

Abstract
The left ventricular outflow tract (LVOTO) malformations, aortic valve stenosis (AVS), coarctation of the aorta (COA), and hypoplastic left heart (HLH) constitute a mechanistically defined subgroup of congenital heart defects that have substantial evidence for a genetic component. Evidence from echocardiography studies has shown that bicuspid aortic valve (BAV) is found frequently in relatives of children with LVOTO defects. However, formal inheritance analysis has not been performed. We ascertained 124 families by an index case with AVS, COA, or HLH. A total of 413 relatives were enrolled in the study, of which 351 had detailed echocardiography exams for structural heart defects and measurements of a variety of aortic arch, left ventricle, and valve structures. LVOTO malformations were noted in 30 relatives (18 BAV, 5 HLH, 3 COA, and 3 AVS), along with significant congenital heart defects (CHD) in 2 others (32/413; 7.7%). Relative risk for first-degree relatives in this group was 36.9, with a heritability of 0.71–0.90. Formal segregation analysis suggests that one or more minor loci with rare dominant alleles may be operative in a subset of families. Multiplex relative risk analysis, which estimates number of loci, had the highest maximum likelihood score in a model with 2 loci (range of 1–6 in the lod-1 support interval). Heritability of several aortic arch measurements and aortic valve was significant. These data support a complex but most likely oligogenic pattern of inheritance. A combination of linkage and association study designs is likely to enable LVOTO risk gene identification. This data can also provide families with important information for screening asymptomatic relatives for potentially harmful cardiac defects.
Keywords: congenital heart defects, hypoplastic left heart syndrome, bicuspid aortic valve, cardiac development, echocardiography, genetics
INTRODUCTION
Congenital heart defects (CHD) are among the most common birth defects, affecting 8–10/1,000 live born infants [Botto et al., 2001; Hoffman and Kaplan, 2002], and are a leading cause of infant mortality [Arias et al., 2003]. The etiology for the majority of these defects is unknown, but both environmental and genetic factors are likely involved in the pathogenesis [Nora, 1993; Burn and Goodship, 1996]. Many structural malformations of the heart occur in the context of more complex birth defect syndromes or as the result of chromosomal disorders [Ferencz et al., 1989; Ferencz, 1993]. Approximately three-fourths of cases occur without other obvious disturbances in other developmental fields, however, and are largely sporadic. Complex genetics combined with the traditional classification of the cardiac malformations by their physiology and anatomy has made investigation of the genetic component of CHD difficult.
Classification of CHD into groups based on the suspected developmental mechanism may help in uncovering the genetic components [Clark, 1996]. The left ventricular outflow tract (LVOTO) obstruction malformations consist of an anatomically varied set of defects across a wide spectrum of clinical severity, including aortic valve stenosis (AVS), bicuspid aortic valve (BAV), coarctation of the aorta (COA), hypoplastic left heart (HLH), Shone complex, and interrupted aortic arch type A (IAAA). Left-sided flow abnormalities account for ~14% of CHD. Rates of isolated LVOTO malformations in Texas for the years 1996–2000 were 7.38/10,000 live births (Kim L. McBride, MD Texas Department of Health unpublished data, 2004). Rates are higher in Europeans compared to Africans and Asians [Botto et al., 2001; Pradat et al., 2003], and there is a relatively consistent excess in males compared to females (ratio 1.5–3) [Muir, 1966; Ferencz, 1993; Samanek, 1994]. The observed sibling recurrence risk for these defects is 1.8–3.2 [Laursen, 1980; Dennis and Warren, 1981; Briard et al., 1984; Maestri et al., 1988; Nora and Nora, 1988]. The paternal recurrence risk is ~3%, while the maternal risk is substantially higher, varying from 8% to 13% [Zetterqvist, 1977; Laursen, 1980; Dennis and Warren, 1981; Briard et al., 1984; Ferencz, 1986; Nora and Nora, 1988; Whittemore et al., 1994].
BAV may be the most common CHD with studies in healthy adults indicating a prevalence of 0.9% [Roberts, 1970; Gray et al., 1995]. This anatomic variant is generally asymptomatic but constitutes an important risk factor for subacute bacterial endocarditis and late onset aortic valve calcification/stenosis in adults [Ward, 2000]. The ~5.5-fold excess of BAV in first-degree relatives of LVOTO cases [McBride et al., 2004] probably represents a mild manifestation of the disease trait and thus, implicates epistasis, environmental factors, or stochastic factors in the expression of severe LVOTO defects.
Accumulating evidence from multiple sources supports a strong genetic involvement in the causation of these defects. This includes multiple case reports detailing recurrences of AVS, COA, and HLH within families [Kojima et al., 1969; Beekman and Robinow, 1985; Menahem, 1990; Grobman and Pergament, 1996; Stoll et al., 1999], high incidence among the chromosomal defects, Turner syndrome (45,X) and Jacobsen syndrome (11q23del) [van Egmond et al., 1988; Mazzanti and Cacciari, 1998; Grossfeld et al., 2004], and occurrence in single gene disorders, such as Holt-Oram syndrome (TBX5) [Basson et al., 1997] and ZIC3 [Ware et al., 2004].
Recent family studies have further elucidated the phenotype of LVOTO malformations, by making use of improvements in echocardiography [Huntington et al., 1997; Loffredo et al., 2004]. Brenner et al. [1989] were the first to note increased frequency of asymptomatic individuals with BAV in families identified by an affected infant with HLH. We have expanded and confirmed the increased incidence of BAV in families with any of the LVOTO malformations, adding further evidence for a genetic etiology and highlighting phenotypic heterogeneity and the importance of searching for mild or asymptomatic anatomical defects among these families [McBride et al., 2004].
