

Abstract
Lymphocytic chorimeningitis virus (LCMV), the prototype arenavirus, and Lassa virus (LASV), causative agent of Lassa hemorrhagic fever (LHF), belong to the Old World group of the Arenaviridae. Both viruses have extensive strain diversity and significant variations in lethality and pathogenicity for man and experimental animals. We have shown that the infection of rhesus macaques with the WE strain of LCMV affects liver functions, induces hepatocyte proliferation, and causes a rise in IL-6 and soluble TNF receptors (sTNFR) concomitant with a rise in viremia. The levels of IL-6 and sTNFR can serve as an additional diagnostic tool for liver involvement in pathogenesis of arenavirus infection. Mucosal inoculation of rhesus macaques with LCMV-WE can result in attenuated infection with a transient viremia and liver enzyme abnormalities.
The ARM strain of LCMV shares 88% amino acid homology with WE. In contrast to LCMV-WE, ARM strain does not induce manifested disease in monkeys, does not affect liver functions, and does not induce hepatocyte proliferation. Previously we demonstrated that LCMV-ARM infection protected rhesus macaques challenged with LCMV-WE. Here we have shown that the protected animals have no signs of hepatitis and hepatocyte proliferation.
Introduction
Arenaviruses represent a fast growing group of single-stranded, two-segmented, negative-sense RNA viruses producing chronic, asymptomatic infections in their natural rodent hosts. The family Arenaviridae includes two phylogenetic groups of viruses: Old World and New World arenaviruses; the latter group has been further divided into A, B, and C lineages. Among all arenaviruses discovered so far six viruses cause serious, sometimes fatal diseases in man and belong to the Old World group (Lymphocytic choriomeningitis virus, LCMV; and Lassa virus, LASV) and to clade B (Junin virus, JUNV; Machupo virus, MACV; Guanarito virus, GUAV; and Sabia virus, SABV) [3, 8, 9, 45–48, 52]. LCMV, the prototype arenavirus with world-wide distribution, induces in man a spectrum of outcomes ranging from subclinical infections to acute aseptic meningitis and congenital malformations. LASV, the etiological agent of Lassa hemorrhagic fever (LHF), causes up to 500,000 annual infections in West Africa, of which approximately 30% result in disease varying from mild influenza-like illness to lethal LHF. The burden and potential threat from LHF in endemic areas is underestimated [6]. Recent deterioration of social and economic conditions in the endemic areas increased incidence and mortality caused by LASV. This virus is also considered a potential biological weapon [7].
Arenaviruses become a public health threat when human beings contact infected rodents and/or their excreta. The natural host for LCMV is Mus musculus found all over the world. The natural hosts for LASV are multimammate rats (Mastomys spp.) widely distributed throughout sub-Saharan Africa. Arenaviruses are transmitted from rodents to humans by direct contact and/or by mucosal exposure. Population-based studies provide clear evidence that inappropriate food storage, catching, cooking and eating rats correlate with LASV infection and LHF [38, 56]. Feeding monkeys neonatal mice that were infected with LCMV caused outbreaks of hepatitis among captive callitrichid monkeys in the U.S. and in Europe [4, 43, 44, 54].
Prevention of contact between rodents and man has been most effective in controlling Bolivian HF [38]. However, vaccination is a broader and more viable control measure [6, 21, 38, 39, 46]. So far, the only arenavirus vaccine is Candid #1 against Argentine HF. This is a safe and effective live-attenuated arenaviral vaccine and application of this vaccine significantly reduced disease cases in the rural areas of Argentine [17, 18].
LASV and LCMV have extensive strain diversity with remarkable biological variations in pathogenicity and lethality [15, 24, 25, 30, 45–48]. The WE strain of LCMV is highly pathogenic for non-human primates and guinea pigs. Rhesus macaques intravenously (i.v.) infected with the WE strain of LCMV developed a severe LHF-like disease providing a valuable model for study of pathogenesis and vaccine development [48]. We have shown recently that hepatitis was one of the most prominent pathophysiological signs of the LCMV-WE infection in macaques and the infection induced strong hepatocyte proliferation [34, 35].
Here we have shown that LCMV-inducible LHF-like disease in rhesus macaques is associated with a broad functional liver damage as assessed by biochemical tests. In contrast to WE, animals infected with the ARM strain of LCMV had no manifested disease and virus was not detectable in plasma and liver. LCMV-WE-mediated liver pathology was associated with up-regulation of markers of liver regeneration: Ki-67 antigen, IL-6 and soluble TNF receptors (sTNFR). IL-6 and sTNFR in plasma were elevated after detection of virus in blood and increased rapidly during the incubation period.
Although WE is more virulent than ARM, oral infection of rhesus macaques with WE or ARM can sometimes protect animals from lethal hepatitis in challenge experiments. Protected animals had no symptoms of the disease and viremia. All liver functional tests and markers of liver regeneration were in normal ranges in the protected monkeys.
Materials and methods
Virus strains, rhesus macaque inoculations, experimental groups
We and Armstrong 53b (ARM) strains of LCMV and virus inoculations of rhesus macaques have been previously described [32]. Fifteen healthy rhesus macaques were housed in a BSL-2/3 facility and were subsequently subdivided into two groups. Group 1 (long-term study) included five animals that have been partially described in previous publications [34, 51]. In order to compare infections with WE and ARM strains we replaced “rh” in the animal names with WE or ARM. Thus, “rh-ig7a” is “WE-ig7a” and others are “WE-ig7b”, “WE-iv3”, “ARM-iv3”, and “ARM-ig8”. Group 2 (short-term study) included ten animals inoculated with LCMV-WE (1 × 103 PFU, i.v.). One or two animals from this group were euthanized each day from day 1 untill day 7 after infection. Two animals, “Rh1a and Rh1b”, were euthanized on day 1; two animals, “Rh2a and Rh2b”, were euthanized on day 2; one animal, “Rh3”, was euthanized on day 3; two animals, “Rh4a and Rh4b”, were euthanized on day 4; two animals, “Rh6a and Rh6b”, on day 6; and one animal, “Rh7”, was euthanized on day 7.
Sample collections, biochemical liver tests
Blood samples were collected at specified intervals from the saphenous vein and submitted for blood counts and PBMC isolation as described [33]. Liver samples were obtained during biopsy or necropsy at the specified time and used for plaque titration to determine viral load and for RNA extraction. Plasma samples were used to assess liver function by common liver tests [12]. As markers of hepatocellular necrosis aspartate- and alanine-aminominotransferases (AST, ALT) were measured. Gamma-glutamyltransferase (GGTP), total bilirubin, and alkaline phosphatase (AP) were measured in plasma for assessment of cholestatis. Synthetic capacity of liver was evaluated by levels of albumin in plasma.
Ki-67 staining
To detect proliferating cells in liver sections immunochemical staining for Ki-67 nuclear antigen was performed as previously described [34]. Briefly, paraffin-embedded liver sections were deparaffinized, rehydrated, treated with pepsin and with Retrievit™-10 (InnoGenex, San Francisco, CA). Anti-Ki-67 antibody (Zymed Laboratories, Inc., San Francisco, CA) was used at a final dilution of 1:50. After incubating with the secondary biotinylated antibody and blocking of endogenous peroxidase (HPR) activity, streptavidin-HPR conjugate was added and signal was developed with diaminobenzidine. Cell proliferation index was determined as the fraction of cells staining for Ki-67 and expressed as a percentage of cells per field (an average of data from 10 fields). For quantitative purposes, Ki-67 control slides (Zymed #09–0040, tonsil) were included in all staining protocols and used as a standard. Positively stained liver cell nuclei were easily identifiable and clearly distinguished from background. Some variations in background staining (necropsy vs. biopsy, healthy animals vs. diseased animals) did not affect the Ki-67 proliferation index score.
