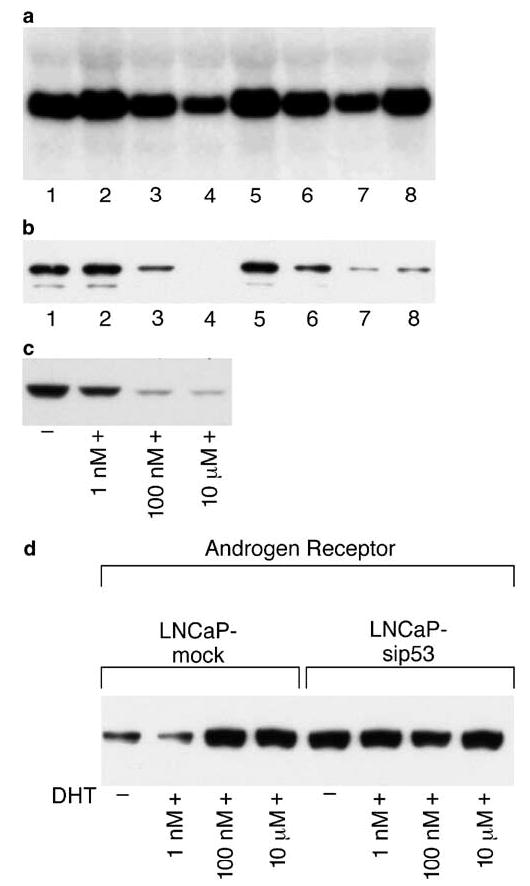
Androgen-dependent expression of p53 and p53-dependent expression of AR in LNCaP. Northern (a) and Western (b) blot analysis were performed after treatment for 3 days with 0.1 pm(#2), 1 nm (#3) and 10 μm (#4) of DHT or with 10 μm DHT for 4 (#5), 8 (#6) and 24 (#7) h. Cells were also cultured for 3 days in SFC (#8). #1 is untreated cells. (c) Cells were cultured for 48 h with indicated concentration of DHT. p53 protein expression was examined by Western blot analysis. (d) Cells were cultured for 24 h with indicated concentrations of DHT and AR expression was examined by Western blot analysis
To examine whether there is a correlation between the levels of p53 and caspase activation, LNCaP was treated with different concentrations of DHT. Figure 5c shows that p53 levels were decreased by 1 nm DHT and further decreased after treatment with 100 nm and 10 μm DHT. As can be seen from Figure 1, 1 nm DHT decreased caspase activity after treatment with TNF-α and TRAIL in the presence of wortmannin. Therefore, as was observed with the activated form of Akt (Figure 2b), there is a direct correlation between p53 levels and caspase activation: decreased levels of p53 are accompanied by decreased caspase activity. These data indicate that sensitivity of prostate cancer cells to TNF-α and TRAIL treatment might be regulated by androgens via p53- and Akt-dependent signaling pathways.
The data described in Figure 5a–c clearly indicate that DHT/AR regulates the expression of p53. However, the response to DHT of LNCaP-si-p53 suggests that p53 can regulate the level of AR. The level of AR was found to be higher in LNCaP-si-p53 compared to LNCaP-mock (Figure 5d). Moreover, DHT increased the level of AR in LNCaP, but in LNCaP-si-p53, the AR remained at the same level regardless of treatment with DHT. Therefore, there is a mutual regulation of expression between p53 and AR. We have subsequently examined the presence of ARE sequences in the p53 gene and found four potential AREs: one is in 5′-flanking region, one is in the 1st exon and two sites in the 1st intron (Table 1).
Table 1.
ARE in p53 and caspase-2 genes
| Position | Region | ARE sequence |
|---|---|---|
| p53 (Accession #U94788) | ||
| 584 | 5′ Flanking | TGCCCT cac AGCTCT |
| 877 | 1 Exon | TGGGCT ccg GGGACA |
| 990 | 1 Intron | CGGGCT ctc GGCTCC |
| 4322 | 1 Intron | GGAACA gac TGGGCG |
| Caspase-2 (Accession #AY219042) | ||
| 2632 | 1 Intron (ARE-1) | GGCTCC gag TGTCCA |
| 15087 | 8 Intron (ARE-2) | GGTACA aac TGTACT |
ARE sequences were searched using nGnnCnnnnnGnnCn as a consensus DNA sequence
Caspase-2 expression is regulated by androgen
During investigation of apoptosis in LNCaP under treatment with different concentrations of DHT, we found that caspase-2 levels are androgen regulated: high concentration of DHT inhibited apoptosis and resulted in decreased levels of caspase-2 (but not other caspases) (Figure 2a). To examine the role of caspase-2 in TNF-α- mediated apoptosis, we transfected LNCaP with a vector containing a mutant, dominant-negative, form of caspase-2 (casp-2-mut). Casp-2-mut was obtained by site-directed mutagenesis by replacing 303Cys to Ser; 303Cys is a crucial amino acid for activity of caspase-2 as a protease, and replacement results in complete inactivation of caspase activity (Lassus et al., 2002). LNCaP with caspase-2-mut had decreased caspase activity after TNF-α treatment (Figure 6). However, we did not find any differences between transfectant and mock cells after treatment with wortmannin and simultaneous treatment with wortmannin + TNF-α or TRAIL (data not shown). These data suggest that either caspase-2 plays a role only in TNF-α-mediated apoptosis or coexpression of dominant-negative and wild-type forms of caspase-2 was not sufficient to inhibit apoptosis after treatment with other TNFR family ligands in the presence of wortmannin.