Formal analyses of the inheritance pattern in LVOTO malformations have not been performed previously. The widespread availability of echocardiography, its non-invasive nature, and the improvement in technical capabilities allowed use of this modality to study the phenotype of LVOTO families in detail. Our aims were to: (1) define the inheritance pattern by segregation analysis, if possible; (2) provide an estimate of the number of genes involved; and (3) identify quantitative measures of various heart and great vessel structures with high heritability. This information will be critical in the design and power analysis for future gene mapping and identification studies.
METHODS
Ascertainment
The families were ascertained during the time period June 1998 to June 2003 through children seen at Texas Children’s Hospital, which receives almost all of the serious cases of CHD in Southeast Texas (single ascertainment). In a few cases, families were referred from outside institutions after contact was made to our research team. We identified children under the care of our practice group in the Department of Pediatric Cardiology and the Texas Children’s Hospital to identify index cases (probands). Identified cases and their families were approached for enrollment into the study. Informed consent was obtained through a protocol approved by the Baylor College of Medicine Institutional Review Board. Each enrolled family was interviewed in person for detailed information on the prenatal course, the diagnosis, and family history. Additional clinical information was obtained through review of the medical records. Most of the probands in this study have been reported previously [McBride et al., 2004], with this study including 11 more families and 131 more relatives.
Index cases were included if they had a non-syndromic diagnosis of AVS, BAV, COA, HLH, or Shone complex. AVS was defined as a critical narrowing or atresia of the aortic valve at the level of the valve. Narrowing of the outflow tract above (supravalvar) or below (subvalvar), the valve was not included. BAV was defined as a bileaflet aortic valve. COA was defined as a discrete narrowing of the aorta at the level of the ductus arteriosus. Index cases with COA could also have a BAV or a ventricular septal defect. HLH was defined as AVS or atresia, mitral valve stenosis or atresia, hypoplastic left ventricle, and hypoplasia of the ascending arch. Shone complex was defined as multiple areas of narrowing, including mitral atresia, aortic atresia, and COA. Diagnoses were confirmed by echocardiography, cardiac catheterization, or open cardiac surgery.
Phenotypic Evaluation by Echocardiography
All available first-degree relatives were approached for echocardiography. Siblings under the age of 5 were included if an echocardiography exam could be obtained without sedation. If abnormalities were found in the parents of the index case, we attempted to enroll the first-degree relatives of the parents. Echocardiography was free of charge to all participants, with billing covered by research funding and not insurance companies.
Echocardiographic examinations were performed using commercial ultrasound systems (Acuson Sequoia, Siemens Medical, Mountain View, CA, or GE Vivid 7, General Electric Ultrasound, Milwaukee, WI) by a trained pediatric cardiac echo sonographer, a pediatric cardiology fellow, or a pediatric cardiologist. Data were obtained by two-dimensional, spectral Doppler and color flow Doppler imaging. Examinations included M mode measurements of a variety of left ventricle dimensions in diastole and systole, measurements at various locations in the aortic arch, and real time studies of the aortic, mitral, pulmonic, and tricuspid valves. Echocardiograms were recorded both digitally (Echo Vacs 2.30 SP3, VMI Medical, Ottawa, Ontario, Canada) and on 1/2-inch VHS videotape and were subsequently analyzed by use of either internal echocardiographic system software (Acuson Sequoia, Siemens Medical or GE Vivid 7, General Electric Ultrasound) or directly from the stored digital images. A board certified pediatric echocardiographer read all studies. A select, random proportion of the studies were overread by a second pediatric echocardiographer; no changes in interpretation were made in the 55 studies reviewed.
Statistical Analysis
All general statistical analysis and data management were performed in Stata 8.2 (Stata Corporation, College Station, TX). The Bonferroni correction was used in all instances of multiple testing.
Segregation Analysis
The segregation analysis was performed using the multivariate logistic regression model of Karunaratne and Elston [1998], in the SEGREG program from the S.A.G.E. 4.4 software package (Statistical Solutions, Cork, Ireland). Multivariate logistic models allow for better correction in single ascertainment study designs of binary outcomes, as the order of the relatives does not affect the likelihood calculations.
Six models were constructed by postulating three different types of individuals (AA, AB, BB) and three different transmission possibilities (τAA, τAB, and τBB) that describe the probability that a parent of a given type transmits the disease causing ‘‘factor’’ A (or allele in the single gene models) to an offspring. The 4 single gene models specify probabilities of transmitting an A allele to an offspring as τAA = 1.0, τAB = 0.5, and τBB = 0. The susceptibilities of the types are constrained in the single gene models to correspond to the Mendelian gene effect being tested: dominant (β AA = β AB, β BB), codominant (β AA, β AB, β BB), and recessive (β AA, β AB =β BB). An environmental model (no transmission) and a no major gene model (frequency of A =1.0) were also tested. Models for polygenic inheritance were attempted, but no maxima could be found using a variety of starting values for number of genes and allele frequencies. Similarly, attempts to model the sex effect by constraining parent-offspring residuals and adding sex as a covariate in these models never converged in these data. These nested models were compared to a general, or full, model where no parameters were constrained. Correction for ascertainment bias was performed by conditioning on the proband sampling frame. Log likelihood values were generated for each model. Hypothesis testing was performed by the likelihood ratio test (LRT), obtained by subtracting the −2ln (likelihood) of the nested model from the general model. This produces a difference that is part of an asymptotically distributed χ2 distribution with degrees of freedom equal to the difference in independent parameters between the 2 models. Parsimony of the models was assessed by the Akaike Information Criterion (AIC).
Multiplex Relative Risk Analysis
Estimation of the number of loci involved was performed by multiplex relative risk analysis [Schliekelman and Slatkin, 2002]. This statistical technique, a form of complex segregation analysis, employs relative risk and disease prevalence, incorporating all affected individuals in a pedigree, to generate a maximum likelihood estimate of the number of loci contributing to the disease. First, the probability of observing each of the various combinations of affected relatives is calculated, based on the number of affected relatives observed in the set of total relatives. The probabilities of observing each set is multiplied together; for example, the probability of 3 sets of sibships containing 1 affected and 2 unaffected individuals, multiplied by the probability of 6 sets of sibships containing 2 affected and 2 unaffected individuals and so on. The product of the multinomial distributions for the number of affected relatives of the probands forms the likelihood function. Second, to find the maximum likelihood, the parameters that affect the probabilities of observing the sets of relatives are varied. These parameters include the number of loci, the disease penetrance, and the dominance effects at the locus. The disease allele frequencies are calculated based on the prevalence (assumed to be 8/10,000) and the other model parameters.