Virus titration, RT/PCR, cytokine ELISAs
Detection of infectious virus in blood samples and liver extracts was performed by plaque assay on monolayers of Vero-E6 cells [33]. Viral RNA from plasma was extracted using QIAamp viral RNA mini spin protocol (Qiagen GmbH, Valencia, CA). RNA from liver tissues was extracted with Trizol (Gibco BRL, Grand Island, NY). RNA was converted to cDNA with AMV RT and random primers using a Roche Molecular Biochemicals kit (Roche Diagnostic Co., Indianapolis, IN) and analyzed using real-time PCR with SYBR green kit from Qiagen as previously described [32, 33]. The detection limit of the real-time PCR was approximately 1,000 copies of viral RNA in 1 ml of plasma.
IL-6 in plasma of rhesus macaques was measured by ELISA using OptEIA™ sets (Pharmingen, San Diego, CA). The detectable IL-6 ELISA level was <5 pg/ml IL-6 was undetectable in plasma of healthy animals. Soluble tumor necrosis factor receptors (sTNFRI and sTNFRII) were measured by ELISA kits (R&D Systems, Minneapolis, MN). In plasma of control animals IL-6 was <5 pg/ml and levels of sTNFRI and sTNFR-II were 150–400 and 130–330 pg/ml, respectively.
Detection of anti-LCMV antibodies
Antibody responses were measured by ELISA and neutralization assays. Viral antigen for ELISA was prepared from serum-free culture medium of Vero cells as described previously [34]. 100 μl of viral antigen in carbonate-bicarbonate buffer was used to cover wells of microtitration plates and incubated overnight at 4 °C. After blocking with 10% FCS in PBS, ten-fold dilutions of monkey sera were added and incubated for 2 h at room temperature. Peroxidase-labeled goat anti-monkey IgG (KPL, Gaithersburg, MD) was used at a final dilution of 1:2000 and substrate solution (Turbo TMB-ELISA, Pierce, Rockford, IL) was added for color development. Neutralization antibody titers were measured by plaque reduction neutralization (PRNT) assay using a constant dose of virus, Vero cell monolayers and serial 1-log dilutions of plasma. Incubation of virus with serum was performed at 37 °C for 1 h. As a control, serum collected before infection was used. End points were calculated from the highest serum dilution inducing 50% plaque reduction.
Challenge experiments
The surviving animals from group 1 (WE-ig7a, WE-ig7b, ARM-iv3, and ARM-ig8) were challenged with 1 × 103 PFU of LCMV-WE (i.v.). Two WE-infected monkeys, WE-ig7a and WE-ig7b, were challenged on day 98 and 112 after the primary infection, respectively. Two ARM-infected monkeys, ARM-iv3 and ARM-ig8, were challenged on day 56 after the primary infection. After challenge animals were observed up to 10 weeks and blood samples and liver biopsies were collected at 2 week intervals. Liver biopsy samples were collected at 4 week intervals. Hepatitis development in challenged animals was evaluated by biochemical liver tests and by measurement of hepatocyte proliferation markers (Ki-67 staining, IL-6, sTNFRI and sTNFRII).
Cell-mediated immunity
CTL and lymphocyte proliferation assays followed previously published protocols [14, 51, 61]. In brief, effector cells (PBMC from infected monkeys) were cultivated in RPMI 1640 with 20% FBS, concanavalin A (Con A) and IL-12 for 3 days, washed and cultivated again for 7 days without Con A. Target cells (autologous B-lymphoblastoid cell lines established by transformation of monkey PBMC with Herpes papio) were infected with recombinant vaccinia (rVV) expressing nucleocapsid (NP) or glycoprotein (GP) genes of LCMV, rVV-NP, rVV-GP [58]. 51Cr-labeled targets cells (2 × 104) were plated in 96-well plates in triplicate for each effector:target ratio, 50:1, 25:1, and 12.5:1. Specific lysis was calculated as follows: [(experimental lysis–spontaneous lysis)/(maximum lysis–spontaneous lysis)] × 100. Spontaneous lysis varied from 10 to 20% and specific lysis was considered positive if it was above mean and standard deviation of background lysis using uninfected or VVwt-infected targets.
For proliferation assay [14] fresh PBMC (105 in 100 μl) were plated in triplicate for each of the different stimuli in 96-well plates. Each well received an additional 100 μl of RPMI 1640 (negative control), RPMI + PHA, 10 μg/ml (mitogen control) or RPMI + inactivated LCMV (WE or ARM, 3 × 105 PFU before inactivation). Plates were set up in duplicate, incubated for 3 days, pulsed with 3H-thymidine and supernatants were harvested 12 hours after labeling. The stimulation index (SI) was determined as the mean number of 3H cpm (counts per minute) incorporated in the presence of LCMV antigen divided by the mean cpm incorporated in the presence of medium control. The SI > 5 was considered as specific response.
Data analysis
Statistical analyses and graphing were performed using the Origin 6.0 package (Microcal Software, Inc., Northampton, MA).
Results
Outcome of LCMV infection of rhesus macaques after primary infection and after challenge
From five monkeys infected with LCMV (Table 1), three animals, WE-ig7a, ARM-iv3, and ARM-ig8, showed no signs of disease. From two diseased animals, WE-iv3 and WE-ig7b, rhesus WE-iv3 had LHF-like manifestations and died on day 11. After necropsy the highest viral titers were found in liver, blood, and spleen. This monkey was a “positive” control for the LCMV-WE viral challenge stock [33].
Table 1.
