

Summary
Phosphorylation of translation initiation factor 2α (eIF2α ) coordinates a translational and transcriptional program known as the integrated stress response (ISR), which adapts cells to endoplasmic reticulum (ER) stress. A screen for small molecule activators of the ISR identified two related compounds that also activated sterol-regulated genes by blocking cholesterol biosynthesis at the level of CYP51. Ketoconazole, a known CYP51 inhibitor, had similar effects, establishing that perturbed flux of precursors to cholesterol activates the ISR. Surprisingly, compound-mediated activation of sterol-regulated genes was enhanced in cells with an ISR-blocking mutation in the regulatory phosphorylation site of eIF2α . Furthermore, induction of the ISR by an artificial drug-activated eIF2α kinase reduced the level of active sterol regulatory element binding protein (SREBP) and sterol-regulated mRNAs. These findings suggest a mechanism by which interactions between sterol metabolism, the ISR, and the SREBP pathway affect lipid metabolism during ER stress.
Introduction
Phosphorylation of the α subunit of eukaryotic translation initiation factor 2 on serine 51 adapts mammalian cells to a variety of stressful conditions (Hinnebusch, 1994; Dever, 2002). eIF2α phosphorylation promotes a stress-resistant state by global attenuation of protein synthesis, which reduces the load of unfolded proteins on chaperone networks and diverts amino acids from energetically costly protein synthesis to other metabolic pathways. In addition to these general effects on protein and amino acid metabolism, eIF2α phosphorylation also activates a gene-expression program that depends in part on the translational upregulation of the transcription factor ATF4 (Harding et al., 2000a; Lu et al., 2004a). This stereotyped response, centered on eIF2α phosphorylation, is activated by diverse stressful conditions and is therefore referred to as an integrated stress response (ISR) (Harding et al., 2003).
Four different kinases are known to phosphorylate eIF2α , and each responds to a distinct upstream activation signal. Amino acid starvation activates the ISR through GCN2, a kinase conserved from yeast to mammals (Hinnebusch and Natarajan, 2002). Viral infection and iron deficiency activate the ISR through the vertebrate-specific kinases PKR (Kaufman, 2000) and HRI (Chen, 2000), respectively, whereas stress from accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER stress) activates the ISR through the ER-localized PERK kinase (Harding et al., 1999).
A single point mutation that precludes eIF2α phosphorylation (eIF2α S51A) sensitizes cells to a wide variety of stressful conditions (Scheuner et al., 2001), whereas loss of specific eIF2α kinase function sensitizes cells selectively to cognate stress. The phenotype associated with loss of the ER-stress-inducible eIF2α kinase PERK is especially severe, as it leads to dysfunction and destruction of multiple secretory cells (Harding et al., 2001; Zhang et al., 2002). The ISR is thus important for dealing with physiological levels of ER stress, and it constitutes one of the three known components of the unfolded protein response (UPR) (Mori, 2000). The UPR’s two other components are mediated by the ER-membrane-associated kinase IRE1 and its downstream effector, the transcription factor XBP-1, and by the ER-membrane bound transcription factor ATF6. Collectively PERK, IRE1, and ATF6 regulate the load of ER client proteins and the cellular apparatus that copes with that load (Mori, 2000; Patil and Walter, 2001; Kaufman, 2002).
The signaling pathway that controls sterol metabolism in mammalian cells is also organized around an ER nexus and is known to share components with the ER-stress-responsive pathways described above (Brown et al., 2000; Ye et al., 2000). Furthermore, there is mounting evidence for a measure of overlap in the readouts of pathways that respond to ER stress and sterol depletion (Cox et al., 1997; Ron and Hampton, 2004; Sriburi et al., 2004). Here we report on a screen for small molecule activators of the ISR that revealed hitherto unsuspected links between sterol synthesis, sterol-mediated signaling, and the ISR.