Quantitative Trait Heritability Analysis
The measurements of left ventricle, valves, and aorta obtained by echocardiography were analyzed for heritability using unaffected siblings of the index cases and the parents. The index cases themselves were excluded from this analysis due to changes in many measurements from secondary effects of the underlying cardiac defect.
Polygenic heritability (H2r) was calculated in the program SOLAR 2.0 [Almasy and Blangero, 1998]. Variance components analysis is very sensitive to deviations from the normal distribution, so all variables were first assessed for normality by the Shapiro-Wilks test in Stata 8.2 (Stata Corporation, College Station, TX). Transformations were attempted for all non-normal distributions; those that could not be successfully transformed were excluded from the heritability analysis. Two analyses were performed for each heart measurement: one for the computed Z score, and one using the raw measurement with covariates sex, age, body mass index, and first order interactions of these covariates. Models were calculated first with all covariates included, and then screened to remove any covariate that did not significantly contribute to the model (P > 0.10).
RESULTS
Based on a simple review of the published sibling recurrence risk data, one may estimate the broad sense heritability of LVOTO lesions (Table I). Crittenden and Falconer [Crittenden, 1961; Falconer, 1965] exploit the simple multi-factorial liability model, but is sensitive to non-normal distribution or differences in variances in the disease families. Edwards [1969] and Reich et al. [1972] use different approaches to correct for these potential sources of bias. It should be noted that there is an approximately twofold increase in the occurrence of all CHD in monozygotic twins and that classical twin studies do not appear useful in this context [Burn and Corney, 1984]. This analysis is incomplete in the sense that extensive data on occurrence in second- and third-degree relatives are not available.
TABLE I.
Sibling Recurrence Relative Risk, and Heritability for LVOTO Malformations
| Heritability
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Probands | Affected sibs | Total sibs | Sibling recurrence (%) | RR (λs) | Crittenden- Falconer | Edwards | Reich | Reference |
| 286 | 13 | 642 | 2.0 | 15 | 0.59–0.74 | 0.61–0.77 | 0.66–0.83 | Nora and Nora [1983] |
| 491 | 26 | 680 | 3.8 | 30 | Briard et al. [1984] | |||
| 406 | 14 | 751 | 1.9 | 14 | Dennis and Warren [1981] | |||
| 624 | 20 | 920 | 2.2 | 16 | Laursen [1980] | |||
| 121 | 13 | 402 | 3.2 | 24 | Maestri et al. [1988] | |||
| 94 | 5 | 283 | 1.8 | 13 | Zetterqvist [1977] | |||
| 113 | 4 | 117 | 3.4 | 34 | 0.81 | 0.77 | 0.90 | Current study |
Descriptive Analysis
Two hundred eleven families participated in acquisition of family history and development of a biological sample research resource. Family history was positive for any type of CHD in an extended family member in 44 of these families (20.9%). Of the 211 families that provided family histories and biologic samples, 124 were enrolled in the echocardiography study. There were no significant differences between enrolled and not enrolled families for index case race, sex, or diagnosis, or for the proportion of multiplex families. A total of 537 individuals including probands and relatives were ascertained from the 124 families in the echocardiography branch of the study. Characteristics of the 124 probands are noted in Table II. Of the 413 relatives enrolled, echocardiography data were unavailable on 41 siblings and 11 parents, resulting in data from 361 relatives, of whom 176 were male, 178 female, and 92 were siblings. The most common reason for the siblings’ lack of data was young age that would have necessitated sedation to adequately perform the echocardiography examination. The average sibship size was 2.07, with 21% of the families having only one child. The percentage of relatives enrolled that were affected with a clinically important cardiac malformation was 7.7% (32/413). LVOTO malformations were noted in 30 of the relatives, of which there were 18 BAV, 5 HLH, 3 COA, and 3 AVS. Two additional relative had other CHD, one with pulmonic valve stenosis (PVS) and one with a hemodynamically significant ventricular septal defect (VSD). Abnormalities were previously noted in all non-BAV defects and for four of the individuals with BAV (symptomatic, leading to valve replacement). Of the 381 first-degree relatives, there were 4 HLH, 2 COA, 2 AVS, 1 PVS, 1 VSD, and 15 BAV, of which 2 were symptomatic. Relatives without echocardiography data were included in the segregation analysis as unknown phenotype.
TABLE II.
Characteristics of the Probands*
| Male | Female | Total | |
|---|---|---|---|
| Ethnicity | |||
| Caucasian | 50 | 23 | 74 |
| Hispanic | 35 | 10 | 45 |
| African American | 4 | 0 | 4 |
| Other | 0 | 1 | 1 |
| Diagnosis | |||
| Aortic valve stenosis | 25 | 9 | 34 |
| Coarctation of the aorta | 44 | 15 | 59 |
| Hypoplastic left heart | 20 | 9 | 30 |
| Bicuspid aortic valve | 0 | 1 | 1 |
| Totals | 89 | 34 | 124 |
One proband (HLH) had unknown sex.
Segregation Analysis
As shown in Table III, all of the 6 nested models gave a poor fit against the general model. Correction of the P-value for multiple testing would elevate the autosomal dominant model to above the cut-off to a P-value of 0.09; this model also has the lowest AIC, making it the most parsimonious of all models. This suggests the existence of one or more minor loci with autosomal dominant inheritance, with low frequency of the causative alleles (~0.002).