Main characteristics of rhesus macaques infected and challenged with LCMV
| Monkey* |
|||||
|---|---|---|---|---|---|
| WE-iv3 | WE-ig7a | WE-ig7b | ARM-iv3 | ARM-ig8 | |
| Primary infection** | |||||
| Outcome | D11 | S | S | S | S |
| Hepatitis | + | − | +/− | − | − |
| Ki-67 | + | − | +/− | − | − |
| IL-6/sTNFRs | + | − | +/− | − | − |
| Viremia | + | − | +/− | − | − |
| IgG ELISA | <2 | <2 | 7.2 | 5.4 | 5.8 |
| PRNT | <1 | <1 | 3.3 | 1.2 | <1 |
| SI | ND | <5 | 25/12 | 13/90 | 7/18 |
| CTL (% Spec. Lysis) | ND | 17/21(10) | 47/64(8) | ND | ND |
| Challenge** | |||||
| Outcome | D14 | S | S | S | |
| Hepatitis | + | − | − | − | |
| Ki-67 | + | − | − | − | |
| IL-6, sTNFRs | + | − | − | − | |
| Viremia | + | − | − | − | |
| IgG ELISA | <2 | 6.0 | 5.2 | 5.3 | |
| PRNT | <1 | 2.0 | 1.9 | <1 | |
| SI | ND | 50/8 | 19/82 | 3/15 | |
| CTL (% Spec. Lysis) | 5/12(6) | 47/44(8) | ND | ND | |
Rhesus macaques were infected with WE or ARM strains of LCMV using intravenous (i.v.) or intragastrical (i.g.) routes. Rhesus WE-iv3 received 1 × 103 PFU of WE strain and died on day 11 after infection. Two monkeys, WE-ig7a and WE-7b, were i.g. inoculated with 1 × 107 PFU of LCMV-WE, and two monkeys, ARM-iv3 and ARM-ig8, were infected with LCMV-ARM, 1 × 103 PFU and 1 × 108 PFU using i.v. and i.g. routes, respectively
“Outcome”, D11, death at day 11 after infection. S, animal survived. “Hepatitis” was evaluated by biochemical liver tests and marked as “+” if liver tests were elevated in be-weekly bleeding samples; “+/−”, transient elevation of liver enzymes on week 4 after infection; “−” no biochemical signs of hepatitis. “Ki-67+”, positive staining on Ki-67 antigen of monthly collected liver biopsy samples or necropsy samples (more than 5% of positive nuclei); “+/−”, transient positive staining at week 4 after infection; “−”, less than 5% positively stained nuclei. “IL6/sTNFR+”, detection of IL-6 in plasma and levels of sTNFRI and RII higher than detectable levels (3.0 and 6.5 pg/ml, respectively) in bi-weekly collected plasma samples; “+/−” transient IL-6 detection and sTNFR elevation, at week 4 after infection; “−”, no IL-6, sTNFRs below detectable levels. “Viremia+”, detection of the virus in plasma; “+/−”, transient viremia in plasma samples collected at 4 weeks after infection; “−”, viremia below detectable level, 1.3 log10 PFU/ml. IgG ELISA and plaque neutralization (PRNT) titers expressed as log10 dilutions. For primary infection animals antibody titers are indicated at day of the challenge, 98 and 112 days for rhesus WE-ig7a and WE-ig7b, respectively, and 56 days for ARM-infected monkeys, ARM-iv3 and ARM-ig8. For challenged animals titers are shown at the week 10 after challenge. “SI”, stimulation index evaluated in lymphocyte proliferation assay and expressed as the mean number of radioactivity incorporated in the presence of autologous and heterologous antigens, WE/ARM (see Methods). The highest levels proliferative responses are shown in the table, 4 weeks after primary infection and 4 weeks after challenge. “CTL”, cytotoxic T lymphocyte assay (see Methods for details). Specific lysis values are shown against targets expressing NP and GP antigens, NP/GP. Although the assays were performed at effector: target ratios of 50:1, 25:1, and 12.5:1, only results from the 50:1 ratio are shown. The highest levels of CTL are shown in the table, time points after primary and challenge infection are indicated in brackets. “ND” = not done
Rhesus WE-ig7b became ill at 21 days after infection and developed a transient fever, weight loss and viremia. Liver biopsy samples taken at week 4 after infection were negative for LCMV antigens by immunohistochemistry but gave strong positive RT/PCR signals with primers to the LCMV-WE GP and NP genes (not shown). The disease lasted for two weeks, followed by full recovery associated with development of strong immune responses (Table 1 and Ref. [51]). Before the challenge anti–LCMV antibodies reached 7.2 and 3.3 log10 as detected by ELISA and plaque neutralization assay, respectively. PBMC from this monkey showed 47% and 64% specific lysis against targets infected with VV-NP and VV-GP, respectively (Table 1).
Rhesus ARM-iv3 and ARM-ig8 did not develop clinical symptoms of the disease and the virus was not detected by RT/PCR in any tested plasma and liver biopsy samples. Asymptomatic ARM infection in these animals was associated with development of immune responses. At 10 weeks after the primary infection titers of anti-LCMV IgG antibodies detected by ELISA were 5.4 and 5.8 log10 for rhesus ARM–iv3 and ARM–ig8, respectively. Low titer neutralizing antibodies were found only in i.v.-inoculated monkey, ARM-iv3 (Table 1). Circulating PBMC isolated from ARM-infected animals developed proliferation responses after incubation with specific antigens. Rhesus WE-ig7a did not develop manifested disease and virological (plaque assay, RT/PCR) and serological (ELISA, PRNT) tests were negative. However, at 10 weeks after infection low but reproducible LCMV-specific cytotoxic activities against targets expressing NP and GP antigens were found in this monkey (Table 1).
Animals surviving after the primary infection were challenged with LCMV-WE using lethal i.v. inoculation (1 × 103 PFU). As seen from Table 1, rhesus WE-ig7a developed fever and viremia and died on day 14 with hemorrhagic and hepatic manifestations similar to those found in rhesus WE-iv3. Rhesus monkeys WE-ig7b, ARM-iv3, and ARM-ig8 survived after challenge. The surviving animals did not develop fever or any other symptoms of the disease. Protection of these animals seems to be not associated with induction of neutralizing antibodies and correlated with cell-mediated immunity (Table 1).
LCMV-WE but not LCMV-ARM affects liver functions in rhesus macaques
Postmortem histological studies of LHF patients and monkeys infected with LASV or LCMV-WE showed that the most important pathological changes are hepatic and that liver disease was a necessary, but not sufficient, condition in the events leading to death [33–35, 41, 46–48, 57]. As seen in Fig. 1, on the day of death levels of aminotransferases, AP, GGTP and bilirubin were significantly elevated in the fatally infected WE-iv3 monkey indicating the development of hepatocellular necrosis and cholestasis. Levels of albumin in plasma deeply dropped after the infection. In plasma samples collected from monkeys involved in a short-term study these liver abnormalities were found only in Rh-6a, Rh6-b and Rh-7 animals (not shown).
Fig. 1.
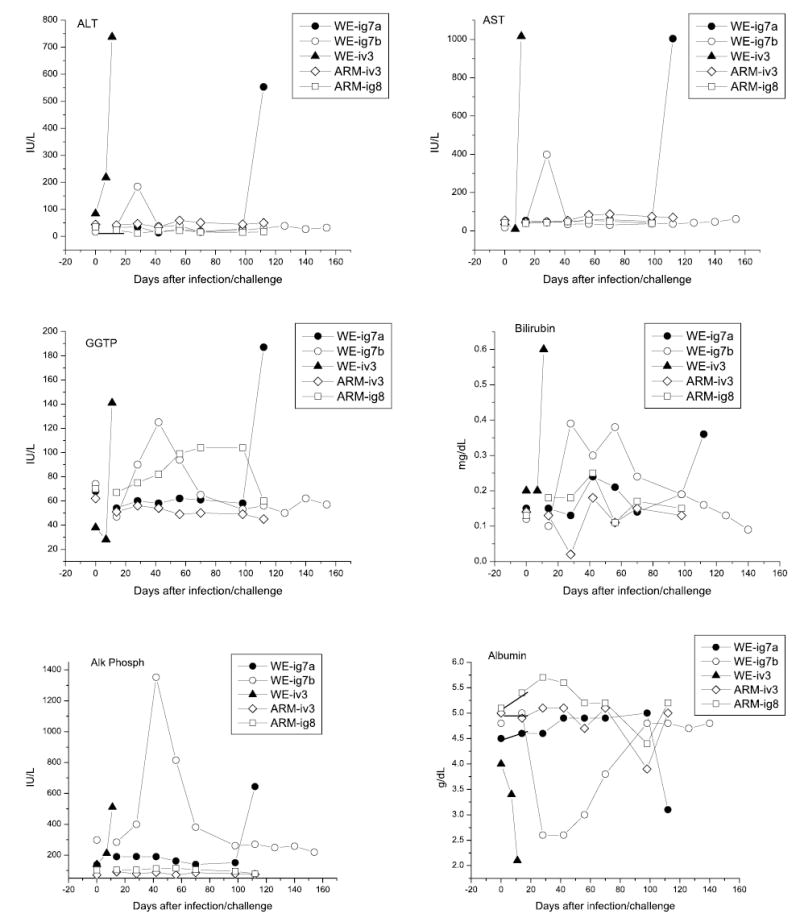
Biochemical liver abnormalities in LCMV-infected rhesus macaques. Markers of hepatocellular necrosis (AST, ALT), cholestatis (GGTP, bilirubin, alkaline phosphatase), and hepatic synthetic activity (albumin) were measured in plasma samples collected from WE- and ARM-infected monkeys. References ranges are 18–82, 22–87, 28–86 IU/L for AST, ALT, and GGTP, respectively; 0.1–<0.3 mg/dL, 30–120 IU/L, and 4.0–6.0 g/dL for bilirubin, alkaline phosphatase and albumin, respectively [12]
In the diseased WE-ig7b animal (long-term study) liver tests showed transient abnormalities. Plasma levels of ALT, AST, GGTP, AP, and bilirubin were elevated on 28–42 days after infection and reversed to normal ranges in 2–3 weeks. Albumin level dropped 2-fold at week 4 and reversed to the normal ranges in 2 months. In WE-ig7a monkey (without apparent disease) and in rhesus macaques infected with ARM strain of LCMV most biochemical liver tests were within the normal ranges.