Figure 6.
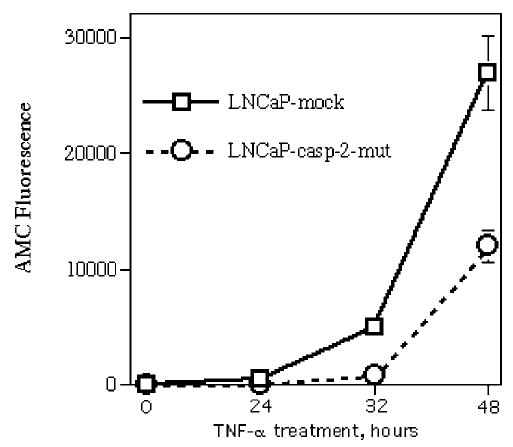
Expression of mutant caspase-2 results in decreased caspase activity under TNF-α treatment. Caspase activity was measured in living cells with the fluorogenic substrate Ac-DEVD-AMC as described in legend to Figure 1. Each point represents mean values of four replicates in one of two experiments, which gave similar results
To clarify the role of caspase-2 in TNFR family-mediated apoptosis, we switched off the caspase-2 expression by using a si-caspase-2 construct. We created two independent transfectants with decreased level of caspase-2. Western blot analysis showed that the levels of caspase-2 were decreased by six- and 33-fold in transfectants #1 and #2, respectively (Figure 7a). We then treated these transfectants and mock cells with TNF-α as well as with anti-Fas Ab ant TRAIL in the presence of wortmannin. As can be seen from Figure 7b, the decreased level of caspase-2 resulted in inhibition of caspase activity after treatment with anti-Fas Ab and TRAIL, suggesting that caspase-2 plays an important role in apoptosis induced by all three investigated TNFR family ligands.
Figure 7.
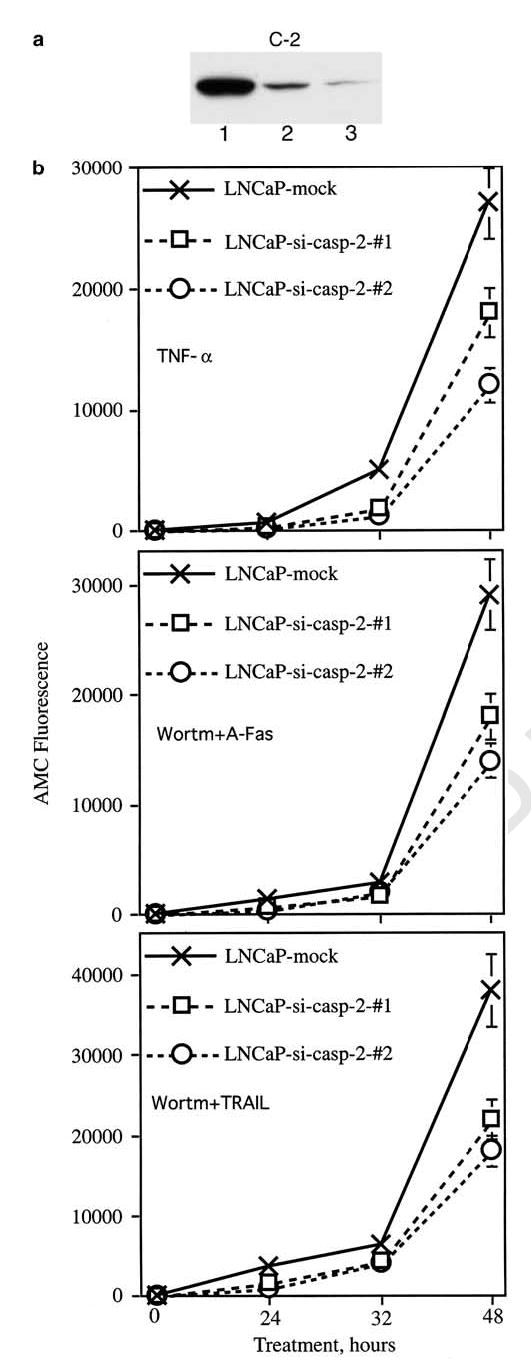
Inhibition of caspase-2 expression in LNCaP results in decreased caspase activity after treatment with TNF-α, as well as with anti-Fas Ab and TRAIL in the presence of wortmannin. (a) Western blot analysis of caspase-2 expression in mock cells (#1) and in two independent LNCaP-si-casp-2 transfectants: #1 (lane 2) and #2 (lane3). (b) Caspase activity was measured in living cells with the fluorogenic substrate Ac-DEVD-AMC as described in legend to Figure 1. Cells were treated with TNF-α (20 ng/ml), wortmannin (1 μm) plus anti-Fas Ab (1 μg/ml) or wortmannin plus TRAIL (200 ng/ml). DEVDase activity was measured after 24, 32 and 48 h of treatment. Each point represents mean values of four replicates in one of two experiments, which gave similar results
To further investigate androgen regulation of caspase-2 expression, LNCaP was treated with different doses of DHT. Figure 8a shows that DHT decreased the level of caspase-2 in a dose-dependent manner in LNCaP. We have also treated with DHT the androgen-independent LNCaP subline C4-2, which express AR and can be stimulated by androgen (Thalman et al., 1994), and found that androgen inhibited the expression of caspase-2 in this cell line (Figure 8a). We then used caspase-2-mut transfectants to investigate whether androgen regulation of caspase-2 occurs at the genomic or protein level. As casp-2-mut contains two different tags, myc and under flag, treatment this allowed us to discern the expression of ectopic from endogenous caspase-2. As can be seen from Figure 8b, caspase-2 is regulated at the genomic level since only endogenous caspase-2 level was decreased under treatment with 10 μm of DHT. To examine whether androgen regulation of caspase-2 expression is cell line-specific, three different PC3 cell lines transfected with AR-WT were treated with DHT and caspase-2 levels were analysed. Figure 8c shows that DHT decreased caspase-2 levels in all three transfectants but not in PC3-mock cells. Thus, androgen-regulated caspase-2 expression is not cell line-specific.