TABLE III.
Segregation Analysis Parameters and Models of AVS, BAV, COA, and HLH Phenotypes in 124 Pedigrees
| General | Dominant | Dominant reduced penetrance | Codominant | Recessive | No major gene | Environmental | |
|---|---|---|---|---|---|---|---|
| β AA | −0.7029 | 0.8012 | 0.9647 | 0.932 | 0.7413 | −2.7637 | −0.6516 |
| β AB | −135.16 | 0.8012 | 0.9647 | 0.9221 | 0.7413 | −2.7637 | −10.624 |
| β BB | −49.20 | −3.4059 | −3.3489 | −3.1933 | 0.5188 | −2.7637 | −104.85 |
| prob AA | 0.0451 | 0 | 0 | 0 | 0.0173 | — | 0.1432 |
| prob AB | 0.3346 | 0.004 | 0.0046 | 0.0051 | 0.2284 | — | 0.4705 |
| prob BB | 0.6203 | 0.9959 | 0.9954 | 0.9949 | 0.7543 | — | 0.3862 |
| Freq A | 0.2124 | 0.002 | 0.0023 | 0.0025 | 0.1315 | [1] | 0.3785 |
| τAA | 0.9888 | [1.0] | [1.0] | [1.0] | [1.0] | — | — |
| τAB | 0.4777 | [0.5] | [0.5] | [0.5] | [0.5] | — | — |
| τBB | 0.4522 | [0.0] | [0.0] | [0.0] | [0.0] | — | — |
| PO SS residual | 2.257 | 30.3419 | 30.3106 | 30.3164 | 30.2422 | 0.1952 | 2.133 |
| −2ln (likelihood) | 142.972 | 155.325 | 154.771 | 154.983 | 158.948 | 162.763 | 158.022 |
| AIC | 158.972 | 163.325 | 164.771 | 164.983 | 166.948 | 166.763 | 168.022 |
| Ind. parameters | 8 | 4 | 5 | 5 | 4 | 2 | 5 |
| χ2 | 12.353 | 11.799 | 12.011 | 15.976 | 19.791 | 15.05 | |
| df | 4 | 3 | 3 | 4 | 6 | 3 | |
| P | 0.015 | 0.008 | 0.007 | 0.003 | 0.003 | 0.002 |
] indicates parameter was fixed in analysis; β AA indicates the susceptibility for homozygote for the high risk allele; τAA indicates the probability that a parent with AA type transmits A to an offspring; PO, parent offspring; SS, sib sib; AIC, Akaike information criteria.
Multiplex Relative Risk
Results of the multiplex relative risk analysis are shown in Figure 1. The maximum likelihood estimates for the LVOTO pedigrees are most consistent with the activity of a small number of loci. The highest likelihood occurs for 2 loci, with the likelihood dropping sharply if more loci are inferred. Between 1 and 6 loci are included in the lod-1 support interval, and are thus considered as possible values. The introduction of a dominance variance parameter does not alter this general trend with highest relative likelihood distributed in the region of less than 6 loci (data not shown).
Fig. 1.
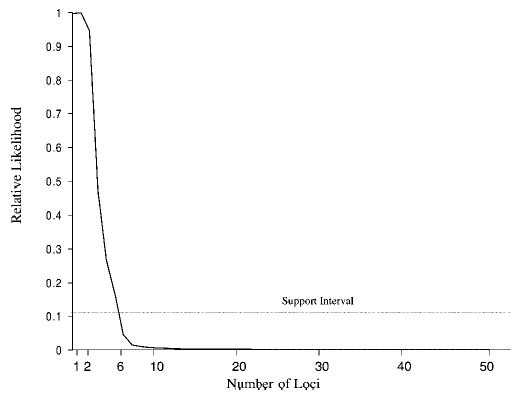
Multiplex relative risk plot of maximum likelihood estimate against number of loci.
Quantitative Trait Heritability Analysis
Heritability analysis results are contained in Table IV. Several of the distributions were not normally distributed, and could not be transformed into a normal distribution. High heritability is demonstrated for several of the aortic arch measures, including the aortic root and sinotubular junction. Heritability of the aortic valve size approaches significance at a P of 0.06. None of the left ventricle measures appears to have strong heritability in this group.
TABLE IV.