WE-infection but not ARM-infection is associated with markers of hepatocyte activation
Histopathological studies performed by McCormick et al. [41] provided clear evidence of hepatocellular mitosis in LHF patients. Recently we have found evidence of hepatocyte proliferation in rhesus macaques with fatal LHF-like disease [33]. As seen in Fig. 2, nuclei of rhesus WE-ig7b hepatocytes were positively stained for proliferation Ki-67 antigen. The cell proliferation index before infection was less than 1% and reached at least 25% for biopsy samples taken at the peak of the disease (4 weeks post infection). A month later the number of positively stained nuclei reversed to the background level. In ARM-infected animals the proliferation index was 1–2% before and after infection.
Fig. 2.
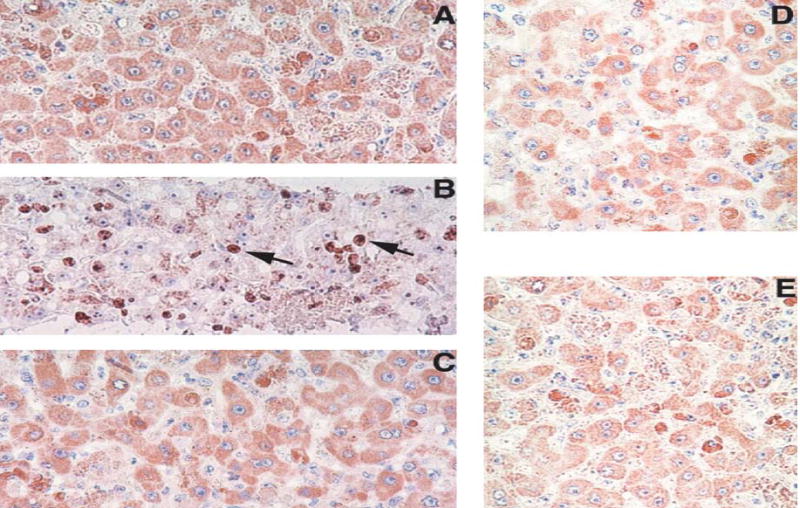
Ki-67 staining of liver sections from rhesus macaques infected with LCMV. A, B, C: Liver biopsies taken from monkey WE-ig7b before infection (A), four weeks after infection (B), and four weeks after challenge (C). D and E: Ki-67 staining of biopsy liver samples taken from ARM-iv3 (D) and ARM-ig8 (E) monkeys at four weeks after infection.
Magnifications: ×300. Note brown staining of Ki-67-positive nuclei (arrowed) in B
A large body of evidence, primarily from murine studies, indicates that IL-6 and sTNFR plays a key role in hepatocyte proliferation after surgery/toxic injury [2, 10, 19, 22, 28, 37, 42, 59, 60, 62]. Virus-mediated hepatocyte proliferation seems to exploit a similar pathway [34, 35, 41, 63]. To evaluate the diagnostic value of IL-6 and soluble TNF receptors in LCMV-inducible liver pathology in rhesus macaques, plasma levels of these markers were measured in two groups of animals, group 1 (long-term study) and group 2 (short-term study). In healthy rhesus macaques IL-6 is not detectable in plasma. In the WE-iv3 animal the level of IL-6 reached 780 pg/ml at the day of death and plasma levels of sTNFRI and sTNFRII were elevated more than 5–10-fold over pre-infection levels (Fig. 3).
Fig. 3.
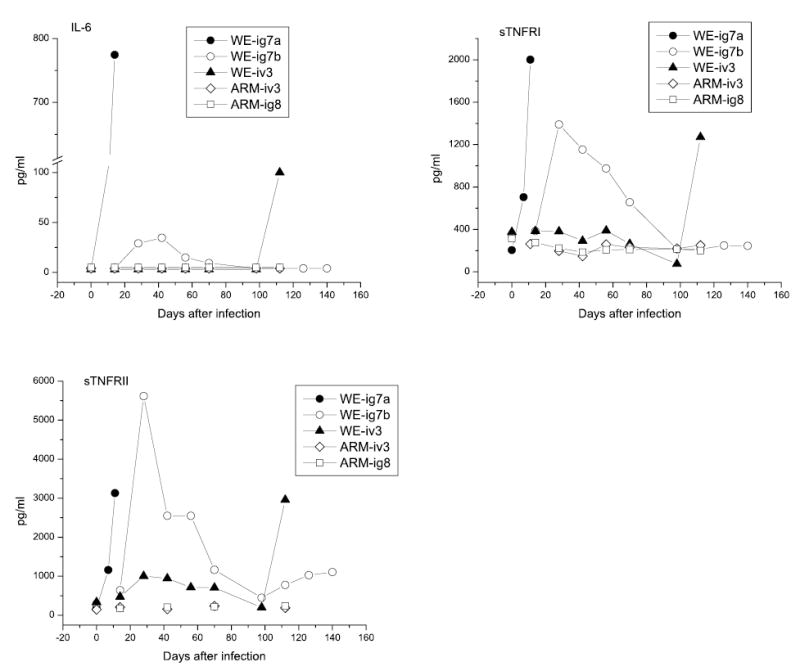
IL-6 and sTNFRs in plasma of LCMV-infected monkeys before and after challenge. Rhesus macaques were infected and challenged as described in “Materials and methods” and in the legend to Fig. 1. Plasma samples were assayed in triplicate and expressed as mean values. The minimum detectable doses were <5.0, 3.0, and 6.5 pg/ml for IL-6, sTNFRI, and sTNFRII, respectively
In rhesus WE-ig7b, the monkey that recovered from i.g. infection, IL-6, sTNFRI and TNFRII were elevated in parallel with viremia, peaking at 4–5 weeks after infection and gradually dropping to undetectable levels (IL-6) or to the normal ranges (sTNFRI and II) in 3 months (Fig. 3). In WE-ig7a (no manifested infection) the measured markers of hepatocyte proliferation were in the normal ranges. In ARM-infected monkeys the infection did not induce detectable levels of IL-6. Plasma levels of sTNFRs were in the normal ranges during the whole observation period.
