

Abstract
By viewing virus development in real time, the experiments reported here reveal novel processes—rapid directional translocations—that are likely to be important elements of virus replication. Herpes simplex virus type 1 (HSV-1) was labeled by the fusion of the green fluorescent protein to a structural protein of its tegument (VP11/12), the product of gene UL46. Infection of cultured cells with this recombinant virus (GHSV-UL46) produced fluorescent particles that were distributed throughout the cytoplasm with concentrations in the perinuclear region; they were absent from the nucleus. Viewing infected cells in real time by means of video microscopy produced a novel dynamic picture of virus development. Most strikingly, some of the fluorescent particles exhibited extremely rapid directional translocations at velocities as great as 5 μm/s. The trajectories and destinations of these particles suggest that the rapid directional translocations serve at least three functions: the rapid transport of viral components to and between cytoplasmic processing stations, the delivery of materials for functions specific to the perinuclear region, and the conveyance of maturing virus particles to the plasma membrane. These rapid directional translocations are novel elements of virus assembly that are likely to be critical for efficient replication.
The herpes simplex virus type 1 (HSV-1) virion comprises three concentric compartments: the capsid (100 nm in diameter, containing the viral DNA), the tegument (a shell surrounding the capsid composed of approximately 15 proteins), and the envelope (approximately 200 nm in diameter, composed of a trilaminar membrane containing 11 glycoproteins) (15). When HSV-1 infects a cell, the envelope fuses with the plasma membrane and the double-stranded viral DNA is delivered to the nucleus; here it directs the synthesis of new viral components (24). The capsid, which is assembled in the nucleus, is enveloped initially at the inner nuclear lamina as it exits the nucleus (23). The intracellular site of addition of the tegument, which resides between the capsid and envelope, is, however, not clear. Certain tegument proteins are located near sites within the nucleus where the capsid is constructed, consistent with the notion that they are incorporated into the virion in the nucleus (7, 29). In contrast, other tegument proteins are located primarily in the cytoplasm, consistent with the ideas that the initial nuclear envelopment of the capsid is transient and that tegument proteins are incorporated and final envelopment occurs in cytoplasmic compartments (4, 9, 10, 12, 13, 16, 30, 33).
Such uncertainties are in part a consequence of two limitations of the techniques that have been available to study the life cycles of viruses. First, their destructive nature limits observation of an individual cell or viral component to a single point in time. The resulting discontinuity of observation sacrifices information that may be required to determine the direction of a process—for example, whether a partially enveloped capsid is in the process of gaining or losing an envelope—rendering functional interpretations ambiguous. A second limitation of these techniques is that their temporal resolution is insufficient to resolve rapid processes. Consequently, there may well be processes that are important for virus replication that have not been appreciated because they occur on a time scale (fractions of seconds to minutes) that cannot be resolved by traditional cytological techniques.
To circumvent these limitations, we monitored the fate of a viral structural component with continuity in real time. For this purpose, the coding sequence of the green fluorescent protein (GFP, an autofluorescent protein of 24 kDa from the jellyfish Aeqorea victoria) was incorporated into the viral gene (UL46) that codes for a major tegument protein designated VP11/12. Observations of cells infected with this labeled virus in real time by video microscopy reveal that virus development involves extremely rapid movements of viral components between different regions of the cytoplasm; the behavior of these rapidly translocating particles suggests that the rapid delivery of developing virus or viral components to intracellular sites—to processing stations in the cytoplasm, to the nuclear membrane, and to the plasma membrane—may be an important principle in ordering the course of viral assembly.
MATERIALS AND METHODS
Reagents.
HSV-1 strain D120 and E5 Vero cells were obtained from Paul Olivo, with permission from Neil Deluca. The wild-type HSV-1 KOS strain and Vero cells were obtained from David Leib. LysoTracker-red (Molecular Probes, Eugene, Oreg.) was a gift from Rick Roberts. A polyclonal anti-GFP antibody was from Clontech (Palo Alto, Calif.), and a monoclonal antibody against the major capsid protein VP5 was from Biodesigns (Kennebunk, Maine). Nocodazole, secondary antibodies, and alkaline phosphatase substrate were from Sigma (St. Louis, Mo.).
Construction of GHSV-UL46.
The GFP coding sequence was introduced into the HSV-1 UL46 gene by homologous recombination between infectious HSV-1 DNA and a recombination vector in which the coding sequence of GFP was flanked on both sides by HSV-1 sequences that flank the target site in UL46. The recombination vector was generated by inserting the GFP coding sequence into an endogenous HindIII restriction site in HSV-1 DNA (Fig. 1B).
FIG. 1.

Schematic diagram of the construction of GHSV-UL46. (A) Position in the HSV-1 genome of a 3.6-kb XbaI-NotI segment that contains the UL46 open reading frame. (B) Details of this segment. Because this region contained an unanticipated NotI site (not1-cryptic), a shorter 1.7- to 1.8-kb fragment (from XbaI to NotI-cryptic) was cloned and used to construct the recombination vector (C) by insertion of GFP DNA into a unique HindIII site. Homologous recombination between this vector and infectious HSV-1 DNA was used to generate recombinant virus GHSV-UL46D. Numbers in parentheses indicate positions in base pairs in the HSV-1 sequence (20).