Heritability Analysis of Aortic, Valve, and left Ventricle Echocardiography Measurements in 190 First-Degree Relatives of Children With a Left Ventricular Outflow Tract Obstruction Malformation
| Shapiro-Wilk W test for normal dataa |
Heritability
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Location | Variable | Data | Obs | W | V | z | Prob >z | No. | H2r | SE | Covar | P |
| Aorta | Aortic root | Raw | 174 | 0.98322 | 2.218 | 1.82 | 0.034 | 99 | 0.74 | 0.2 | 0.67 | 0.001 |
| Z score | 71 | 0.98997 | 0.624 | 31.026 | 0.847 | 70 | 0.96 | 0.15 | — | 0.016 | ||
| Sinotubular junction | Raw | 168 | 0.98785 | 1.558 | 1.011 | 0.156 | 94 | 0.40 | 0.2 | 0.7 | 0.022 | |
| Z score | 71 | 0.96085 | 2.437 | 1.939 | 0.026 | 70 | 0.72 | 0.34 | — | 0.036 | ||
| Aortic isthmus | Raw | 59 | 0.71138 | 15.479 | 5.899 | 0.0001 | c | |||||
| Z score | 49 | 0.9349 | 3.013 | 2.35 | 0.009 | c | ||||||
| Ascending aorta | Raw | 60 | 0.97804 | 1.194 | 0.381 | 0.351 | 47 | 0.19 | 0.41 | 0.67 | 0.321 | |
| Z score | 35 | 0.97423 | 0.92 | 30.175 | 0.569 | 34 | 0.67 | 0.7 | — | 0.210 | ||
| Transverse arch | Raw | 159 | 0.98412 | 1.942 | 1.51 | 0.066 | 89 | 0.06 | 0.35 | 0.68 | 0.428 | |
| Z score | 64 | 0.97295 | 1.548 | 0.946 | 0.172 | d | ||||||
| Descending aorta | Raw | 66 | 0.97874 | 1.248 | 0.48 | 0.316 | 44 | 0.50 | 0.45 | 0.29 | 0.166 | |
| Z score | 2 | — | — | — | — | c | ||||||
| Valves | Aortic valve | Raw | 174 | 0.98007 | 2.635 | 2.213 | 0.013 | 98 | 0.07 | 0.25 | 0.62 | 0.386 |
| Z score | 72 | 0.97785 | 1.395 | 0.725 | 0.234 | 71 | 0.57 | 0.25 | — | 0.064 | ||
| Pulmonic valve | Raw | 17 | 0.93052 | 1.468 | 0.765 | 0.222 | c | |||||
| Z score | 12 | 0.89183 | 1.807 | 1.153 | 0.124 | c | ||||||
| Mitral valve, lateral cusp | Raw | 28 | 0.98688 | 0.396 | 31.906 | 0.972 | 24 | 0.28 | 0.75 | 0.72 | 0.363 | |
| Z score | 14 | 0.87091 | 2.389 | 1.714 | 0.043 | c | ||||||
| Mitral valve, ap cusp | Raw | 17 | 0.9496 | 1.065 | 0.125 | 0.450 | c | |||||
| Z score | 8 | 0.80052 | 2.779 | 1.896 | 0.029 | c | ||||||
| Left ventricle | LVED dimension | Raw | 171 | 0.98776 | 1.594 | 1.064 | 0.144 | 98 | 0.09 | 0.27 | 0.75 | 0.373 |
| Z score | 145 | 0.9799 | 2.272 | 1.857 | 0.032 | 144 | 0.01 | 0.15 | — | 0.475 | ||
| LVES dimension | Raw | 169 | 0.98301 | 2.19 | 1.788 | 0.037 | d | |||||
| Z score | 144 | 0.99184 | 0.917 | 30.196 | 0.578 | d | ||||||
| LVED thickness | Raw | 162 | 0.97834 | 2.692 | 2.254 | 0.012 | 91 | 0.17 | 0.32 | 0.62 | 0.296 | |
| Z score | 141 | 0.98331 | 1.841 | 1.38 | 0.084 | 139 | 0.20 | 0.2 | — | 0.158 | ||
| LVES thickness | Raw | 161 | 0.06366 | 115.774 | 10.812 | 0.0001 | c | |||||
| Z score | 64 | 0.97215 | 1.594 | 1.009 | 0.157 | d | ||||||
| LVED septal thickness | Raw | 157 | 0.98759 | 1.502 | 0.924 | 0.178 | 92 | 0.15 | 0.23 | 0.6 | 0.253 | |
| Z score | 124 | 0.99441 | 0.553 | 31.328 | 0.908 | 122 | 0.19 | 0.19 | — | 0.164 | ||
| LVES septal thickness | Raw | 157 | 0.98652 | 1.631 | 1.111 | 0.133 | d | |||||
| Z score | 123 | 0.98169 | 1.799 | 1.317 | 0.094 | d | ||||||
| LV mass | Raw | 150 | 0.23255 | 89.297 | 10.184 | 0.0001 | d | |||||
| Z score | 44 | 0.97595 | 1.023 | 0.049 | 0.481 | 43 | 0.67 | 0.58 | — | 0.150 | ||
| Fractional shortening | Raw | 163 | 0.77235 | 28.449 | 7.623 | 0.0001 | c | |||||
| Z score | 52 | 0.94958 | 2.446 | 1.912 | 0.028 | c | ||||||
LV, left ventricle; LVED, left ventricular end diastolic; LVES, left ventricular end systolic.
W is the Shapiro-Wilk statistic, and V is the Shapiro-Francis statistic. Prob >Z is the probability the data are normal; P >0.05 rejects the hypothesis the data are normal.
No. is the number of observations in the pedigrees that could be used for heritability analysis; H2r is the polygenic heritability, and the SE is the H2r standard error, covar is the proportion of variance that is attributable to the covariates sex, age, and body mass index for raw data. P-values are corrected for multiple testing (Bonferroni correction).
Data could not be transformed to a normal distribution.
Large standard errors for heritability calculations, H2r cannot be estimated.
DISCUSSION
The heritable component in the causation of LVOTO malformations appears to be considerable. Examination of previously published estimates of sibling relative risk indicates a substantial tendency to familial clustering (Table I). Using pedigree information in this study, we found that there is at least one affected relative in approximately 20% of families and the first-degree relative risk (λ R) for AVS, COA, and HLH, based on Texas rates of 7.38/10,000 live births is 36.9. The higher first-degree relative risk and sibling recurrence rates observed in this study compared to the literature reflect a lower rate of isolated LVOTO malformations in the general Texas population, and a larger number of single offspring families, respectively. We have previously estimated the heritability of BAV, by itself, at 0.5 [McBride et al., 2004] even though this is an exceptionally common cardiac anomaly. Familial clustering may be due to genetic factors, but may also be due to shared exposures, nutrition, or other environmental components. It is therefore interesting to compare the several methods for assessing LVOTO inheritance.
Formal segregation analysis showed the expected result, that a single major locus is not the most credible scenario. Although most of the Mendelian models were rejected, there is a suggestion that, similar to the breast cancer and prostate cancer stories, there may be a small number of families in which a single gene may be causative. However, pure sporadic or environmental models were also rejected in this sample, indicating the most likely explanation may be the action of more than one disease locus, with or without interacting environmental factors. An estimate of the number of genes involved could not be found with this dataset, as the polygenic models could not find plausible likelihood maxima.