Figure 8.
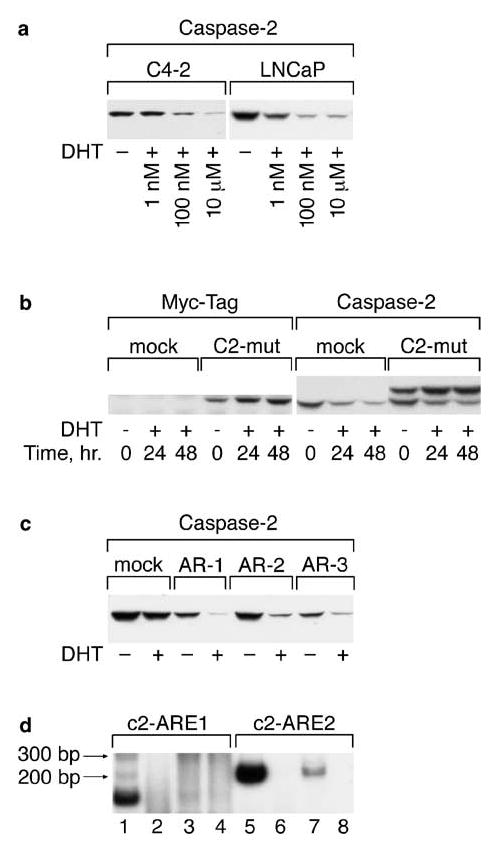
Caspase-2 expression is regulated by DHT. (a) C4-2 and LNCaP were cultured for 48 h in the presence of the indicated doses of DHT. Caspase-2 expression was estimated by Western blot analysis. (b) DHT regulates the expression of endogenous but not ectopically expressed caspase-2. LNCaP-mock and LNCaP-casp-2-mut cells were treated for 24 and 48 h with 10 μm of DHT. Caspase-2 expression was detected by Western blot analysis. (c) PC3-Hygro (mock) and three different PC3-transfected cell lines with wild-type AR were treated with 10 μm DHT and caspase-2 expression was examined by Western blot analysis. (d) The caspase-2 gene contains ARE in intron regions and binds to the AR in vivo. PCR was performed on DNA specifically complexed to the AR protein. Two primer sets were designed to cover the c2-ARE1 and c2-ARE2 that should produce amplicons of 146 and 210 bp in size. PCR was performed in the presence of [α-32P]-dATP, using primers for c2-ARE1 and c2-ARE2, on DNA extracted after ChIP procedure with antibodies to AR. Samples were separated on 6% PAGE and gels were dried and exposed on X-ray film for 12 h at room temperature. 1 and 5 – positive control with non complexed DNA before immunoprecipitation; 2 and 6 – control immunoprecipitation procedure without antibodies; 3 and 7 – chromatin DNA immunocomplexed with AR antibody; 4 and 8 – chromatin immunoprecipitation with mouse IgG
We searched for AREs in the caspase-2 gene and two AREs have been found in the 1st (ARE-1) and 8th introns (ARE-2) (Table 1). To investigate whether there is a direct interaction in vivo between AR and the caspase-2 gene, chromatin immunoprecipitation (ChIP) experiments were performed followed by PCR on chromatin DNA specifically complexed to the AR protein. Two primer sets were designed to cover the c2-ARE1 and c2-ARE2 and produce amplicons of 146 and 210 bp in size, respectively. PCR was performed on AR-immunocomplexed DNA obtained from LNCaP cells. As can be seen from Figure 8d, PCR with both primer sets produced bands of the expected size from the control (nonimmunoprecipitated) DNA (lanes 1 and 5). PCR with the primer set for c2-ARE2 region (lane 7) but not with primers for c2-ARE1 (lane 3) produced a signal when AR-immunocomplexed DNA were used as a template. DNA after immunoprecipitation procedure without antibodies (lanes 2 and 6) or with nonspecific mouse IgG (lanes 4 and 8) did not reveal any bands with primers for c2-ARE1 as well as for c2-ARE2. These results suggest that AR interacts in vivo with the ARE in intron 8 resulting in direct regulation of caspase-2 expression.