A detailed description of recombination vector construction is as follows. HSV-1 DNA, purified by cesium chloride equilibrium density gradient centrifugation, was digested with XbaI and NotI, and a 1.7-kb fragment containing the 3′ portion of the UL46 coding sequence was separated by agarose gel electrophoresis in low-melting-temperature agarose. The fragment was ligated between the NotI and SpeI sites of a Bluescript KS(+) vector (Stratagene, La Jolla, Calif.) whose HindIII site had been removed previously by excision of an XhoI-BamHI fragment. The 1.7-kb NotI-XbaI HSV-1 fragment was 1.6 kb shorter than expected as a consequence of a NotI site (designated NotI-cryptic in Fig. 1B and C) in this KOS strain DNA that was not predicted by the sequence derived from strain 19 (20). The identity of the 1.7-kb NotI-XbaI piece of HSV-1 DNA was established by its ability to serve as a template for producing an 800-bp PCR product with primers (primers 11 and 12, below) complementary to sequences in this region of the HSV-1 genome, as well by restriction mapping, which showed that the piece contained a single HindIII site at the expected position.
The GFP coding sequence was amplified from a plasmid (pEGFP-N1; Clontech) by PCR using primers (primers 1 and 2, described below) that added a HindIII site to the 5′ end and a diagnostic BfrI site followed by a HindIII site at the 3′ end. The amplified GFP DNA was purified by electrophoresis on low-melting-temperature agarose and ligated into the HindIII site of a modified Bluescript (BS-FS) whose XhoI site had previously been filled in order to shift the reading frame of downstream sites by one base. Plasmids were purified from transfectants that emitted green fluorescence when observed with a fluorescence microscope. This precaution insured that a functional copy of GFP was selected.
The HindIII fragment containing GFP coding sequence was excised and ligated into the HindIII site of the 1.7-kb XbaI-NotI HSV-1 fragment. Inserts of the proper orientation were identified by means of digestion of the diagnostic BfrI site at the 3′ end of the GFP coding sequence and flanking restriction sites of Bluescript. In the final construct, the GFP coding sequence was flanked by 1.2 kb of HSV-1 on one side and approximately 0.6 kb on the other side. To confirm that the reading frame had been preserved, a 1,443-kb BamHI fragment containing 144 nucleotides of UL46 upstream of the GFP coding sequence and the entire GFP coding sequence was excised and ligated into the BamHI site of BS-FS; 39% of the ampicillin-resistant colonies expressed GFP, as judged by fluorescence microscopy, indicating preservation of the reading frame.
The recombination vector was cotransfected with HSV-1 infectious DNA (strain D120, in which ICP4 is deleted [5], purified from infected cells by CsCl equilibrium density gradient centrifugation) into the E5 strain of Vero cells (a strain that is stably transfected with ICP4 [5]). When the resulting virus was assayed on E5 cells, approximately 0.05% of the plaques emitted green fluorescence when observed with a fluorescence microscope. Green plaques were picked and replated three times sequentially. After the third plating, all of the plaques identified in a phase microscope were green when observed in the fluorescence mode (Fig. 2A). Viral stocks were grown from single plaques and purified by differential centrifugation. This recombinant virus is designated GHSV-UL46D.
FIG.2.

Characteristics of GHSV-UL46. (A) Green fluorescent plaques generated by GHSV-UL46 viewed with a fluorescence microscope 4 days after a monolayer of Vero cells was infected with dilute GHSV-UL46. (B) Ethidium bromide-stained agarose gel showing a segment of DNA amplified from wild-type HSV-1 or GHSV-UL46 DNA with primers 9 and 10 (lanes un-cut) and restriction digests of the amplified band cut with BfrI or NotI. The primers for the PCR were complementary to sequences on each side of the targeted site of insertion of GFP (Fig. 1). S, standards of the indicated lengths (base pairs). (C) Western blot showing that partially purified preparations of GHSV-UL46 contain a polypeptide (electrophoretic mobility corresponding to an apparent molecular mass of 110 to 120 kDa) reactive with an antibody specific for GFP that is absent in wild-type HSV-1 (left two lanes). Both preparations contain a polypeptide that reacts with an antibody against the major capsid protein VP5 (right two lanes). (D) Density gradient equilibrium sedimentation illustrating similar buoyant-density distributions of the VP11/12-GFP fusion protein and VP5, the major capsid protein that served as a marker for virions. VP11/12-GFP and VP5 were identified by the staining of a Western blot of the sucrose gradient fractions with specific antibodies against GFP and VP5. The discontinuous gradient was formed from steps (from the bottom) of 77, 50, 40, 30, and 20% (wt/vol) sucrose. (E) One-step growth curve comparing growth of wild-type HSV-1 to that of GHSV-UL46. Subconfluent Vero cells were infected at a multiplicity of 7 PFU/cell. At 3-h intervals, total PFU were measured.
Subsequently, the recombinant gene was moved to a wild-type background by coinfection of Vero cells with the recombinant D120 virus and wild-type HSV-1. Wild-type recombinants, designated GHSV-UL46, were identified as viruses that produced green plaques on Vero cells that do not support the growth of HSV-1 strain D120. Virus from green plaques was subjected to three cycles of plaque purification; the plaques produced by the resulting virus preparation were homogeneously fluorescent.
Virus preparation.
The experiments reported here used virus that was released from monolayers of infected cells, and partially purified by differential centrifugation. The infected cells were scraped into the medium (Dulbecco modified Eagle medium [DMEM] with 10% fetal calf serum) and collected by centrifugation at 2.7 × 103 × g for 20 min. The virus that was released was concentrated from the supernatant by centrifugation at 1.6 × 104 × g for 2 h. After resuspension, this released virus was used in the experiments described here.
Observation of infected cells.