The multiplex relative risk approach is a new complementary method for assessing the most likely genetic mechanisms involved in diseases with complex genetics. Multiplex relative risk assumes a multiplicative action between loci, while the standard polygenic model assumes additive interactions, accounting for the difference in results between the two methods. Multiplicative models may be more realistic, based on previous theoretical work by Risch [1990], who demonstrated that multiplicative models more closely resemble diseases with multiple interacting loci than additive models.
Some disorders with complex inheritance result from variation in several integrated physiological pathways. The genetic components may be best understood by delineation of endophenotypes, which themselves may have several genetic components. It is hoped that by defining phenotypes that underlie the liability to severe disease that one can more easily identify discrete genetic loci that have a major role. Quantitative measures of the aortic arch in this study appear to have a strong genetic influence among the LVOTO families. Genetic influence on aortic arch measurements has also been established in Native American Indians as part of a hypertension and cardiac disease study [Bella et al., 2002]. The lack of genetic influence on measurements of the left ventricle in this study is not surprising, as these measures are more sensitive to age, sex, hypertension, and activity levels, among other confounders. Previous studies have generally been in agreement with our findings [Bielen et al., 1990, 1991], although careful controlling for covariates may be able to identify genetic influences [Bella et al., 2004].
The etiology for the variability in aortic arch measurements from family to family is unknown. One hypothesis could be subtle inherited differences in blood flow through the LVOTO, or possibly a heritable response of the vessel to flow pressures. Supporting evidence has recently been observed in a zebrafish model of obstructed cardiac blood flow. This model demonstrated cardiac and great vessel growth is dependant on intracardiac fluidic forces [Hove et al., 2003].
All these analyses support the concept that genetic components of LVOTO liability are quantitatively important but complex. This raises the issue of what strategies for finding such genes are most likely to be effective. The segregation and multiple relative risk analyses suggest perhaps one, but more likely several, genes may be involved. Traditional approaches of collecting many multiplex families for genome wide linkage studies may not prove fruitful, as the ability to detect a significant signal decreases with the number of genes involved. This has been confirmed by our preliminary analysis of 14 families, where maximum lod scores have not been >3.0. However, finding a single large family with multiple affected members (including the use of echocardiography to find asymptomatic BAV) does have the potential to succeed. Increasing the number of families and use of new very high density SNP marker sets may increase power to detect linkage [Middleton et al., 2004]. Affected sib pair or relative pair approaches may be effective, but collection of adequate samples will require a concerted multicenter collaboration.
The sizeable heritability of several measures of aortic growth suggests that these phenotypes might be used as quantitative trait loci (QTL) in gene mapping experiments. QTL mapping, particularly the variance components methods, are typically more powerful than traditional affected sib pair approaches in common diseases, and are as powerful in lower prevalence diseases when heritability is high (>0.4). This is important in LVOTO malformations, as the ability to collect several hundred sibpairs, even in multicenter collaborations, could be prohibitive, whereas echocardiography studies of family members of an isolated case would not. What is most appealing for the LVOTO malformations is the ability of this approach to accommodate multiple loci simultaneously in a true oligogenic model.
Family-based association studies, built on carefully selected candidate genes and single nucleotide polymorphisms with high minor allele frequency (>0.05), may be powerful enough to find significant results, and the potential for studying interactions between genes is growing with newer statistical methods. Preliminary data from our group has found significant associations in several genes utilizing a candidate pathway approach. Direct sequencing of candidate genes can uncover mutations in affected cases. However, demonstrating causality of the mutation in isolated cases is difficult, as it may not be possible to differentiate a mutation from a rare variant, and in vitro functional studies may not be representative of in vivo action in the early embryo during cardiogenesis.
Improvements in our understanding of normal cardiac development and new very high throughput genotyping and sequencing technologies are now making it feasible to address the genetic etiology of LVOTO malformations [Shendure et al., 2004]. Further progress will require large well-designed studies that integrate carefully collected phenotype data with information about transmission of both common and rare variants.
These data provide important information to first-degree relatives of individuals with AVS, CoA, and HLH. The genetic contribution to these diseases is higher than previously realized. Screening by echocardiography should be offered to relatives, as the chance of finding significant silent malformations is substantial, and the regimen to prevent morbidity and mortality is easy to implement (e.g., prophylactic antiobiotics to prevent endocarditis for BAV). Additional data, and hopefully, elucidation of the genes, will ultimately provide these families with more solid recommendations than current empiric recurrence risk estimates.
Acknowledgments
This research was supported by grants from the March of Dimes and NIH RO1 HD39056 (J.W.B., J.A.T.), and NIH K23 HL70823 and K12 HD41648 (K.L.M.). Some of the results of this project were obtained by using the program package S.A.G.E., which is supported by a U.S. Public Health Service Resource Grant (RR03655) from the National Center for Research Resources.
Footnotes
Kim L. McBride’s present address is Department of Molecular and Human Genetics, Children’s Research Institute, Columbus, OH 43205
Grant sponsor: The March of Dimes; Grant sponsor: NIH; Grant numbers: RO1 HD39056, K23 HL70823, K12 HD41648; Grant sponsor: National Center for Research Resources; Grant number: RR03655.