Result
Identification of small molecule activators of the ISR
To screen for compounds that activate the ISR, a reporter cell line was created by stably transfecting CHO-K1 cells with a fusion of the regulatory sequences from the ISR-inducible CHOP gene (Novoa et al., 2001) and a luciferase reporter (Figure 1A). The reporter gene was activated by thapsigargin and tunicamycin, agents that cause ER stress and activate the ISR through PERK, and by proteotoxic/oxidative stress induced by sodium arsenite, as expected (Figure 1B). Three thousand compounds from a space-filling combinatorial library (Khersonsky et al., 2003; Uttamchandani et al., 2005) were screened at a concentration of 10 μM in a 96-well format. Two structurally related compounds, TGD31 and TGD45, reproducibly induced the reporter ~2-fold when added to the culture media (data not shown). Benzoyl-modified derivatives of these compounds (NH2→ NHBz) with increased cell permeability were synthesized. TGD31BZ and TGD45BZ proved to be more potent activators of the ISR reporter gene than the parent compounds (Figures 1C and 1D) and, because of their indistinguishable activity profile and structural similarity, were used interchangeably for subsequent studies.
Figure 1.
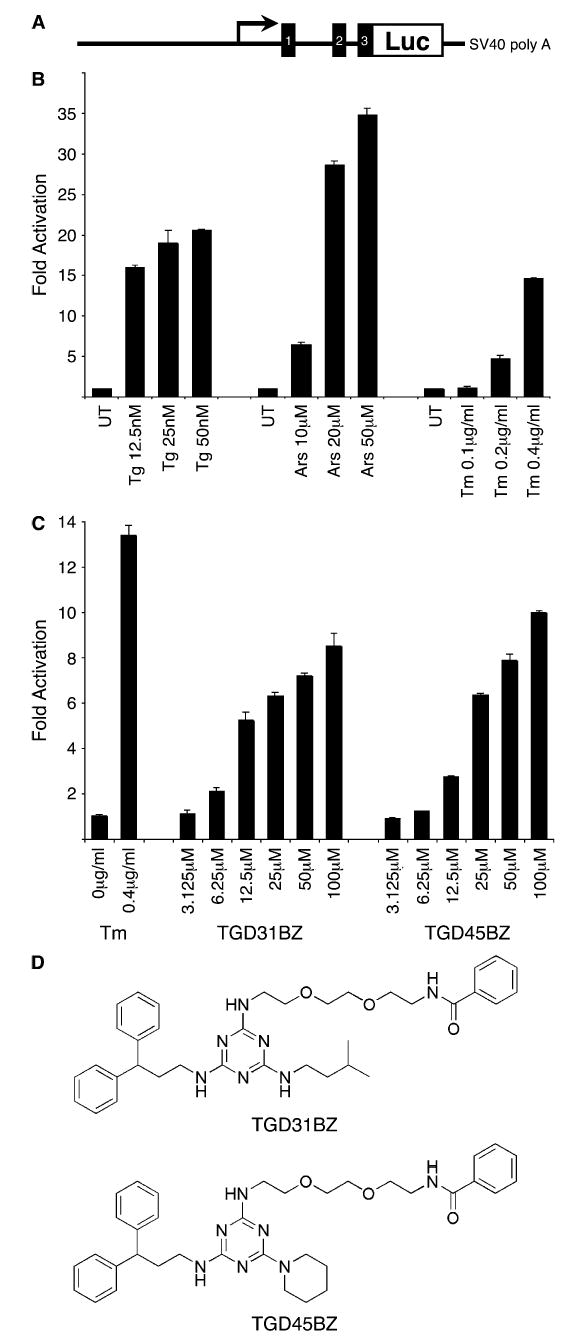
Identification of chemical activators of the integrated stress response
A) Cartoon depicting the structure of the CHOP::luciferase reporter. The CHOP gene is fused to the luciferase protein coding region at exon 3, immediately upstream of the start codon.
B) Fold increase of the luciferase activity of a CHOP::luciferase-expressing stable CHO cell line after 16 hr exposure to thapsigargin (Tg), sodium arsenite (Ars), or tunicamycin (Tm). The mean ± range of luciferase activity normalized for protein content in duplicate samples is displayed.
C) Fold increase in luciferase activity of the aforementioned cell line following 16 hr exposure to the benzoyl-modified derivatives of the two ISR activators identified in our screen (TGD31 and TGD45). Luciferase activity of cells exposed to the known ISR inducer tunicamycin is provided as a reference.
D) Chemical structure of TGD31BZ and TGD45BZ.