Trypsinized Vero cells were introduced into glass-bottomed wells (18 mm in diameter) in petri dishes in 0.5 ml of medium (DMEM with 10% fetal calf serum) and grown for 12 to 24 h. Each well received approximately 6 × 104 cells; the cells were at 0.4 to 0.8% of confluence at the time of observation. The medium was removed and replaced with 0.3 ml of medium containing virus dilutions to produce the desired multiplicity of infection. At various times after infection, medium was removed by aspiration and replaced with 0.4 ml of medium lacking phenol red. In some cases this medium contained LysoTracker-red (Molecular Probes) at a dilution of 1:15,000. A second coverslip was affixed to the top of the well with vacuum grease, forming a closed, medium-filled chamber. To record movements, the chamber was viewed with a Nikon Diaphot III inverted microscope equipped with a 60× lens (numerical aperture, 1.4) and coupled to a Dage-MTI (Michigan City, Ind.) Sit-66 camera. The camera's output was processed by a Hammamatsu Argus 20 image processor and monitored with a video monitor such that the magnification at the screen was ×5,500. The images were recorded on SVHS video tape with a Sony SVO-9500MD video recorder. The atmosphere surrounding the stage and objective was maintained at 35°C.
Processing of video information.
Sequences from video tape were digitized by means of a Scion frame buffer, converted to movies with IPLAB, and viewed with QuickTime Player, version 5, to determine the paths of particles. For static presentation, frames at 0.5-s intervals were selected and the positions of individual moving particles were marked on consecutive frames. Ambiguities were resolved by reference to the movies that maintained the continuity of the paths of particles. To create the sequential-difference plots showing trajectories in Fig. 7, the MAX function of the National Institutes of Health (NIH) Image image-processing program was sequentially applied to each frame and the previous resultant. This served to display moving particles as sequences of dots on a background of the microscopic field.
FIG. 7.


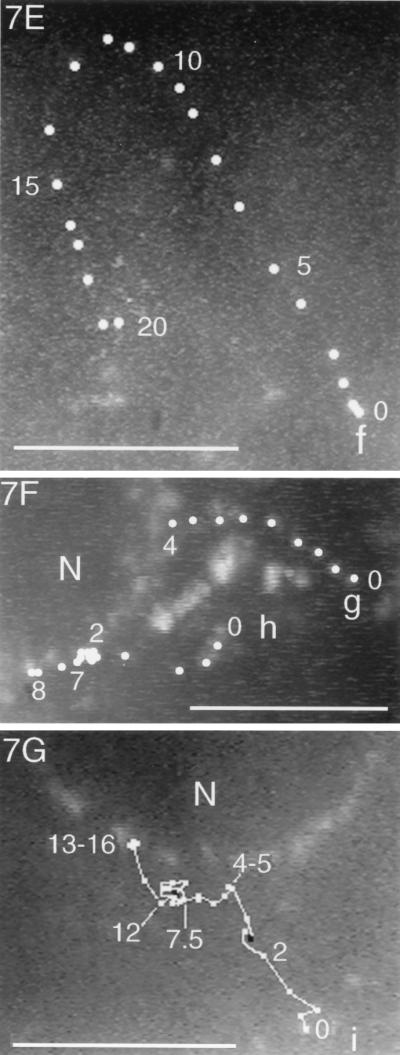

Trajectories of VP11/12-GFP-labeled particles that underwent sustained rapid directional translocations in GHSV-UL46-infected cells. (A) Single video frame showing the initial positions (arrowheads) of four fluorescent particles whose subsequent translocations are shown in panel B. (B and D to J) Series of video images at 0.5-s intervals that were sequentially subtracted so that the trajectories of moving particles appeared as trails of white dots (arrowheads in panel B mark the beginnings of four trails). In panels D to J the trajectories are highlighted. Numbers show the elapsed times (in seconds) from the beginning of observation. (C) Trajectory of particle a (see panels A and B), which transiently fused with other particles at three points (asterisks). The change in colors and symbols demarcate changes in direction. (F and G) Three particles that moved from the cytoplasm to the nuclear membrane. (H to J) Three particles in the same field that moved toward the tip (arrowheads) of a thin cellular process. Black lines indicate transient reversals of direction. Bars, 10 μm. N, nucleus. The video information that is the basis of this figure is available at http://thalamus.wustl.edu/willardlab/hsv.html.
Confocal microscopy.
To produce static confocal pictures, the chamber containing infected cells was inverted and viewed (at ambient temperature) through the 60× objective of an Olympus BX50W1 microscope coupled to a Bio-Rad MRC 1024ES scanning confocal microscope with aperture set at 2. Vertical series of sections through a cell were collected at 20- to 40-nm intervals. Images were flattened with the Bio-Rad LaserSharp program and transferred via NIH Image to Adobe Photoshop. GFP was viewed with the 522DF32 emission filter, and LysoTracker-red was viewed with the 605DF35 filter.
Plaque assays.
Virus was diluted in DMEM with 10% fetal calf serum and assayed on monolayers of Vero or E5 cells. After adsorption for 2 h at 37°C, either 4 ml of low-melting-temperature agarose (0.75%) in DMEM with 2% fetal calf serum or 3 ml of DMEM (10% fetal calf serum) containing human immunoglobulin was added. After 3 to 4 days, medium (or agarose) was removed, cells were rinsed with phosphate-buffered saline (PBS), fixed for 7 min with a solution of methanol-acetic acid (3:1), stained with crystal violet in methanol (50%), and rinsed with H2O. Plaques were counted with the aid of a dissecting microscope.
Buoyant-density analysis.