Discussion
The major finding in our study is that the apoptotic response to TNFR family ligands treatment is regulated by DHT in dose-dependent manner. Apparently, DHT/AR interaction results in different coactivator/corepressor cross-talk depending on androgen concentration. Several distinct signaling pathways involved in the differential response to androgen have been described, but only a few studies have addressed the role of androgen in apoptosis. Coffey et al. 2002 found that DHT protected LNCaP from radiation- and etoposide-induced apoptosis. In these experiments, LNCaP was pretreated for 72 h with 1 nm DHT and decreased levels of caspase-3 and -7 as well as Bik, Bak and Bax were found. In another study (Huang et al., 2004a), forkhead transcription factor FKHR-induced death of LNCaP was blocked by the synthetic androgen R1881. In these experiments, LNCaP was pretreated for 48 h with 1 nm of R1881 and resulted in a reduction in transcriptional activity of FKHR. A similar inhibitory effect of androgen on transactivation of FKHR was observed in LNCaP treated with LY294002, a chemical inhibitor of PI3K. Treatment of LNCaP with R1881 led to a decrease in the intact FKHR protein since androgen induced increased activity of an acidic cysteine protease that in turn cleaves FKHR. Of note, we have measured protease activity using the fluorogenic protease substrate LLVY-AMC. In accordance with the experiments mentioned above, we have found that DHT treatment of LNCaP resulted in increased LLVYase activity, while decreased DEVDase activity was found (data not shown). Apparently, there is inverse correlation between protease and caspase activity under androgen treatment. It has also been observed that treatment of LNCaP with R1881 decreased the levels of Bcl-2, Bax and the transcriptional factor E2F1, while activation of Rb protein and elevated levels of the cyclin-dependent kinase inhibitors (CDKIs) p151NK4B and p27 KIP1 were found. These authors showed that androgens suppress Bcl-2 expression by negatively modulating the activity of the E2F site in the Bcl-2 promoter by activating the CDKI-Rb axis (Huang et al., 2004b).
There are many apoptosis-modulating proteins, such as Akt and p53, that may impact several apoptotic pathways simultaneously, possibly at multiple points. We have investigated Akt, p53 and caspase activation pathways during androgen-regulated apoptotic response to TNFR family ligands.
Akt is an important regulator of cell proliferation and survival. Pathological elevation in Akt activity is common occurrence in tumors due to loss of the tumor suppressor phosphatase PTEN that is able to dephosphorylate the lipid second messenger phosphotidylinositol (PI) 3, 4, 5-phosphate and negatively regulates survival signaling through the PI3K/Akt pathway. There are three mammalian isoforms of this enzyme, Akt1, Akt2 and Akt3 (Chan et al., 1999). The activation of all three isoforms is similar and requires the phosphorylation of two sites. The activation of PI3K and Akt promotes cell survival through multiple mechanisms. Akt directly phosphorylates multiple protein targets of relevance to apoptosis, such as caspase-9 and Bad, suppressing cell death within the intrinsic pathway. In addition to acting as a kinase for many substrates, Akt forms complexes with other proteins that act as modulators of Akt activity and function (Brazil et al., 2002). Amplification of genes encoding Akt isoforms has been found in several types of human cancers (Hill and Hemmimgs, 2002). It has also been recently shown that AR interacts with the p85α regulatory subunit of PI3K, and its binding affinity is increased after androgen stimulation (Sun et al., 2003). These authors further demonstrated that the complex between AR, p85α and the oncoprotein Src is required for androgen-stimulated PI3K/Akt activation. It has also been demonstrated that Akt phosphorylates the AR at Ser-210 and Ser-790 and results in suppression of AR transactivation. Taken together, these data suggest that there is the crosstalk between these two signaling pathways and that it is finely regulated.
We have previously shown that simultaneous treatment of LNCaP with wortmannin, an inhibitor of the PI3K/Akt pathway, and TRAIL converted the phenotype of LNCaP from TRAIL resistant to sensitive (Rokhlin et al., 2002a, b). Simultaneous treatment with TRAIL and wortmannin induced activation of caspase-2, -3, -7, -8 and -9 as well as the release of cytochrome c. Overexpression of FADD-DN completely blocked caspase activation and apoptosis. Therefore, caspase-8 activation is necessary but not sufficient for TRAIL-mediated apoptosis and is presumably blocked by the PI3K/Akt pathway downstream of caspase-8. Androgen withdrawal has been shown to result in an increase of PI3K/Akt pathway activity and supports survival after androgen ablation (Murillo et al., 2001). We have also found that androgen deprivation increased the levels of Akt protein and phosphoactive Akt, suppressed the formation of the TRAIL-DISC, and that these could be reversed with DHT (Rokhlin et al., 2002a, b). In this study, we found that, in agreement with DHT-mediated inhibition of TNF-α and TRAIL-induced caspase activity in LNCaP, the DHT can sharply increase the level of Akt-Thr308 (Figure 2b). Thus, there is a direct androgen-dependent correlation between the levels of Akt-Thr308 and caspase inhibition under treatment with TNF-α and TRAIL even in the presence of an inhibitor of the PI3K/Akt pathway. Obviously, DHT can over-ride the apoptosis-inducing effect of wortmannin since the inhibitory effect of DHT was observed under treatment with ligands in the presence of wortmannin. Apparently, depending on the steroid milieu, AR is capable (or incapable) of interacting with its coactivators/corepressors and other AR-interacting proteins.
p53 is a regulator of genotoxic stress that plays an important role in DNA damage response, DNA repair, cell cycle regulation and in triggering apoptosis after cell injury. p53 regulates the expression of a variety of apoptosis-related genes that affect both the intrinsic (Bax, Noxa, Puma, Bid, Bcl-2, Bcl-XL) and extrinsic (Fas, TRAIL-R2, PIDD, DcR1, DcR2) pathways (reviewed by Reed, 2003). p53 can directly engage each of the major apoptosis pathways in the cell, stimulating both death receptor signaling and mitochondrial perturbation, including cytochrome c release. Transcriptionally independent activities of p53 induce relocalization of death receptors from the Golgi to the cell surface (Bennet et al., 1998). It has been described that p53 transcriptional targets that encode proteins localized to the mitochondria and effect mitochondrial membrane potential (Oda et al., 2000). In addition, p53 itself can localize to mitochondria and contribute to apoptosis by modulating signals in mitochondria (Marchenko et al., 2000). Lastly, the level of p53 can be controlled by another apoptosis multipathway regulator – Akt (Mayo and Donner, 2002).