In a short-term study plasma samples were collected to measure viral RNA load, infectivity, levels of IL-6 and sTNFRs. Semi-quantitative PCR studies showed that LCMV-WE viral RNA in plasma was detected as early as day 4 after infection. As seen in Fig. 4, RNA samples from plasma of monkey Rh-4b had Ct < 35 in semi-quantitative PCR and specificity of PCR was confirmed by nested PCR (not shown). The virus was also recovered from these samples by biological amplification in tissue culture. For monkey Rh–4a the Ct value was well below the detection limit (>37) and the virus was not recovered in tissue culture. The end of incubation period and the appearance of the first clinical symptoms [33] were associated with rapid accumulation of viral RNA and infectious virus in plasma leading to the lethal outcome on day 11 (Fig. 4). The plasma viremia on the day of death was more than 7 log10 PFU/ml.
Fig. 4.
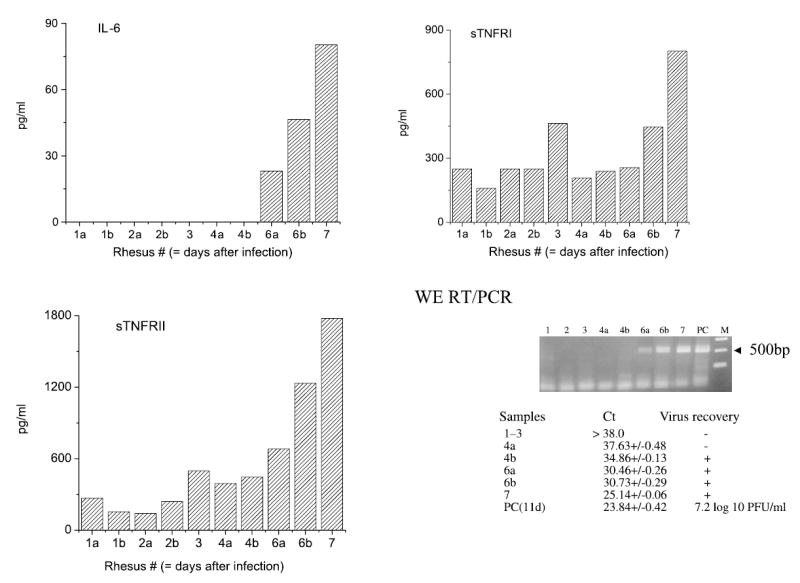
Viremia, IL-6 and soluble TNF receptors at the early stage of LCMV-WE-infection Ten rhesus macaques were i.v. inoculated with 1 × 103 PFU of LCMV-WE and euthanized at day 1 (animals Rh-1a, b); 2 (Rh-2a, b); 3 (Rh-3); 4 (Rh-4a, b); 6 (Rh-6a, b); and day 7 (Rh-7). IL-6 and sTNFRI and II were measured in plasma as described in “Materials and methods”. Viral RNA from plasma was extracted, converted into cDNA and quantitative PCR with GP primers 1–19 and 499–480 (499 bp PCR product) was performed as previously described [33]. Virus was recovered by cultivation on Vero E6 cells and by plaque assay. PC, positive control, viral RNA from plasma of rhesus WE-iv3
IL-6 in plasma of animals from group 2 (a short-term study) was detected on day 6 and increased until 80 pg/ml at day 7. Concentrations of sTNFRI and II were elevated at day 6 after infection. Marginal elevation of these receptors was also noted on day 3. On day 6 and 7, levels of these receptors were 2–4-fold over pre-infection ranges.
Vaccinated rhesus macaques protected against lethal challenge had no hepatitis and hepatocyte activation
In a monkey that survived after lethal challenge (rhesus WE-ig7b, ARM-iv3, and ARM-ig8) all liver tests were in the normal ranges (Fig. 1). IL-6 in plasma was not detectable and levels of soluble TNF receptors were also in the normal ranges. Marginal elevation of sTNFRII was observed in the WE-ig7b monkey. Ki-67 staining of WE-ig7b liver biopsy samples taken at 4 weeks after challenge revealed no more than 1–2% positive nuclei. Liver biopsy samples taken from ARM-vaccinated animals, ARM-iv3 and arARM-ig8, were also negative on Ki-67 nuclear antigen.
Monkey WE-ig7a was not protected against WE challenge and died on day 14 with LHF manifestations. The animal had viremia, virus was detected in liver and all measured markers of liver function, ALT, AST, GGTP, total bilirubin, and alkaline phosphatase were significantly elevated. The level of plasma albumin was lowered (Fig. 1). Markers of hepatocyte proliferation, IL-6, sTNFRI, II (Fig. 2) and Ki-67 (not shown) were strongly elevated in this animal.
Discussion
Postmortem histological studies of LHF patients and LASV-infected experimental animals indicate that the liver is one of the most affected organs participating in a systemic breakdown [16, 20, 24–27, 38–41, 48, 57]. Three categories of morphological changes were found in liver of fatal LHF patients: active hepatocellular injury, continued damage and early recovery, and regenerative phase with high mitotic activity of hepatocytes [41]. Despite the substantial liver dysfunction, there were few indications of microscopic pathology in livers from LHF patients and experimentally infected animals. The relationship between the liver pathology, hematological and endothelial dysfunctions leading to shock and death remains unclear.
Rhesus macaques inoculated with LCMV-WE rapidly develop a fatal LHF-like disease providing a valuable model for human LHF [24, 33, 48]. We have observed substantial hepatocyte proliferation in both LCMV i.v.- and i.g.- inoculated rhesus macaques and have shown that during peak disease at least 25–40% of hepatocyte nuclei in liver biopsy or necropsy samples stained positively for proliferation antigen Ki-67 [34]. In this communication we have shown that the WE, but not the ARM strain of LCMV, affects biochemical, excretory, and perhaps synthetic functions of liver and that only the WE strain induces hepatocyte proliferation accompanied by high levels of IL-6 and sTNFRs in plasma.
LASV infection elevates hepatic aminotransferase levels that seem to correlate with viremia and disease progression in man and in experimental animals [24, 25, 30, 34, 35, 40, 41]. Commonly used tests for evaluation of excretory liver functions include total bilirubin, GGTP, and alkaline phosphatase [12]. We have found elevation of these tests in the WE-infected monkeys (Fig. 1). Clinically no jaundice was observed in LHF patients and bilirubin levels were near normal [41]. In contrast, callitrichid hepatitis in New World monkeys caused by LCMV-CH was associated with jaundice, extremely high levels of GGTP and bilirubin in plasma [43, 44, 53].
Albumin plasma levels dropped in fatally infected LCMV-WE monkeys (Fig. 1). No significant renal failure was mentioned as judged by levels of urea nitrogen and creatinine (not shown). In rhesus WE-ig7b the initial stage of the disease was also associated with decreased concentrations of albumin in plasma. However, during the recovery stage the albumin concentration increased to normal ranges. In fatally infected monkeys endothelial dysfunction seems to be an additional factor contributing to albumin decrease.