Approximately 108 PFU of GHSV-UL46, partially purified by differential sedimentation as described above, were applied in a volume of 0.05 ml to the top of a discontinuous sucrose gradient with steps of 77, 50, 40, 30, 20, and 10% (wt/vol) sucrose dissolved in Na-PBS (0.1 M NaPO4, 0.15 M NaCl, pH 7.4) in a 0.65-ml centrifuge tube and subjected to density gradient equilibrium centrifugation for 20 h at 30,000 rpm in a Beckman SW50.1 rotor with adapters. Six 3-drop fractions were collected from the bottom of the tube, diluted with 0.5 ml of Na-PBS, and centrifuged for 5 h in the same rotor at 30,000 rpm. After removal of the supernatants, the pellets were frozen and subsequently dissolved in 0.1 ml of sodium dodecyl sulfate sample buffer, and electrophoresed through a 7.5% Bio-Rad Ready Gel in a Bio-Rad Mini Protean II gel system. The separated polypeptides were transferred to an Immobilon-F (polyvinylidene difluoride) membrane (Millipore, Bedford, Mass.). After the membrane was blocked, it was probed with anti-VP5 (Biodesigns), incubated with a secondary antibody conjugated to alkaline phosphatase (Sigma), and stained with the Sigma FAST substrate for alkaline phosphatase. An unreactive strip of the membrane below the reactive VP5 band was excised and probed similarly with a polyclonal anti-GFP antibody (Clontech). The pieces of the blot were reassembled and digitized by a laser scanner. A second identical blot was probed initially with the anti-GFP antibody; this blot demonstrated that the presumptive VP11/12-GFP band is the only reactive band on the blot.
One-step growth curve.
Vero cells (1.7 × 104 per 11.1-mm-diameter well of a 48-well tissue culture dish) were infected at a multiplicity of infection of 7 with either wild type HSV-1 or GHSV-UL46 in a volume of 0.5 ml. The number of cells was determined by counting cells in equivalent wells that had been fixed with methanol-acetic acid (3:1) and stained with crystal violet in methanol (50%). At 3-h intervals, cells were scraped into the medium, rapidly frozen and thawed three times, exposed to sonic oscillations for 1 min in a bath sonicator, and stored frozen until they were evaluated by a plaque assay on Vero cells, with human immunoglobulin G used to prevent secondary plaque formation.
Primers.
The sequences of primers used in these experiments are the following: 1, GTC AAA GCT TAA GAT GGT GAG CAA GG; 2, CTT GAA GCT TCT TGT ACA GCT CGT CC; 9, ATTA CTC GAG CCT GCG GCT CGT GGC GTC TC; 10, ATTA AAG CTT CCG GCT CCG GCG TCCTTC; 11, ATTA AGA TCT TGA ACG CCT CCG CCC GTG CTG; 12, ATTAACT TAG TCC TCC CCC TGA GCC GCT TTC.
RESULTS
Characteristics of GHSV-UL46.
GHSV-UL46 is a recombinant HSV-1 in which GFP (EGFP, Clontech) is spliced into the tegument protein VP11/12, 10 amino acids from its C terminus. This virus was constructed by insertion of the coding sequence for GFP into the UL46 open reading frame between the codons for threonine 708 and lysine 709 (Fig. 1). Restriction digests of amplified segments of GHSV-UL46 DNA confirmed that GFP DNA was inserted into the UL46 gene (Fig. 2B). When Vero cells were infected with GHSV-UL46, the virus generated green fluorescent plaques, demonstrating that the recombinant protein is expressed during replication (Fig. 2A). The expressed protein reacted with an anti-GFP antibody on Western blots of partially purified GHSV-UL46, generating a novel band corresponding to an apparent molecular mass of 110 to 120 kDa, approximately the sum of VP11/12 (∼90 kDa) and GFP (24 kDa) (Fig. 2C). Together with the evidence that the GFP coding sequence was inserted into the UL46 gene (Fig. 2B), this result provides compelling evidence that the expressed fusion protein is VP11/12-GFP. When partially purified GHSV-UL46 was subjected to equilibrium density gradient sedimentation, VP11/12-GFP manifested the same buoyant-density distribution as the major capsid protein, which served as a marker for virions (Fig. 2D). This indicates that VP11/12 is incorporated into virions. The time course of replication of the recombinant virus in Vero cells was similar to that of the parental virus (Fig. 2E), showing that the expression and incorporation of the recombinant protein did not significantly alter virus development.
Incoming virus-associated VP11/12-GFP decorates the plasma membrane during initial stages of infection.
The fate of the VP11/12-GFP fusion protein was monitored during the course of infection by growing Vero cells on glass coverslips and infecting them with GHSV-UL46. At intervals, coverslips were incorporated into a closed medium-filled chamber maintained at 35°C and observed either by conventional fluorescence microscopy or confocal microscopy. Fifteen minutes after infection at high multiplicity (50 PFU per cell), the cells were decorated with fluorescent particles, presumably virions, adsorbed to their plasma membranes (Fig. 3A). This pattern of decoration persisted for at least 3 h (Fig. 3B), indicating that most of the incoming VP11/12-GFP remains associated with the plasma membrane and is not transported to the nucleus.
FIG. 3.

Flattened confocal images of a living Vero cell 15 min (A) and 3 h (B) after infection with GHSV-UL46 at a high multiplicity (50 PFU/cell) showing attachment of incoming fluorescent virus to the plasma membrane. Bars, 10 μm. N, nucleus.
Appearance and intracellular distribution of newly synthesized VP11/12-GFP during infection.