We have previously analysed the role of p53 in TNF-α-mediated apoptosis in LNCaP by generating a derivative of LNCaP, LN-56, that expressed a dominant-negative element of p53, GSE56 (Rokhlin et al., 2000). The expression of GSE56, which corresponds to the C-terminal portion of p53, resulted in the accumulation of p53 in the cytosol and inhibition of p53 transactivation. We have reinvestigated the role of p53 in TNF-α-induced caspase activation using LNCaP-si-p53 instead of LN-56. The reason for this reinvestigation is that the transactivation activity of p53 in LN-56 is suppressed, but the large amount of p53 sequestered in the cytosol of LN-56 might mediate the interaction of p53 with different target proteins. We have indeed found that LN-si-p53 was substantially more resistant to TNF-α treatment compared to LN-56; caspase activity was inhibited by 6–10-fold in LNCaP-si-p53 compared to LNCaP (Figure 4), whereas in LN-56, this inhibition never exceeded threefold (Rokhlin et al., 2000). In addition, wortmannin + TRAIL treatment decreased caspase activity twofold in LNCaP-si-p53 compared to LNCaP-mock.
It is extremely interesting that there is mutual regulation of expression between p53 and AR. We have found that DHT decreased the level of p53 protein in dose-dependent manner. At the same time, culturing LNCaP in SFC decreased the level of p53 to the same extent as treatment with high concentrations of DHT. Thus, the interaction of AR with high concentration of DHT as well as the absence of androgen in SFC resulted in decreased levels of p53 protein. Confirmatory data show that higher concentrations of DHT decreased p53 mRNA levels. A time course of DHT treatment on mRNA p53 levels showed that 4 h did not have any effect, 8 h resulted in a slight decrease and 24 h decreased the mRNA p53 to the same level as 3 days of treatment. However, culturing of LNCaP for 3 days in SFC did not decrease the level of p53 mRNA. Therefore, there are at least two different regulatory mechanisms of p53 expression by androgens: high concentrations of DHT regulate p53 expression at the gene level, whereas androgen deprivation resulted in decreased protein levels of p53. These data indicate that AR regulates the expression of p53. We have subsequently examined the presence of ARE sequences in the p53 gene and found four potential AREs: one is in 5′ region, one in the 1st exon and two sites are in the 1st intron. However, the response to DHT and culturing in SFC of LN-si-p53 indicates that the p53 in turn can regulate the activity of AR. The level of AR was found to be higher in LN-si-p53 compared to LNCaP. Moreover, DHT regulates the level of AR in LNCaP, but in LN-si-p53, the AR remained on the same level regardless of treatment with DHT.
There is a direct correlation between the sensitivity of LNCaP to apoptotic stimuli under DHT treatment and the effects of DHT on the levels of p53 and the activated form of Akt. These data indicate that the sensitivity of prostatic carcinoma cells might be regulated by androgens via p53- and Akt-dependent signaling pathways.
Androgen regulation of different pathways in prostate cells is likely much more complicated and inter-related than is currently thought. For example, Nantermet et al. 2004 analysed genes that rapidly respond to DHT activation of the AR in the rat ventral prostate gland. Using large-scale gene expression analysis of ventral prostate samples collected 6 and 24 h after DHT treatment in castrated animals, 243 genes were shown to be androgen-regulated prior to the onset of cell proliferation; approximately 200 of these genes were not previously known to be androgen responsive.
One of the unexpected, undescribed previously, findings in our study is that the caspase-2 expression is regulated by AR. Caspase-2 is the most unusual member of the caspase family. For example, caspase-2 exhibits features of both an initiator as well as an executioner caspase. As an initiator caspase, caspase-2 contains a CARD domain and it has been recently shown that activation of caspase-2 occurs in a complex that contains the death-domain containing protein PIDD and the adapter protein RAIDD (Tinel and Tschopp, 2004). Unlike other initiator caspases, caspase-2 cannot activate other caspase zymogens, although it can cleave Bid (Guo et al., 2002). Another distinguishing feature of caspase-2 is its ability to localize in nuclei and participate in the late stages of apoptosis (Paroni et al., 2002). As an executioner caspase, caspase-2 has the same substrate preference (DXXD) and cleavage site between the large and small subunits that are present in other executioner caspases. It has been shown that caspase-2 plays a prominent role in different cell death systems (reviewed by Degterev et al., 2003) including the release of apoptogenic factors from mitochondria directly (Guo et al., 2002; Lassus et al., 2002). It is interesting that processed caspase-2 can induce mitochondria-mediated apoptosis independent of its enzymatic activity (Robertson et al., 2004).
We have previously found several new virtues of caspase-2: N-α-tosyl-l-phenylalanyl chloromethyl ketone (TPCK) induced degradation of caspase-2, whereas it did not affect other caspases; TPCK-induced degradation of caspase-2 was partially protected by Bcl-2 overexpression, although direct interaction between caspase-2 and Bcl-2 was not detected (Rokhlin et al., 2004). In this study, we have shown that the inhibition of caspase-2 expression resulted in decreased caspase activation after treatment with TNFR family ligands. Caspase-2 expression was found to be regulated by androgen and two AREs were detected within the caspase-2 gene. More directly, ChIP analysis showed that the AR binds to an ARE of the caspase-2 gene. We have also found that expression of caspase-2 can be regulated by androgen not only in AR-positive cell lines LNCaP and C4-2 but also in PC3 transfected with AR-WT, which indicates that androgen-regulated caspase-2 expression is not cell line-specific.