References
- Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E, Anderson RN, Kung HC, Murphy SL, Kochanek KD. Deaths: Final data for 2001. Natl Vital Stat Rep. 2003;52:1–115. [PubMed] [Google Scholar]
- Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, Renault B, Kucherlapati R, Seidman JG, Seidman CE. Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet. 1997;15:30–35. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- Beekman RH, Robinow M. Coarctation of the aorta inherited as an autosomal dominant trait. Am J Cardiol. 1985;56:818–819. doi: 10.1016/0002-9149(85)91156-7. [DOI] [PubMed] [Google Scholar]
- Bella JN, MacCluer JW, Roman MJ, Almasy L, North KE, Welty TK, Lee ET, Fabsitz RR, Howard BV, Devereux RB. Genetic influences onaortic root size in American Indians: The Strong Heart Study. Arterioscler Thromb Vasc Biol. 2002;22:1008–1011. doi: 10.1161/01.atv.0000017473.78775.f6. [DOI] [PubMed] [Google Scholar]
- Bella JN, MacCluer JW, Roman MJ, Almasy L, North KE, Best LG, Lee ET, Fabsitz RR, Howard BV, Devereux RB. Heritability of left ventricular dimensions and mass in American Indians: The Strong Heart Study. J Hypertens. 2004;22:281–286. doi: 10.1097/00004872-200402000-00011. [DOI] [PubMed] [Google Scholar]
- Bielen E, Fagard R, Amery A. Inheritance of heart structure and physical exercise capacity: A study of left ventricular structure and exercise capacity in 7-year-old twins. Eur Heart J. 1990;11:7–16. doi: 10.1093/oxfordjournals.eurheartj.a059595. [DOI] [PubMed] [Google Scholar]
- Bielen E, Fagard R, Amery A. The inheritance of left ventricular structure and function assessed by imaging and Doppler echocardiography. Am Heart J. 1991;121:1743–1749. doi: 10.1016/0002-8703(91)90021-9. [DOI] [PubMed] [Google Scholar]
- Botto LD, Correa A, Erickson JD. Racial and temporal variations in the prevalence of heart defects. Pediatrics. 2001;107:E32. doi: 10.1542/peds.107.3.e32. [DOI] [PubMed] [Google Scholar]
- Brenner JI, Berg KA, Schneider DS, Clark EB, Boughman JA. Cardiac malformations in relatives of infants with hypoplastic left- heart syndrome. Am J Dis Child. 1989;143:1492–1494. doi: 10.1001/archpedi.1989.02150240114030. [DOI] [PubMed] [Google Scholar]
- Briard ML, Chauvet ML, Le Merrer M, Frezal J. [Epidemiological and genetic study of 3 congenital cardiopathies with neonatal disclosure] Arch Fr Pediatr. 1984;41:313–321. [PubMed] [Google Scholar]
- Burn J, Corney G. Congenital heart defects and twinning. Acta Genet Med Gemellol. 1984;33:61–69. doi: 10.1017/s0001566000007510. [DOI] [PubMed] [Google Scholar]
- Burn J, Goodship J. Developmental genetics of the heart. Curr Opin Genet Dev. 1996;6:322–325. doi: 10.1016/s0959-437x(96)80009-8. [DOI] [PubMed] [Google Scholar]
- Clark EB. Pathogenetic mechanisms of congenital cardiovascular malformations revisited. Semin Perinatol. 1996;20:465–472. doi: 10.1016/s0146-0005(96)80062-0. [DOI] [PubMed] [Google Scholar]
- Crittenden L. Interpretation of familial aggregation based on multiple genetic and environmental factors. Ann NY Acad Sci. 1961;91:769–780. doi: 10.1111/j.1749-6632.1961.tb31106.x. [DOI] [PubMed] [Google Scholar]
- Dennis NR, Warren J. Risks to the offspring of patients with some common congenital heart defects. J Med Genet. 1981;18:8–16. doi: 10.1136/jmg.18.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JH. Familial predisposition in man. Br Med Bull. 1969;25:58–64. doi: 10.1093/oxfordjournals.bmb.a070672. [DOI] [PubMed] [Google Scholar]
- Falconer D. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann Hum Genet. 1965;29:51–71. [Google Scholar]
- Ferencz C. Offspring of fathers with cardiovascular malformations. Am Heart J. 1986;111:1212–1213. doi: 10.1016/0002-8703(86)90034-7. [DOI] [PubMed] [Google Scholar]
- Ferencz C. 1993. Epidemiology of congenital heart disease: The Baltimore-Washington infant study 1981–1989. Mount Kisco, NY: Futura Publishing. xix, 353 p.
- Ferencz C, Neill CA, Boughman JA, Rubin JD, Brenner JI, Perry LW. Congenital cardiovascular malformations associated with chromosome abnormalities: An epidemiologic study. J Pediatr. 1989;114:79–86. doi: 10.1016/s0022-3476(89)80605-5. [DOI] [PubMed] [Google Scholar]
- Gray GW, Salisbury DA, Gulino AM. Echocardiographic and color flow Doppler findings in military pilot applicants. Aviat Space Environ Med. 1995;66:32–34. [PubMed] [Google Scholar]
- Grobman W, Pergament E. Isolated hypoplastic left heart syndrome in three siblings. Obstet Gynecol. 1996;88:673–675. doi: 10.1016/0029-7844(96)00178-0. [DOI] [PubMed] [Google Scholar]
- Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, Cotter F, Jones C. The 11q terminal deletion disorder: A prospective study of 110 cases. Am J Med Genet. 2004;129A:51–61. doi: 10.1002/ajmg.a.30090. [DOI] [PubMed] [Google Scholar]
- Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- Hove JR, Koster RW, Forouhar AS, Acevedo-Bolton G, Fraser SE, Gharib M. Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature. 2003;421:172–177. doi: 10.1038/nature01282. [DOI] [PubMed] [Google Scholar]
- Huntington K, Hunter AG, Chan KL. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol. 1997;30:1809–1812. doi: 10.1016/s0735-1097(97)00372-0. [DOI] [PubMed] [Google Scholar]
- Karunaratne PM, Elston RC. A multivariate logistic model (MLM) for analyzing binary family data. Am J Med Genet. 1998;76:428–437. doi: 10.1002/(sici)1096-8628(19980413)76:5<428::aid-ajmg12>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Kojima H, Ogimi Y, Mizutani K, Nishimura Y. Hypoplastic-left-heart syndrome in siblings. Lancet. 1969;2:701. doi: 10.1016/s0140-6736(69)90418-8. [DOI] [PubMed] [Google Scholar]