To study the appearance of newly synthesized VP11/12-GFP, Vero cells were infected at low multiplicities (0.1 to 0.5 PFU/cell) such that most infected cells received only 1 PFU; thus, the incoming virus did not contribute significantly to the fluorescence. Fluorescence was first observed in these cells between 5 and 6 h after infection; the cytoplasm initially acquired a green sheen (Fig. 4A). After 6 h, VP11/12-GFP was predominantly associated with particles with apparent diameters ranging between approximately 0.4 and 1.2 μm. The number, size, and brightness of particles increased for several hours and persisted through observations at 21 h (Fig. 4B to D). Concentrations of particles near the nuclear envelopes of some cells formed a perinuclear ring (Fig. 4D); in contrast, the nucleoplasm was devoid of fluorescent particles throughout the infection.
FIG. 4.

Newly synthesized VP11/12-GFP 7 (A), 11 (B), 12.5 (C), and 19 (D) h after infection at low multiplicity (0.1 to 0.5) with GHSV-UL46. (A to C) Flattened confocal images; (D) conventional micrograph. Bars, 10 μm.
VP11/12-GFP is not associated with lysosomes.
To address the possibility that the fluorescent particles might represent the accumulation of fusion proteins in lysosomes, infected cells were exposed to LysoTracker-red (Molecular Probes), a vital dye that labels lysosomes. The confocal microscopic section in Fig. 5 shows that the green (GFP) fluorescence and red (lysosomal) fluorescence occupy different compartments.
FIG. 5.

Single confocal section of a living Vero cell infected with GHSV-UL46 and exposed to the vital dye LysoTracker-red to label lysosomes 10 h after infection with GHSV-UL46. Bar, 10 μm.
Dynamics of fluorescent particles.
Cells infected with GHSV-UL46 for 6 to 24 h provided a novel dynamic picture of viral development when viewed in real time and recorded by fluorescence video microscopy at a rate of 30 frames per s. The situation is best appreciated by viewing the video data available at http://thalamus.wustl.edu/willardlab/hsv.html. At a given moment, these infected cells contained fluorescent particles in three dynamic states. First, many particles, including those that formed the perinuclear rings, were stationary, suggesting that they are anchored to stationary cellular structures such as the cytoskeleton. These are illustrated by the yellow particles (e.g., particle labeled S) in Fig. 6. This figure shows the superimposition of two video frames, colored red and green, separated by a 2-s interval; particles that did not move during this interval appear yellow. Second, other particles oscillated about a central point with displacements of as much as 1 μm (variegated red-green-yellow particles, e.g., particles labeled O in Fig. 6). Third, some particles underwent rapid directional translocations for distances of several micrometers for periods of more than 1 s. For example, the particle labeled RDT in Fig. 6 moved from the green position to the red position during this 2-s interval. During the course of longer observations (Fig. 7), particles made transitions between these dynamic states (e.g., rapidly translocating particles became oscillating or stationary particles and vice-versa).
FIG. 6.

. Movement of fluorescent particles in a living Vero cell 12 h after infection with GHSV-UL46 at low multiplicity of infection (0.5 PFU/cell). A single digitized video frame was false colored green and superimposed with a second frame (false colored red) taken 2 s later. S, stationary (yellow) particle; O, two oscillating (variegated) particles; RDT, particle that moved directionally from the green position to the red position during this 2-s interval. The image is based on video information available at http://thalamus.wustl.edu/willardlab/hsv.html. Bar, 10 μm.
Trajectories, frequency, and speed of rapid directional translocations.
Because these rapid directional translocations may represent a novel process important for virus development, they were analyzed in detail. The paths of individual rapidly translocating particles were tracked by marking their positions at 0.5-s intervals. Figure 7 illustrates the variety of trajectories; these include relatively straight paths, long curving paths (Fig. 7E), and paths that undergo reversals of direction (Fig. 7D, particle c). The directions of movement relative to intracellular organelles were also diverse; in some cases different particles moved simultaneously toward, away from, and parallel to the nucleus (Fig. 7D, particles c, d, and e).
The frequency of these rapid directional translocations is illustrated in Fig. 7B and D; four particles in each of these fields moved rapidly and directionally during the period of observation, approximately 20 s. The duration of rapid directional translocations varied from several seconds to more than 20 s, during which continuous path lengths as long as 49 μm were recorded. The speed of movement of individual particles varied with time. Figure 8 shows the average speeds during consecutive 0.5-s intervals of the particles whose paths are illustrated in Fig. 7. The maximum speed recorded was more than 5 μm/s, whereas the average speed of the long excursion of particle a, shown in Fig. 7C, was approximately 2 μm/s.
FIG. 8.

Average speeds of fluorescent particles containing VP11/12-GFP during sequential 0.5-s intervals. Panels A to L show speeds for particles a to l, respectively, whose trajectories are shown in Fig. 7.
In cells in which the lysosomes were labeled with LysoTracker-red, the moving green particles were distinct from the red-labeled lysosomes (not shown).
Rapid directional translocations are inhibited by nocodazole.
The addition of nocodazole (10 μg/ml), a drug that causes the depolymerization of microtubules, to cultures 12 h after infection with GHSV-UL46 completely eliminated rapid directional translocations. It did not diminish the frequency of the oscillatory movements.
Rapidly directionally translocating particles have at least three destinations: other fluorescent particles, the nuclear membrane, and the plasma membrane.
Whereas the destinations of many rapidly directionally translocating particles were not apparent (Fig. 7E), the trajectories of other particles indicated specific destinations. First certain particles moved to positions where they coincided with oscillating or stationary fluorescent particles, appearing to merge with them, either transiently or for the duration of observation. For example, the particle labeled RDT in Fig. 6 merged with an oscillating particle whose initial position is marked by the green edge of red particle RDT. Strikingly, certain particles underwent multiple mergers during the course of observation. For example particle a (Fig. 7A to C) moved back and forth between oscillating particles (asterisks in Fig. 7C), merging transiently with them at each extreme of its trajectory.