In conclusion, our study indicates that androgen-dependent outcome of apoptotic treatment can occur, at least in part, via the caspase-2-, Akt- and p53-mediated pathways.
Materials and methods
Cell lines and estimation of cell viability
The human prostatic carcinoma cell lines LNCaP, PC3 and DU145 were cultured in RPMI 1640, as has been described previously (Rokhlin et al., 1997). The C4-2 cell line was obtained from K Gurova (Lerner Research Institute, CCF, Cleveland, OH, USA). A plasmid with myc-tagged mutated caspase-2 (IE-C2) was obtained from Y Lazebnik (Cold Spring Harbor Lab). TNF-α was purchased from R&D Systems (Minneapolis, MN, USA), human TRAIL was purchased from PeproTech (Rocky Hill, NJ, USA), and agonistic anti-Fas (IgM) mAb was used as described previously (Rokhlin et al., 1998). To measure cell viability, we used the calcein AM assay (Molecular Probes, Eugene, OR, USA), as described previously (Rokhlin et al., 2002a). Cells were incubated and treated in 96-well flat-bottomed plates and cell death was estimated after 24, 48 or 72 h.
Caspase activity measurement in cell lysates and in live cells
Caspase activity in cell lysates was measured as described previously (Rokhlin et al., 2001). Briefly, cell lysates were prepared in 1% Triton X-100 buffer, pH 7.2, containing protease inhibitors. Protein lysate (40 μg) was incubated for 60 min in assay buffer (20 mm PIPES, pH 7.2, 100 mm NaCl, 10 mm DTT, 1 mm EDTA, 0.1% CHAPS and 10% sucrose) with 40 μm of fluorescent substrates Ac-DEVD-AMC (Bio-Mol, Plymouth Meeting, PA, USA). Caspase activity in intact (living) cells was measured with the fluorogenic substrates Ac-DEVD-AMC as described previously (Rokhlin et al., 2004). Briefly, the substrates (20 μm final concentration) were mixed with cells in growth medium, cells were plated on 96- (7000 per well) or 24-well (50 000 per well) plates and incubated with the substrates for 24 h to make cells adherent. Cells were then treated with death-inducing ligands or drugs and substrate hydrolysis was monitored using a fluorescence reading system set to 360 nm for excitation and 460 nm for emission.
Western blot analysis
Western blot detection of proteins was performed as described previously (Rokhlin et al., 2001). Briefly, 20 μg of proteins were separated on 4–20% gradient SDS–PAGE, and blotted onto a nitrocellulose membrane (Novex, San Diego, CA, USA). Equal loading was controlled routinely by reversible staining of the membrane with Ponceau S solution or with antibodies to actin (Sigma, St Louis, MO, USA). Membranes were blocked with 5% nonfat dry milk in PBS containing 0.1% Tween-20 and then incubated with the corresponding mouse monoclonal or rabbit polyclonal antibodies: anti-TRAIL-R1, anti-TRAIL-R2, anti-caspase-7, anti-caspase-9, anti-p53 (Oncogene, Uniondale, NY, USA), anti-caspase-8, (Upstate, Lake Placid, NY), anti-Akt, anti-Akt-Thr308 (Cell Signaling, Beverly, MA, USA), anti-caspase-3 (Transduction Laboratories, San Diego, CA, USA), anti-caspase-2 (R&D System, Minneapolis, MN, USA). The blots were counterstained with goat anti-mouse or anti-rabbit IgG conjugated with HRP (Pierce, Rockford, IL, USA). The immunoreactive bands were visualized by incubation of the membrane with enchanced chemiluminescence reagent (Pierce).
Generation of LNCaP with decreased expression of p53 (LNCaP-si-p53) and caspase-2 (LNCaP-si-casp-2)
The expression of endogenous p53 was inhibited by infection with recombinant lentivirus constructs pLSL-puro-expressing siRNA hairpin under control of the RNA H1 promoter (Budanov et al., 2004). The structure of the 19 bp siRNA complementary to human p53 mRNA was as follows: 5′-GACTCCAGTGGTAATCTAC-3′. The structure of the control siRNA derived from the HPV18 E6 gene was as follows: 5′-CTAACACTGGGTTATACAA-3′. LNCaP was infected with lentivirus with siE6 or si-p53 followed by puromycin selection. The effect of siRNA expression was verified by assessing p53 mRNA levels on Northern and p53 protein expression by Western blot analyses. For Northern analysis, 10 μg of total RNA was separated on 1% agarose gel, transferred to Hybond-N membrane and hybridized to 32P-labeled cDNA probes corresponding to 492 bp fragment representing complete protein-coding region of p21WAF1/CIP1, or 1.2 kb cDNA fragment encoding human p53. Hybridization with human GAPDH cDNA probe was used as a normalization control. The vector used for silencing caspase-2 expresses the hairpin form of caspase-2 siRNA under the control of human H1 RNA Pol III promoter and Hygromycin resistance gene under the control of SV40 promoter. This vector was constructed in the following steps. An H1 promoter-driven expression cassette encoding a hairpin siRNA against caspase-2 was inserted in the pBluescript II SK (Stratagene) using the XbaI and BamHI sites. Second, KpnI restriction site were used to insert PCR amplified Hygromycin resistance cassette from pcDNA3.1 Hygro (Invitrogen). The sequence of the oligonucleotides, encoding the caspase-2 hairpin (top strand), was 5′-GATCCCCACAGCTGTTGTTGAGCGAA TTCAAGAGA TTCGCTCAACAACAGCTGTTTTTTGGAAA-3′. The underlined sequences were used as described (Lassus et al., 2002).