- Laursen HB. Some epidemiological aspects of congenital heart disease in Denmark. Acta Paediatr Scand. 1980;69:619–624. doi: 10.1111/j.1651-2227.1980.tb07332.x. [DOI] [PubMed] [Google Scholar]
- Loffredo CA, Chokkalingam A, Sill AM, Boughman JA, Clark EB, Scheel J, Brenner JI. Prevalence of congenital cardiovascular malformations among relatives of infants with hypoplastic left heart, coarctation of the aorta, and d-transposition of the great arteries. Am J Med Genet. 2004;124A:225–230. doi: 10.1002/ajmg.a.20366. [DOI] [PubMed] [Google Scholar]
- Maestri NE, Beaty TH, Liang KY, Boughman JA, Ferencz C. Assessing familial aggregation of congenital cardiovascular malformations in case-control studies. Genet Epidemiol. 1988;5:343–354. doi: 10.1002/gepi.1370050505. [DOI] [PubMed] [Google Scholar]
- Mazzanti L, Cacciari E. Congenital heart disease in patients with Turner’s syndrome. Italian Study Group for Turner Syndrome (ISGTS) J Pediatr. 1998;133:688–692. doi: 10.1016/s0022-3476(98)70119-2. [DOI] [PubMed] [Google Scholar]
- McBride KL, Lewin M, Pignatelli R, Fernbach SD, Combes A, Menesses A, Lam W, Bezold L, Kaplan N, Towbin JA, Belmont JW. Echocardiographic evaluation of asymptomatic parental and sibling cardiovascular anomalies associated with congenital left ventricular outflow tract lesions. Pediatrics. 2004;114:691–696. doi: 10.1542/peds.2003-0782-L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menahem S. Familial aggregation of defects of the left-sided structures of the heart. Int J Cardiol. 1990;29:239–240. doi: 10.1016/0167-5273(90)90227-v. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Pato MT, Gentile KL, Morley CP, Zhao X, Eisener AF, Brown A, Petryshen TL, Kirby AN, Medeiros H, Carvalho C, Macedo A, Dourado A, Coelho I, Valente J, Soares MJ, Ferreira CP, Lei M, Azevedo MH, Kennedy JL, Daly MJ, Sklar P, Pato CN. Genomewide linkage analysis of bipolar disorder by use of a high-density single-nucleotide-polymorphism (SNP) genotyping assay: A comparison with microsatellite marker assays and finding of significant linkage to chromosome 6q22. Am J Hum Genet. 2004;74:886–897. doi: 10.1086/420775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir C. Incidence of congenital heart disease in Singapore. Brit Heart J. 1966;22:243–254. doi: 10.1136/hrt.22.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora JJ. Causes of congenital heart diseases: Old and new modes, mechanisms, and models. Am Heart J. 1993;125:1409–1419. doi: 10.1016/0002-8703(93)91014-6. [DOI] [PubMed] [Google Scholar]
- Nora JJ, Nora AH. Genetic epidemiology of congenital heart diseases. Prog Med Genet. 1983;5:91–137. [PubMed] [Google Scholar]
- Nora JJ, Nora AH. Update on counseling the family with a first-degree relative with a congenital heart defect. Am J Med Genet. 1988;29:137–142. doi: 10.1002/ajmg.1320290117. [DOI] [PubMed] [Google Scholar]
- Pradat P, Francannet C, Harris JA, Robert E. The epidemiology of cardiovascular defects, part I: A study based on data from three large registries of congenital malformations. Pediatr Cardiol. 2003;24:195–221. doi: 10.1007/s00246-002-9401-6. [DOI] [PubMed] [Google Scholar]
- Reich T, James JW, Morris CA. The use of multiple thresholds in determining the mode of transmission of semi-continuous traits. Ann Hum Genet. 1972;36:163–184. doi: 10.1111/j.1469-1809.1972.tb00767.x. [DOI] [PubMed] [Google Scholar]
- Risch N. Linkage strategies for genetically complex traits. I. Multi-locus models. Am J Hum Genet. 1990;46:222–228. [PMC free article] [PubMed] [Google Scholar]
- Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol. 1970;26:72–83. doi: 10.1016/0002-9149(70)90761-7. [DOI] [PubMed] [Google Scholar]
- Samanek M. Boy:girl ratio in children born with different forms of cardiac malformation: A population-based study. Pediatr Cardiol. 1994;15:53–57. doi: 10.1007/BF00817606. [DOI] [PubMed] [Google Scholar]
- Schliekelman P, Slatkin M. Multiplex relative risk and estimation of the number of loci underlying an inherited disease. Am J Hum Genet. 2002;71:1369–1385. doi: 10.1086/344779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Mitra RD, Varma C, Church GM. Advanced sequencing technologies: Methods and goals. Nat Rev Genet. 2004;5:335–344. doi: 10.1038/nrg1325. [DOI] [PubMed] [Google Scholar]
- Stoll C, Alembik Y, Dott B. Familial coarctation of the aorta in three generations. Ann Genet. 1999;42:174–176. [PubMed] [Google Scholar]
- van Egmond H, Orye E, Praet M, Coppens M, Devloo-Blancquaert A. Hypoplastic left heart syndrome and 45X karyotype. Br Heart J. 1988;60:69–71. doi: 10.1136/hrt.60.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C. Clinical significance of the bicuspid aortic valve. Heart. 2000;83:81–85. doi: 10.1136/heart.83.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware SM, Peng J, Zhu L, Fernbach S, Colicos S, Casey B, Towbin J, Belmont JW. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74:93–105. doi: 10.1086/380998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittemore R, Wells JA, Castellsague X. A second-generation study of 427 probands with congenital heart defects and their 837 children. J Am Coll Cardiol. 1994;23:1459–1467. doi: 10.1016/0735-1097(94)90392-1. [DOI] [PubMed] [Google Scholar]
- Zetterqvist P. Recurrence risk in CHD. Circulation. 1977;55:555–556. doi: 10.1161/01.cir.55.3.555. [DOI] [PubMed] [Google Scholar]