The trajectories of a second group of particles ended at the nuclear membrane (Fig. 7F and G), where the particles became stationary and contributed to the perinuclear fluorescence.
Third, in long thin processes of cells, fluorescent particles moved persistently toward the extremities, accumulating at the tip. Three such particles are illustrated in Fig. 7H to J.
These three destinations suggest potential functions of the rapidly directionally translocating particles, which are discussed below.
DISCUSSION
Two shortcomings of traditional techniques for studying virus replication—limited temporal resolution and lack of continuity of observation—are addressed by the use of a vital marker such as GFP to label individual components of the virus. This paradigm provides the potential for observation of virus replication (or other macromolecular assembly processes) with continuity in real time from the point of view of each labeled component. Using this approach, several recent studies have demonstrated significant improvement in temporal resolution and continuity of observation (6, 7, 9). For example, Elliott and O'Hare employed time-lapse confocal microscopy to record individual infected cells at 5-min intervals throughout the course of infection with an HSV-1 virus labeled with GFP in VP22, another major tegument protein (9).
The present experiments represent an additional increase in temporal resolution of more than 3 orders of magnitude. They resolve redistributions of viral components that occur on a subsecond time scale inaccessible to traditional cytological techniques. In so doing, they reveal that virus development involves the rapid directional translocations of viral components between different regions of the cytoplasm, presumably mediating processes that are important for replication. Moreover, they permit resolution of the trajectories and, in some cases, the destinations of components undergoing rapid directional translocations; these destinations suggest their functions. We consider here first certain implications of the results with regard to the reporter protein, VP11/12, and second the potential functional significance of rapid directional translocations for virus development.
We employed an HSV-1 virus, GHSV-UL46, in which GFP was incorporated into the tegument protein VP11/12, which served as the reporter protein. VP11/12 is a phosphoprotein with multiple electrophoretically distinct phosphorylation states (16, 32). Its precise function is obscure. It has been reported to augment the function of another tegument protein, VP16, in its well-characterized role as trans-activator of immediate-early genes (16, 21); it is not essential for replication in cultured Vero cells (21). The abundance of this protein (more than 1,000 copies per virion [32]), recommended it as a suitable marker for the tegument, one of the three major compartments of the virus. The value of VP11/12-GFP as a reporter protein depends on its retention of the targeting signals and the behavior of native VP11/12. The following considerations support the conclusion that VP11/12-GFP is targeted correctly. (i) GFP was inserted into the C-terminal portion of VP11/12 with minimal alteration of its sequence. (ii) Newly synthesized VP11/12-GFP became associated with particles, indicating that it was subject to some form of targeting. (iii) The intracellular distribution of newly synthesized VP11/12-GFP is similar to the distribution of native VP11/12 determined by antibody staining of cells infected with HSV-2 (16); this indicates that the fusion protein is targeted to the same intracellular compartments as VP11/12 from HSV-2. (iv) VP11/12-GFP did not accumulate in lysosomes, as might occur if it were mistargeted. (v) VP11/12 associated with an organelle that copurified with virions, had the same buoyant density as virions, and adsorbed to cells like virions. This indicates that VP11/12-GFP retains all of the targeting signals required for its incorporation into the virus. Its behavior is thus likely to reflect closely that of native VP11/12.
In the initial stages of infection, Vero cells exposed to GHSV-UL46 at high multiplicities of infection became decorated with fluorescent particles, presumably marking the attachment of virions to the cell membranes. The decoration persisted through the first 3 h of infection, a period during which the capsid and certain other tegument proteins such as VP16, which is required for transactivation of immediate-early genes, would be expected to migrate to the nucleus. Thus, most of the VP11/12-GFP does not appear to participate in this nuclear migration. This shows that different tegument proteins may experience different fates in the early stages of infection; it also raises the question of how VP11/12 influences the function of VP16, as previous experiments have indicated (16, 21). However, the migration to the nucleus of a small fraction of the VP11/12-GFP would likely not have been detected and cannot be ruled out.
Newly synthesized VP11/12-GFP appeared in the cytoplasm of infected cells between 5 and 6 h after exposure to GHSV-UL46. Its distribution—discrete fluorescent particles widely distributed through the cytoplasm, perinuclear rings, and absence from the nucleoplasm—closely resembled the distribution of VP22, another major protein of the tegument (9). In contrast, two other major tegument proteins, VP13/14 and VP16, are located principally or partially within the nucleus (7, 16).
Observations of infected cells in real time produced a novel dynamic picture of virus development that is best appreciated by viewing the video information available at http://thalamus.wustl.edu/willardlab/hsv.html. This picture reveals that particles containing VP11/12-GFP are engaged in complicated patterns of motion that are evident only when viewed with subsecond temporal resolution. In addition to states of quiescence and oscillation, some particles executed rapid directional translocation over distances of more than 40 μm, attaining velocities in excess of 5 μm per s. These rapid directional translocations were inhibited by nocodazole, a drug that causes the depolymerization of microtubules. This indicates that they require intact microtubules and suggests that the particles are propelled along microtubules by microtubule-dependent motors such as kinesin and dynein.