Permanent transfections of PC3 with wild-type AR
The human prostatic carcinoma cell line PC3 was maintained in RPMI 1640 supplemented with 5% charcoal-stripped fetal bovine serum. Effectene Transfection Reagent (Qiagen Inc., Valencia, CA, USA) was used for transfection according to the manufacturer’s instruction. The pCEP4/hAR expression construct containing wild-type AR and cell line PC3(AR)2 was obtained from Dr Theodore Brown (Heisler et al., 1997). In addition, PC3 cells were transfected with the pCEP4/hAR followed by hygromycin selection.
ChIP and PCR analysis
ChIP was performed as described by Gnanapragasam et al. (2003). Briefly, LNCaP was treated with formaldehyde (1% final concentration), for 10 min at room temperature. Cells were scraped in ice-cold PBS, the pellet was lysed in lysis buffer (50 mm Tris-HCl, pH 8.1, 1% SDS, 10 mm EDTA, 1 mm PMSF, protease inhibitors cocktail (Sigma)) and sonicated to shear DNA to 200–1000 bp fragments. Samples were spun down at 10 000 g for 10 min, supernatant diluted 10-fold in dilution buffer (25 mm Tris, pH 8.1, 140 mm NaCl, 1% SDS, 3 mm EDTA, 1 mm PMSF, protease inhibitors cocktail). To preclear the chromatin solution, 60 μl salmon sperm DNA/protein G agarose beads (Upstate Biotechnology) was added to each sample and incubated for 30 min at 4°C. For immunoprecipitation, 2 μg of AR antibody was added to 1 ml of the purified chromatin sample and incubated overnight at 4°C. Immunocomplexes were recovered by adding 60 μl salmon sperm DNA/protein G agarose for 1 h at 4°C. Beads were washed sequentially with three different buffers: buffer I-TSE (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1) plus 150 mm NaCl, buffer II-TSE plus 500 mm NaCl and buffer III (0.25 m LiAc, 1% Nonidet P-40, 1% deoxycholate, 1 mm EDTA, 10 mm Tris-HCl, pH 8.1), and then washed three times with TE buffer (pH 8). Immunocomplexes were eluted by adding 250 μl elution buffer (1% SDS and 0.1 m NaHCO3) to beads and subsequently heated for 4 h at 64°C to reverse formaldehyde-induced crosslinks. To verify AR immunoprecipitation, sample aliquots were taken for Western blot analysis after elution. AR-immunocomplexed DNA was then recovered by phenol/chloroform extraction, ethanol precipitation and resuspended in 50 μl TE. Two primers sets were designed to cover the ARE1 and ARE2 and produced amplification bands 146 and 210 bp, respectively. Primer set for c2-ARE1: forward 5′-ATC GAT TGT ATC ATG ATC TGC ATG G-3′; reverse 5′-GGT TTG ACC GGG GCA GAG GCA TGG-3′. Primer set c2-ARE2: forward 5′-GCC CTG CAA CAT GAC TCA GAC TAC-3′; reverse 5′-ATG ATC TAC TCT TAC TGC ATA CTC C-3′. PCR was performed with 10 μl of eluted AR-immunocomplexed DNA, BioTaq DNA polymerase (Perkin-Elmer) and 32P-dATP (NEN). PCR was also performed on unprecipitated chromatin as a positive control. Amplification was carried out for 35 cycles with denaturation at 94°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 1.5 min. PCR products were resolved in 6% PAGE, dried and then exposed to X-ray film for 12 h at room temperature.
Acknowledgments
We thank Y Lazebnik (Cold Spring Harbor Lab) for monoclonal and polyclonal antibodies to caspase-2 and the expression vector with mutated caspase-2. We thank TJ Brown (The University of Toronto, Canada) for the PC-3(AR)2 cell line and expression vector pCEP4/hAR. This work is supported by NIH Grant CA 87717.