The first observation of hepatocellular mitosis in LHF patients was provided by McCormick et al. [41] using H&E staining of liver samples taken within 1–2 hrs after death. As it has been mentioned, LCMV-WE induces a LHF-like disease in rhesus macaques. Using liver biopsy and necropsy samples from WE-infected monkeys we have found that 25–40% of hepatocyte nuclei from liver of LCMV-WE-infected rhesus macaques were positively stained for Ki-67 [34, 35]. Here we showed that recovery from the disease was accompanied by disappearance of Ki-67 staining. Successfully vaccinated and protected monkeys showed no increase in Ki-67 staining after challenge with LCMV-WE (Fig. 2). It suggests that hepatocyte proliferation is marker of the disease in this model.
Among various growth factors and cytokines, IL-6 and its signaling cascade through STAT3, are both important for liver regeneration and for hematopoietic cell proliferation [2, 10, 19, 23, 29, 37, 49, 55]. Severe bone marrow depletion and thrombocytopenia were found in LCMV-WE-infected monkeys [33]. Thrombocytopenia and platelet function depression were also involved in pathogenesis human LHF [11, 20]. It is possible that IL-6 and its signaling are playing a role in hematopoiesis reparation.
TNF-α and its receptors are other important players in initiation of liver regeneration [1, 19, 42, 62]. Interestingly, Mahanty et al. [36] have found that the levels of sTNFRs were significantly higher in patients with fatal LHF than in those with a less severe disease. In HCV patients, levels of sTNFRs did not correlate with TNF-α, but do correlate well with aminotransferase levels [63]. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C viral infection revealed the most notable changes in numerous interferon response genes. In addition, IL6R and TNFR genes were upregulated 5–8 fold [5].
We have not found elevation of TNF-α in plasma of LCMV-WE infected monkeys [34]. It confirms data on LHF patients [36] and our previously published results on human monocytes/macrophages and endothelial cells infected with LASV [32]. Levels of sTNFRs have been elevated in LCMV-WE infected rhesus macaques (Fig. 3). It is not clear whether elevated levels of sTNFRs reflect overexpression or enhanced shedding of these receptors or both. Experiments on mice lacking sTNFRI demonstrated the importance of this type of receptor in liver regeneration [19, 59, 60]. Recently Xia et al. [70] have found that liver regeneration was associated with apoptosis of hepatocytes which is mediated by sTNFRI and proposed that membrane-anchored metalloproteases could play a role in shedding membranous TNFRI.
Results from our short-term study demonstrate that IL-6 and sTNFRs elevation is an early event following viremia (Fig. 4). In contrast to sTNFRs, IL-6 is not detectable in plasma of naïve, healthy rhesus macaques. This suggests that detection of IL-6 in plasma is an accurate pathophysiological marker of liver involvement at the early stage of the disease.
One of the main conclusions of this study is that the infection with two LCMV strains, WE and ARM, has opposite influence on liver pathology in rhesus macaques. These strains also differ in their ability to cause a lethal disease in guinea pigs. Reassortant studies mapped pathogenic potential to the larger (L) of the two viral genomic RNA segments [31, 50]. We have completed the nucleotide sequences for both the WE and ARM strains of LCMV and have found them to be 84% homologous (L RNA) [13, 52]. At the amino acid level similarity between the two viruses is even higher, 87–88% (for Z and L proteins). The most divergent regions were found in the N-terminal parts of the L and Z proteins and these regions are most likely to account for differences in pathogenic potential [13].
Our working hypothesis is that the virus must first replicate to a certain level before tropism and other factors begin to affect virulence. The highest level of replication of LASV and LCMV-WE (but not ARM) was found in liver tissues. Preliminary in vitro results have shown that the WE strain of LCMV replicates more efficiently in primary monkey hepatocytes in comparison with the ARM strain (Lukashevich et al., unpublished observations). Host and viral factors responsible for high replication of WE in liver tissues and mechanisms of virus-inducible hepatocyte proliferation remain to be elucidated.
Acknowledgments
This work was supported by grants RO1-RR13980, R21-AI52367 (to I.S.L.) and AI53619, AI53620 (to M.S.S.) from National Institutes of Health. This work was presented in part at the 11th International Symposium on Viral Hepatitis & Liver Disease, Sydney, Australia, April 6–10, 2003 (abstract 65).
References
- 1.Aggarwal BB, Eessalu TE, Hass PE. Characterization of receptors for human tumor necrosis factor and their regulation by gamma-interferon. Nature. 1985;318:667–667. doi: 10.1038/318665a0. [DOI] [PubMed] [Google Scholar]
- 2.Aldeguer X, Debonera F, Shaked A, Krasinkas AM, Gelman AE, Que X, Zamir GA, Hiroyasu S, Kovalovich KK, Taub R, Olthof KM. Interleukin-6 from intrahepatic cells of bone marrow origin is required for normal murine liver regeneration. Hepatol. 2000;35:40–48. doi: 10.1053/jhep.2002.30081. [DOI] [PubMed] [Google Scholar]
- 3.Archer AM, Rico-Hesse R. High genetic divergence and recombination in arenaviruses from the Americas. Virology. 2000;304:274–281. doi: 10.1006/viro.2002.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asper M, Hofmann P, Osmann C, Funk J, Metzger C, Bruns M, Kaup F-J, Schmitz H, Gunther S. First outbreak of Callitrichid hepatitis in Germany: genetic characterization of the causative lymphocytic choriomeningitis virus strains. Virology. 2001;284:203–213. doi: 10.1006/viro.2001.0909. [DOI] [PubMed] [Google Scholar]
- 5.Bigger CB, Brasky KM, Lanford RE. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C viral infection. J Virol. 2001;75:7059–7066. doi: 10.1128/JVI.75.15.7059-7066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birmingham K, Kenyon G. Lassa fever is unheralded problem in West Africa. Nat Med. 2001;7:788. doi: 10.1038/90892. [DOI] [PubMed] [Google Scholar]
- 7.Borio L, Inglesby T, Peters CJ, Schmaljohn AL, Hughes JM, Jahrling PB, Ksiazek T, Johnson KM, Meyerhoff A, O’Toole T, Ascher MS, Bartlett J, Breman JG, Eitzen EM, Jr, Hamburg M, Hauer J, Henderson DA, Johnson RT, Kwik G, Layton M, Lillibridge S, Nabel GJ, Osterholm MT, Perl TM, Russell P, Tonat K. Hemorrhagic fever viruses as biological weapons. JAMA. 2002;287:2391–2405. doi: 10.1001/jama.287.18.2391. [DOI] [PubMed] [Google Scholar]