These rapid directional translocations are novel processes that are likely to be important elements of viral replication. Because it is known that the fluorescent particles contain viral components and that viral assembly is in progress in these infected cells, it is most likely that these rapid directional translocations serve to convey viral components rapidly to destinations that are functionally significant in virus development. An analysis of the movements of individual rapidly directionally translocating particles identified three such destinations:
First, certain rapidly translocating particles moved to positions where they coincided with other fluorescent particles, appearing to merge with them. It is likely that such merging particles engage in physical contact. This is because the depth of field of the microscope lens is similar to the apparent diameters of the particles. Thus, the centers of two merging particles that are in focus are likely to be separated by a distance of less than one particle diameter. In view of this proximity, it is plausible to consider that these merging particles interact functionally. Specifically, the rapidly translocating particles may serve to deliver the developing virus or its components to cytoplasmic processing sites, represented by the particles with which they merge, where sequential steps in the assembly of the virus or the modification of its components are performed. This possibility is illustrated dramatically by particle a (Fig. 7C), which moved back and forth between oscillating particles (asterisks), merging transiently with them at the extremes of its trajectory prior to reversing direction. Such a particle could serve to shuttle developing virus or its components between two processing stations at which consecutive processing or assembly events occur. These considerations suggest that the assembly of the virus in the cytoplasm may be a highly ordered process mediated by the rapidly translocating particles.
A second functional destination is illustrated by those rapidly directionally translocating particles that followed trajectories ending near the nuclear membrane (Fig. 7F and G). These particles presumably account for the perinuclear rings of fluorescence observed in static representations of infected cells (Fig. 4D). The contents of these particles are likely to be involved in functions in viral replication that are specific to the perinuclear region. Among the candidates for such functions are the delivery to the nucleus of materials involved in capsid assembly and events related to (or following) the exit of the capsid from the nucleus, such as removal of the lamina-derived envelope. It is noteworthy that such perinuclear accumulation is a characteristic shared with proteins VP13/14, VP16, and VP22 (7, 9, 16), which together with VP11/12 account for most of the mass of the tegument (19); this region of the cell would thus be a convenient location for the incorporation of these tegument proteins into the virion.
A third class of rapidly translocating particles moved resolutely toward the periphery of long cellular processes (Fig. 7H to J). These most likely serve to transport viral components that are in the final stages of maturation to the plasma membrane in preparation for release from the cell. Previously, static techniques have been used to study such terminal transport in the axons of neurons (3, 11, 17, 22, 27, 31). Electron micrographs have shown that the unenveloped capsid, in close association with microtubules, is transported to the axon terminals, indicating that final envelopment occurs after terminal transport (22). Using rapid confocal microscopy of neurons infected with pseuodorabies virus (a related herpesvirus), a recent study observed GFP-labeled capsids undergoing transport in axons at velocities similar to those reported here (25). The rapidly directionally translocating particles observed here in real time (Fig. 7H to J) are presumably the vehicles that are the basis for this terminal transport process in nonneuronal cells.
These three functions—communication between cytoplasmic processing stations, delivery of components for perinuclear functions, and terminal transport of maturing virions to the plasma membrane—are almost certainly an incomplete inventory of the functions performed by rapid directional translocations. Our experiments identified destinations for only a fraction of the rapidly directionally translocating particles, and only for those particles containing the reporter protein VP11/12-GFP as one of their constituents. There may well be additional rapidly directionally translocating particles lacking VP11/12 that serve additional functions; these could be observed by using viruses labeled with GFP in other reporter proteins.
We presume that the rapidly directionally translocating particles that serve different functions are composed of different collections of proteins, reflecting different stages in the maturation of the virion or its components. For example, those that deliver components to and between cytoplasmic processing stations would be expected to contain a less mature collection of viral components than those serving the function of terminal transport to the membrane. The techniques used here could be extended to define these differences in composition by employing viruses with several different structural proteins labeled with spectrally distinguishable variants of fluorescent proteins. Such multiply labeled viruses could be used to determine how the composition of a rapidly directionally translocating particle changes with time and whether changes occur when two particles appear to merge, as would be expected if they exchange viral components. In this manner the question of where within the cell and at what point in the life cycle different structural components of the virus (e.g., capsid and tegument proteins) become associated could be addressed directly. These techniques should also be useful for studying the assembly and interactions of the components of other viruses and other macromolecular systems.
The revelation of rapid directional translocations by real-time observation of virus development raises the question of their importance for efficient virus replication. If they indeed play an important role, it would be expected that their inhibition by agents such as nocodazole would have a deleterious effect on virus development. It is notable that the data of Avitabile et al. indicate that nocodazole, applied 4 h after infection, inhibits the production of HSV-1 by 50 to 85% in cultured cells (2). This may be a conservative estimate considering that HSV-1 infection has been reported to stabilize microtubules significantly against nocodazole-mediated depolymerization (8). Although nocodazole may also interfere with the delivery of the incoming HSV-1 genetic material to the nucleus (1, 14, 18, 26, 28), this process would have been completed by the time (4 h after infection) that nocodazole was introduced in the experiments of Avitabile et al. (2). These considerations suggest that rapid directional translocations may be critical for efficient virus development.
Acknowledgments
I am grateful to David Leib, Paul Bridgeman, Paul Olivo, Bob Wilkinson, Neil DeLuca, Jeff Lichtman, Paul Taghert, Leslie Hallick, Rick Roberts, and Brenda McClusky for their contributions of essential equipment, reagents, and advice and to David Leib, John Cooper, Larry Salkoff, David Gottlieb, and Margaret Jones for their comments on the manuscript.
A preliminary phase of this work was supported by a grant from the McDonnell Center for Molecular Neurobiology.