References
- Bennet M, Macdonald K, Chan SW, Luzio JP, Simari R, Weissberg P. Science. 1998;282:290–293. doi: 10.1126/science.282.5387.290. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Park J, Hemmings BA. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- Chan TO, Rittenhouse SE, Tsichlis PN. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Coffey RNT, Watson RWG, O’Neill AJ, Mc Eleny K, Fitzpatrick JM. Prostate. 2002;53:300–309. doi: 10.1002/pros.10159. [DOI] [PubMed] [Google Scholar]
- Degterev A, Boyce M, Yuan J. Oncogene. 2003;22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- Gerhold D, Freedman LP, Ray WJ. J Biol Chem. 2004;279:1310–1322. doi: 10.1074/jbc.M310206200. [DOI] [PubMed] [Google Scholar]
- Gnanapragasam VJ, Robinson MC, Marsh C, Robson CN, Hamdy FC, Leung HY. Br J Cancer. 2003;88:1432–1438. doi: 10.1038/sj.bjc.6600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. J Biol Chem. 2002;277:13430–13437. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- Guseva NV, Taghiyev AF, Rokhlin OW, Cohen MB. J Cell Biochem. 2004;91:70–99. doi: 10.1002/jcb.10707. [DOI] [PubMed] [Google Scholar]
- Heisler LE, Evangelou A, Lew AM, Trachtenberg J, Elsholtz HP, Brown TJ. Mol Cell Endocrinol. 1997;126:59–73. doi: 10.1016/s0303-7207(96)03970-6. [DOI] [PubMed] [Google Scholar]
- Hill MM, Hemmimgs BA. Pharmacol Ther. 2002;93:243–251. doi: 10.1016/s0163-7258(02)00193-6. [DOI] [PubMed] [Google Scholar]
- Huang H, Muddiman DC, Tindall DJ. J Biol Chem. 2004a;279:13866–13877. doi: 10.1074/jbc.M314143200. [DOI] [PubMed] [Google Scholar]
- Huang H, Zegarra-Moro OL, Benson D, Tindall DJ. Oncogene. 2004b;23:2161–2176. doi: 10.1038/sj.onc.1207326. [DOI] [PubMed] [Google Scholar]
- Isaacs JT, Isaacs WB. Nat Med. 2004;10:26–27. doi: 10.1038/nm0104-26. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Lassus P, Opitz-Araya X, Lazebnik Y. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- Linja MJ, Savinainen KJ, Saramaki OR, Tamella TLJ, Vessella RL, Visakorpi T. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- Marchenko ND, Zaika A, Moll UM. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- Mayo LD, Donner DB. Trends Biochem Sci. 2002;27:462–467. doi: 10.1016/s0968-0004(02)02166-7. [DOI] [PubMed] [Google Scholar]
- Murillo H, Huang H, Schmidt LJ, Smith DI, Tindall DJ. Endocrinology. 2001;142:4795–4805. doi: 10.1210/endo.142.11.8467. [DOI] [PubMed] [Google Scholar]
- Nantermet PV, Xu J, Yu Y, Hodor P, Holder D, Adamski S, Gentile MA, Kimmel DB, Harada S-I, Tinel A, Tschopp J. Science. 2004;304:843–846. [Google Scholar]
- Nelson WG, De Marzo AM, Isaacs WB. N Engl J Med. 2003;349:366–381. doi: 10.1056/NEJMra021562. [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Tanigushi T, Tanaka N. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Paroni G, Henderson C, Schneider C, Brancolini C. J Biol Chem. 2002;277:15147–15161. doi: 10.1074/jbc.M112338200. [DOI] [PubMed] [Google Scholar]
- Pinski J, Parikh A, Bova S, Isaacs JT. Cancer Res. 2001;61:6372–6376. [PubMed] [Google Scholar]
- Reed JC. Cancer Cell. 2003;3:17–22. doi: 10.1016/s1535-6108(02)00241-6. [DOI] [PubMed] [Google Scholar]
- Robertson JD, Gogvadze V, Kropotov A, Vakifahmetoglu H, Zhivotovsky B, Orrenius S. EMBO Rep. 2004;5:643–648. doi: 10.1038/sj.embor.7400153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokhlin OW, Bishop GA, Hostager BS, Waldschmidt TJ, Sidorenko SP, Pavloff N, Kiefer MC, Umansky SR, Glover RA, Cohen MB. Cancer Res. 1997;57:1758–1768. [PubMed] [Google Scholar]
- Rokhlin OW, Glover RA, Cohen MB. Cancer Res. 1998;58:5870–5875. [PubMed] [Google Scholar]
- Rokhlin OW, Glover RA, Taghiyev AF, Guseva NV, Seftor REB, Shyshynova I, Gudkov AV, Cohen MB. J Biol Chem. 2002a;277:33213–33219. doi: 10.1074/jbc.M204612200. [DOI] [PubMed] [Google Scholar]
- Rokhlin OW, Gudkov AV, Kwek S, Glover RA, Gewies AS, Cohen MB. Oncogene. 2000;19:1959–1968. doi: 10.1038/sj.onc.1203453. [DOI] [PubMed] [Google Scholar]
- Rokhlin OW, Guseva N, Taghiyev A, Knudson CM, Cohen MB. Oncogene. 2001;20:2836–2843. doi: 10.1038/sj.onc.1204410. [DOI] [PubMed] [Google Scholar]
- Rokhlin OW, Guseva NV, Taghiyev AF, Glover RA, Cohen MB. Cancer Biol Ther. 2004;3:761–768. doi: 10.4161/cbt.3.8.970. [DOI] [PubMed] [Google Scholar]
- Rokhlin OW, Taghiyev AF, Guseva NV, Glover RA, Syrbu SI, Cohen MB. Cancer Biol Ther. 2002b;1:631–637. doi: 10.4161/cbt.311. [DOI] [PubMed] [Google Scholar]
- Sonnenschein C, Olea N, Pasanen ME, Soto AM. Cancer Res. 1989;49:3474–3481. [PubMed] [Google Scholar]
- Sun M, Yang L, Feldman RI, Sun X-M, Bhalla KN, Jove R, Nicosia SV, Cheng JQ. J Biol Chem. 2003;278:42992–43000. doi: 10.1074/jbc.M306295200. [DOI] [PubMed] [Google Scholar]
- Thalman GN, Anezinis PA, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, von Eschenbach AC, Chung LWK. Cancer Res. 1994;54:2577–2781. [PubMed] [Google Scholar]
- Zornig M, Hueber A, Baum W, Evan G. Biochem Biopys Acta. 2001;1551:F1–F37. doi: 10.1016/s0304-419x(01)00031-2. [DOI] [PubMed] [Google Scholar]