- 8.Clegg JCS. Molecular phylogeny of the arenaviruses. Curr Top Microbiol Immunol. 2002;262:1–24. doi: 10.1007/978-3-642-56029-3_1. [DOI] [PubMed] [Google Scholar]
- 9.Clegg JCS, Bowen MD, Buchmeier MJ, Gonzalez J-P, Lukashevich IS, Peters CJ, Rico-Hesse R, Romanowski V (2000) Family Arenaviridae In: Regenmortel MHV van et al. (eds) Virus taxonomy, 7th Report of the ICTV. Academic Press, Orlando
- 10.Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Traub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 11.Cummins D, Fisher-Hoch SP, Walshe KJ, Mackie IJ, McCormick JB, Bennett D, Perez, Farrar B, Machin SJ. A plasma inhibitor of platelet aggregation in patients with Lassa fever. Br J Haematol. 1989;72:543–548. doi: 10.1111/j.1365-2141.1989.tb04321.x. [DOI] [PubMed] [Google Scholar]
- 12.Davern II TJ, Scharschmidt BF (1998) Biochemical liver tests. In: Feldman M, Scharschmidt BF, Sleisenger MH (eds) Sleisenger and Fordtran’s gastrointestinal and liver disease: Pathophysiology/diagnosis/management, Saunders, Philadelphia, pp 345–367
- 13.Djavani M, Lukashevich IS, Salvato MS. Sequence comparison of the large genomic RNA segments of two strains of lymphocytic choriomeningitis virus differing in pathogenic potential for guinea pigs. Virus Genes. 1998;17:151–152. doi: 10.1023/a:1008016724243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Djavani M, Yin C, Lukashevich IS, Rodas J, Rai SK, Salvato MS. Mucosal immunization with S. typhimurium expressing Lassa virus NP cross-protects mice from lethal challenge with LCMV. J Hum Virol. 2001;4:103–108. [PMC free article] [PubMed] [Google Scholar]
- 15.Dutko FJ, Oldstone MB. Genomic and biological variations among commonly used lymphocytic choriomeningitis virus strains. J Gen Virol. 1983;64:1689–1698. doi: 10.1099/0022-1317-64-8-1689. [DOI] [PubMed] [Google Scholar]
- 16.Edington G, White H. The pathology of Lassa fever. Trans R Soc Med Hyg. 1972;66:381–389. doi: 10.1016/0035-9203(72)90268-4. [DOI] [PubMed] [Google Scholar]
- 17.Enria DA, Barrera Oro JG. Junin virus vaccines. Curr Top Med Microbiol Immunol. 2002;263:239–261. doi: 10.1007/978-3-642-56055-2_12. [DOI] [PubMed] [Google Scholar]
- 18.Enria DA, Feuillade MR, Levis SC, Briggiler AM, Ambrosio AM, Saavedra MC, Becker JL, Riera N, Calderon G, Pini N, Sottosanti J, Aviles G, Garcia J, Sabattini M (1999) Impact of vaccination of high risk population for AHF with a live attenuated Junin virus vaccine. In: Saluzzo JJ, Dodet B (eds) Factors in the emergence and control to rodent borne diseases. Elsevier, Amsterdam, pp 273–280
- 19.Fausto N. Lessons from genetically engineered animal models. V. Knocking out genes to study liver regeneration: present and future. Am J Physiol. 1999;277:G917–G921. doi: 10.1152/ajpgi.1999.277.5.G917. [DOI] [PubMed] [Google Scholar]
- 20.Fisher-Hoch SP (1993) Arenavirus pathophysiology. In: Salvato MS (ed) The Arenaviridae Plenum Press, New York, pp 299–323
- 21.Fisher-Hoch SP, Hutwagner L, Brown B, McCormick JB. Effective vaccine for lassa fever. J Virol. 2000;74:6777–6783. doi: 10.1128/jvi.74.15.6777-6783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galun E, Axelrod JH. The role of cytokines in liver failure and regeneration: potential new molecular therapies. Biochim Biophys Acta. 2002;1592:345–358. doi: 10.1016/s0167-4889(02)00326-9. [DOI] [PubMed] [Google Scholar]
- 23.Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–2556. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 24.Jahrling PB, Hesse RA, Eddy GA, Johnson KM, Callis RT, Stephen EL. Lassa virus infection of rhesus monkeys: pathogenesis and treatment with ribavirin. J Infect Dis. 1980;141:580–589. doi: 10.1093/infdis/141.5.580. [DOI] [PubMed] [Google Scholar]
- 25.Jahrling PB, Smith S, Hesse RA, Rhoderick JB. Pathogenesis of Lassa virus infection in guinea pigs. Infect Immunol. 1982;37:771–778. doi: 10.1128/iai.37.2.771-778.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jahrling PB, Frame JD, Smith SB, Monson MH. Endemic Lassa fever in Liberia: III. Characterization of Lassa virus isolates. Trans R Soc Trop Med Hyg. 1985;79:374–379. doi: 10.1016/0035-9203(85)90386-4. [DOI] [PubMed] [Google Scholar]
- 27.Johnson KM, McCormick JB, Webb PA, Smith ES, Elliott LH, King IJ. Clinical virology of Lassa fever in hospitalized patients. J Infect Dis. 1987;155:134–145. doi: 10.1093/infdis/155.3.456. [DOI] [PubMed] [Google Scholar]
- 28.Kovalovich K, DeAngelis R, Li W, Furth E, Gilberto G, Taub R. Increased toxin-induced liver injury and fibrosis in interleukin-6 deficient mice. Hepatol. 2000;31:149–159. doi: 10.1002/hep.510310123. [DOI] [PubMed] [Google Scholar]
- 29.Li W, Liang X, Leu JI, Kovalovich K, Gilberto G, Taub R. Global changes in interleukin-6-dependent gene expression patterns in mouse livers after partial hepatectomy. Hepatol. 2001;33:1377–1386. doi: 10.1053/jhep.2001.24431. [DOI] [PubMed] [Google Scholar]
- 30.Lucia HL, Coppenhaver DH, Harrison RL, Baron S. The effect of an arenavirus infection on liver morphology and function. Am J Trop Med Hyg. 1990;43:93–98. doi: 10.4269/ajtmh.1990.43.93. [DOI] [PubMed] [Google Scholar]
- 31.Lukashevich IS. Generation of reassortants between African arenaviruses. Virology. 1992;188:600–605. doi: 10.1016/0042-6822(92)90514-p. [DOI] [PubMed] [Google Scholar]
- 32.Lukashevich IS, Maryankova R, Vladyko AS, Nashkevich N, Koleda S, Djavani M, Horejsh, Voitenok N, Salvato M. Lassa and Mopeia replication in human monocyte/macrophages and in endothelial cells: different effects on IL-8 and TNF-a gene expression. J Med Virol. 1999;59:552–560. [PMC free article] [PubMed] [Google Scholar]
- 33.Lukashevich IS, Djavani M, Rodas JD, Zapata JC, Usborne C, Emerson C, Mitchen J, Jahrling PB, Salvato MS. Hemorrhagic fever occurs after intravenous, but not after intragastric, inoculation of rhesus macaques with lymphocytic choriomeningitis virus. J Med Virol. 2002;67:171–186. doi: 10.1002/jmv.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukashevich IS, Tikhonov I, Rodas JD, Zapata JC, Yang Y, Djavani M, Salvato MS. Arenavirus-mediated liver pathology: acute LCMV infection of rhesus macaques is characterized by high IL-6 expression and hepatocyte proliferation. J Virol. 2003;77:1727–1737. doi: 10.1128/JVI.77.3.1727-1737.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lukashevich IS, Rodas JD, Tikhonov I, Zapata JC, Yang Y, Djavani M, Salvato MS (2004) Arenavirus-mediated liver pathology in rhesus macaques. In: Viral hepatitis and liver disease. Proc, Eleventh International Symposium. Sydney, Australia