REFERENCES
- 1.Aniento, F., N. Emanns, G. Griffiths, and J. Gruenberg. 1993. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol. 123:1373-1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avitabile, E., G. S. Di, M. R. Torrisi, P. L. Ward, B. Roizman, and F. G. Campadelli. 1995. Redistribution of microtubules and Golgi apparatus in herpes simplex virus-infected cells and their role in viral exocytosis. J. Virol. 69:7472-7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnett, E. M., G. D. Evans, N. Sun, S. Perlman, and M. D. Cassell. 1995. Anterograde tracing of trigeminal afferent pathways from the murine tooth pulp to cortex using herpes simplex virus type 1. J. Neurosci. 15:2972-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Card, J. P., L. Rinaman, R. B. Lynn, V. H. Lee, R. P. Meade, R. R. Miselis, and L. W. Enquist. 1993. Pseudorabies virus infection of the rat nervous system; ultrastructural characterization of viral replication, transport and pathogenesis. J. Neurosci. 13:2515-2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donnelly, M., and G. Elliott. 2001. Fluorescent tagging of herpes simplex virus tegument protein VP13/14 in virus infection. J. Virol. 75:2575-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elliott, G., and P. O'Hare. 1998. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 72:6448-6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elliott, G., and P. O'Hare. 1999. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J. Virol. 73:4110-4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs, W. B., B. G. Klupp, H. Granzow, H. J. Rziha, and T. C. Mettenleiter. 1996. Identification and characterization of the pseudorabies virus UL3.5 protein, which is involved in virus egress. J. Virol. 70:3517-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garner, J. A., and J. H. LaVail. 1999. Differential anterograde transport of HSV type 1 viral strains in the murine optic pathway. J. Neurovirol. 5:140-150. [DOI] [PubMed] [Google Scholar]
- 12.Gershon, A. A., D. L. Sherman, Z. Zhu, C. A. Gabel, R. T. Ambron, and M. D. Gershon. 1994. Intracellular transport of newly synthesized varicella-zoster virus: final envelopment in the trans-Golgi network. J. Virol. 68:6372-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Granzow, H., F. Weiland, A. Jons, G. Klupp, A. Karger, and T. C. Mettenleiter. 1997. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: a reassessment. J. Virol. 71:2072-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo-Jie, Y., K. T. Vaughan, R. B. Vallee, and B. Roizman. 2000. The herpes simplex virus 1 UL34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 74:1355-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haarr, L., and S. Skulstad. 1994. The herpes simplex virus type 1 particle: structure and molecular functions. APMIS 102:321-346. [DOI] [PubMed] [Google Scholar]
- 16.Kato, K., T. Daikoku, F. Goshima, H. Kume, K. Yamaki, and Y. Nishiyama. 2000. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145:2149-2162. [DOI] [PubMed] [Google Scholar]
- 17.Kristensson, K., B. Ghetti, and H. M. Wisniewski. 1974. Study on the propagation of herpes simplex virus (type 2) into the brain after intraocular injection. Brain Res. 69:189-201. [DOI] [PubMed] [Google Scholar]
- 18.Kristensson, K., E. Lycke, M. Roytta, B. Svennerholm, and A. Vahlne. 1986. Neuritic transport of herpes simplex virus in rat sensory neurons in vitro. Effects of substances interacting with microtubular function and axonal flow [nocodazole, taxol and erythro-9-3-(2-hydroxynonyl)adenine]. J. Gen. Virol. 67:2023-2028. [DOI] [PubMed] [Google Scholar]
- 19.Leslie, J., F. J. Rixon, and J. McLauchlan. 1996. Overexpression of the herpes simplex virus type 1 tegument protein VP22 increases its incorporation into virus particles. Virology 220:60-68. [DOI] [PubMed] [Google Scholar]
- 20.McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574. [DOI] [PubMed] [Google Scholar]
- 21.McKnight, J. L. C., P. E. Pellett, F. J. Jenkins, and B. Roizman. 1987. Characterization and nucleotide sequence of two herpes simplex virus 1 genes whose products modulate α-trans-inducing factor-dependent activation of α genes. J. Virol. 61:992-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penfold, M. E., P. Armati, and A. L. Cunningham. 1994. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. USA 91:6529-6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roizman, B., and A. E. Sears. 1993. Herpes simplex viruses and their replication, p. 11-69. In B. Roizman, R. Whitley, and C. Lopez (ed.), The human herpesviruses. Raven Press, New York, N.Y.
- 24.Roizman, B., and A. E. Sears. 1990. Herpes simplex viruses and their replication, p. 1795-1842. In B. N. Fields and D. M. Knipe (ed.), Fields virology, 2nd ed. Raven Press, New York, N.Y.
- 25.Smith, G. A., S. P. Gross, and L. W. Enquist. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA 98:3466-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sodeik, B., M. W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun, N., M. D. Cassell, and S. P. Perlman. 1996. Anterograde, transneuronal transport of herpes simplex virus type 1 strain H129 in the murine visual system. J. Virol. 70:5405-5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Topp, K. S., L. B. Meade, and J. H. LaVail. 1994. Microtubule polarity in the peripheral processes of trigeminal ganglion cells: relevance for the retrograde transport of herpes simplex virus. J. Neurosci. 14:318-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward, P. L., W. O. Ogle, and B. Roizman. 1996. Assemblons: nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 70:4623-4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whealy, M. E., J. P. Card, R. P. Meade, A. K. Robbins, and L. W. Enquist. 1991. Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J. Virol. 65:1066-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zemanick, M. C., P. L. Strick, and R. D. Dix. 1991. Direction of transneu-ronal transport of herpes simplex virus 1 in the primate motor system is strain-dependent. Proc. Natl. Acad. Sci. USA 88:8048-8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang, Y., and J. L. McKnight. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu, Z., M. D. Gershon, Y. Hao, R. T. Ambron, C. A. Gabel, and A. A. Gershon. 1995. Envelopment of varicella-zoster virus: targeting of viral glycoproteins to the trans-Golgi network. J. Virol. 69:7951-7959. [DOI] [PMC free article] [PubMed] [Google Scholar]