- 36.Mahanty S, Bausch DG, Thomas RL, Goba A, Bah A, Peters J, Rollin PE. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J Infect Dis. 2001;183:1713–1721. doi: 10.1086/320722. [DOI] [PubMed] [Google Scholar]
- 37.Maione D, Di Carlo E, Li W, Musiani P, Modesti A, Peters M, Rose-John S, Della Rocca C, Tripodi M, Lazzaro D, Taub R, Savino R, Ciliberto G. Coexpression of IL-6 and soluble IL-6R causes nodular regenerative hyperplasia and adenomas of the liver. EMBO J. 1998;17:5588–5597. doi: 10.1093/emboj/17.19.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormick J (1999) Lassa Fever. In: Saluzzo JJ, Dodet B (eds) Factors in the emergence and control to rodent borne diseases. Elsevier, Amsterdam, pp 177–195
- 39.McCormick JB, Fisher-Hoch SP. Lassa fever. Curr Top Med Microbiol Immunol. 262:75–109. doi: 10.1007/978-3-642-56029-3_4. [DOI] [PubMed] [Google Scholar]
- 40.McCormick JB, King IJ, Webb PA, Scribner CL, Crave RB, Johnson KM. Lassa fever: effective therapy with ribavirin. N Engl J Med. 1986;314:20–26. doi: 10.1056/NEJM198601023140104. [DOI] [PubMed] [Google Scholar]
- 41.McCormick JB, Walker DH, King IJ, Webb PA, Elliott LH, Whitfield SG, Johnson KM. Lassa virus hepatitis: a study of fatal Lassa fever in humans. Am J Trop Med Hyg. 1987;35:401–407. doi: 10.4269/ajtmh.1986.35.401. [DOI] [PubMed] [Google Scholar]
- 42.Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1987;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 43.Montali RJ, Connolly BM, Armstrong DL, Scanga A, Holmes KV. Pathology and immunohistochemistry of callitrichid hepatitis, an emerging disease of captive New World primates caused by lymphocytic choriomeningitis virus. Am J Pathol. 1995;147:1441–1449. [PMC free article] [PubMed] [Google Scholar]
- 44.Montali RJ, Scanga CA, Pernikoff D, Wessner DR, Ward R, Holmes KV. A common-source outbreak of callitrichid hepatitis in captive tamarins and marmosets. J Infect Dis. 1993;167:946–950. doi: 10.1093/infdis/167.4.946. [DOI] [PubMed] [Google Scholar]
- 45.Parekh BS, Buchmeier MJ. Proteins of lymphocytic choriomeningitis virus: antigenic topography of the viral glycoproteins. Virology. 1986;53:168–178. doi: 10.1016/0042-6822(86)90020-6. [DOI] [PubMed] [Google Scholar]
- 46.Peters CJ (1995)Arenavirus diseases. In: Portfield L (ed) Exotic viral infections. Chapman and Hall Medical, London, pp 227–246
- 47.Peters CJ. Human infection with arenaviruses in the Americas. Curr Top Microbiol Immunol. 2002;262:65–74. doi: 10.1007/978-3-642-56029-3_3. [DOI] [PubMed] [Google Scholar]
- 48.Peters CJ, Jahrling PB, Lui CT, Kenyon RH, McKee KT, Barrera Oro JG. Experimental studies of arenaviral hemorrhagic fevers. Curr Top Microbiol Immunol. 1987;134:5–68. doi: 10.1007/978-3-642-71726-0_2. [DOI] [PubMed] [Google Scholar]
- 49.Peters M, Blinn G, Jostock T, Schirmacher P, Meyer zum Buschenfelde KH, Galle PR, Rose-John S. Combined interleukin 6 and soluble interleukin 6 receptor accelerates murine liver regeneration. Gastroenterol. 2000;119:1663–1671. doi: 10.1053/gast.2000.20236. [DOI] [PubMed] [Google Scholar]
- 50.Riviere Y, Ahmed R, Southern PJ, Buchmeier MJ, Oldstone MBA. Genetic mapping of lymphocytic choriomeningitis virus pathogenicity: virulence in guinea pigs is associated with the L RNA segment. J Virol. 1985;55:704–708. doi: 10.1128/jvi.55.3.704-709.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rodas JD, Lukashevich IS, Zapata JC, Cairo C, Tikhonov I, Djavani M, Pauza CD, Salvato MS. Mucosal arenavirus infection of primates can protect them from lethal hemorrhagic fever. J Med Virol. 2004;72:424–435. doi: 10.1002/jmv.20000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salvato MS, Lukashevich IS (2001) Arenavirus. In: Tidona CA, Darai G (eds) The Springer index of viruses. Springer, Berlin Heidelberg New York
- 53.Salvato MS, Shimomaye EM. The completed sequence of lymphocytic choriomeningitis virus reveals a unique RNA structure and a gene for a zinc finger protein. Virology. 1989;173:1–10. doi: 10.1016/0042-6822(89)90216-x. [DOI] [PubMed] [Google Scholar]
- 54.Stephensen CB, Park JY, Blount SR. cDNA sequence analysis confirms that the etiologic agent of callitrichid hepatitis is lymphocytic choriomeningitis virus. J Virol. 1995;69:1349–1352. doi: 10.1128/jvi.69.2.1349-1352.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Streetz KL, Wustefeld T, Klein C, Manns MP, Trautwein C. Mediators of inflammation and acute phase response in the liver. Cell Mol Biol. 2001;47:661–673. [PubMed] [Google Scholar]
- 56.ter Meulen J, Lukashevich IS, Sidibe K, Inapogui AP, Marx M, Dorlemann A, Yansane ML, Koulemou K, Chang-Claude J, Schmitz H. Hunting of peridomestic rodents and consumption of their meat as possible risk factors for rodent-to-human transmission of Lassa virus in the Republic of Guinea. Am J Trop Med Hyg. 1996;55:661–666. doi: 10.4269/ajtmh.1996.55.661. [DOI] [PubMed] [Google Scholar]
- 57.Walker DH, McCormick JB, Johnson KM, Webb PA, Komba-Kono G, Elliott LH, Gardner JJ. Pathologic and virologic study of fatal Lassa fever in man. Am J Pathol. 1982;107:349–356. [PMC free article] [PubMed] [Google Scholar]
- 58.Whitton JL, Southern PJ, Oldstone MBA. Analysis of the cytotoxic T lymphocyte responses to glycoprotein and nucleoprotein components of lymphocytic choriomeningitis virus. Virology. 1988;162:321–327. doi: 10.1016/0042-6822(88)90471-0. [DOI] [PubMed] [Google Scholar]
- 59.Xia M, Xue SB, Xu CS. Shedding of TNFR1 in regenerative liver can be induced with TNF alpha and PMA. World J Gastroenterol. 2002;8:1129–1133. doi: 10.3748/wjg.v8.i6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamada Y, Webber EM, Kirillova I, Peschon JJ, Fausto N. Analysis of liver regeneration in mice lacking type 1 or type 2 tumor necrosis factor receptor: requirement for type 1 but not type 2 receptor. Hepatol. 1998;28:959–570. doi: 10.1002/hep.510280410. [DOI] [PubMed] [Google Scholar]
- 61.Yin C, Wu M, Pauza CD, Salvato MS. High major histocompatibility complex-unrestricted lysis of simian immunodeficiency virus envelope-expressing cells predisposes macaques to rapid AIDS progression. J Virol. 1999;73:3692–3701. doi: 10.1128/jvi.73.5.3692-3701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zimmermann A. Liver regeneration: the emergence of new pathways. Med Sci Monit. 2002;8:RA53–RA63. [PubMed] [Google Scholar]
- 63.Zylberberg H, Rimaniol AC, Pol S, Masson A, De Groote D, Berthelot P, Bach JF, Brechot C, Zavala F. Soluble tumor necrosis factor receptors in chronic hepatitis C: a correlation with histological fibrosis and activity. J Hepatol. 1999;30:185–191. doi: 10.1016/s0168-8278(99)80060-9. [DOI] [PubMed] [Google Scholar]