To determine whether these compounds also activated endogenous markers of the ISR, we measured the levels of endogenous phosphorylated eIF2α , ATF4, and CHOP in compound-treated cells. The compound-dependent increase in activity of all three markers observed in wild-type mouse embryonic fibro-blasts was blocked by a mutation (eIF2α S51A) that eliminates the regulatory phosphorylation site (Scheuner et al., 2001), indicating that CHOP and ATF4 induction depended on eIF2α phosphorylation (Figure 2A). The essential role of eIF2α phosphorylation in mediating the biological effects of the compounds was further substantiated by gene-expression profiling, which revealed that the eIF2α S51A mutation blocked the induction of most of their target genes (Figure 2B; see also Table S1 in the Supplemental Data available with this article online). Only 21 of the 157 genes whose mRNA levels increased more than 2-fold in compound-treated wild-type cells were also inducible in the mutant. Furthermore, most of the eIF2α -phosphorylation-independent genes (induced by the compounds in both wild-type and eIF2α S51A mutant cells) were not previously identified as ISR targets (Harding et al., 2003; Lu et al., 2004b); the significance of this last observation will be clarified below.
Figure 2.

Compound-mediated ISR activation requires eIF2α phosphorylation and correlates with activation of PERK
A) Immunoblots of phosphorylated eIF2α (eIF2α -P), total eIF2α , ATF4, and CHOP from wild-type and mutant eIF2α S51A cells that were untreated or treated with sodium arsenite (Ars) or 35 μM TGD45BZ for 4, 8, or 16 hr.
B) Relative expression levels of the subset of genes induced at least 2-fold by TGD45BZ in wild-type cells compared to their level of expression in identically treated eIF2α S51A mutant cells. ‘‘Expression level’’ is defined as the ratio of the average signal strength of each gene at each time point to the median signal strength of that gene derived from all experimental time points.
C) Immunoblots of CHOP, GCN2, and PERK from extracts of wild-type and eIF2α S51A cells that were untreated or treated with tunicamycin (Tm), medium lacking nonessential amino acids and leucine (− NEAA, Leu), or 35 μM TGD45BZ for 4, 8, or 16 hr. CHOP was detected by immunoblot of nuclear extracts, whereas GCN2 and PERK were immunoprecipitated first from detergent-solubilized cell lysates. The GCN2 immunoprecipitates were sequentially reacted with antisera specific to the phosphorylated form of GCN2 (GCN2-P) and then total GCN2, whereas PERK immunoprecipitates were probed with antisera that recognize both the phosphorylated (PERK-P) and nonphosphorylated forms of the protein.
Next we examined the activity of individual eIF2α kinases in compound-treated cells. GCN2, the highly expressed eIF2α kinase responsive to amino acid starvation, remained inactive in compound-treated cells, as measured by reactivity to a phospho-GCN2 antiserum. However, PERK activity as measured by the phosphorylation-dependent decrease in the protein’s mobility on SDS-PAGE (Harding et al., 1999) was modestly but reproducibly enhanced in compound-treated cells (Figure 2C). This observation suggests that the compounds cause some level of ER stress; however, the magnitude of that perturbation is modest compared with potent activators of the ER stress response (such as tunicamycin) as it did not lead to activation of other ER stress pathways, such as XBP-1 processing (Figure S1A).
Due to its low level of expression in fibroblasts, we were unable to assay HRI activation, but PKR, which was readily induced by viral infection in these cells, was not measurably activated by TGD45BZ (negative data not shown). Despite these observations, which point selectively to PERK activation by the compounds, ISR markers remained inducible in Perk−/− (and Pkr−/−, Gcn2−/−, and Hri−/−) MEFs. Thus, no single eIF2α kinase was required for the induction of the ISR by the compound (Figure S1B). The compounds thus promote the ISR by both activation of PERK and additional mechanisms that function in the absence of that kinase.
Consistent with experiments conducted with other ISR activators (Jousse et al., 2003; Lu et al., 2004b), we found that pre-treatment with TGD31BZ (or TGD45BZ) strongly protected HT22 cells from oxidative glutamate toxicity and had more modest protective effects against stress induced by protein misfolding induced by tunicamycin or azetadine-2-carboxylic acid (Figures S2C and S2D). The pervasive role of eIF2α phosphorylation in compound-mediated gene activation (Figure 2B) led us to expect that their protective effects would also be blocked by the eIF2α S51A mutation. Surprisingly, the compounds specifically protected the eIF2α S51A mutant cells against tunicamycin (Figures 3A and 4E), and the magnitude of the protective effect was greater in the mutant cells than in the wild-type (compare Figures S2D and S2E with Figure 3A).
Figure 3.
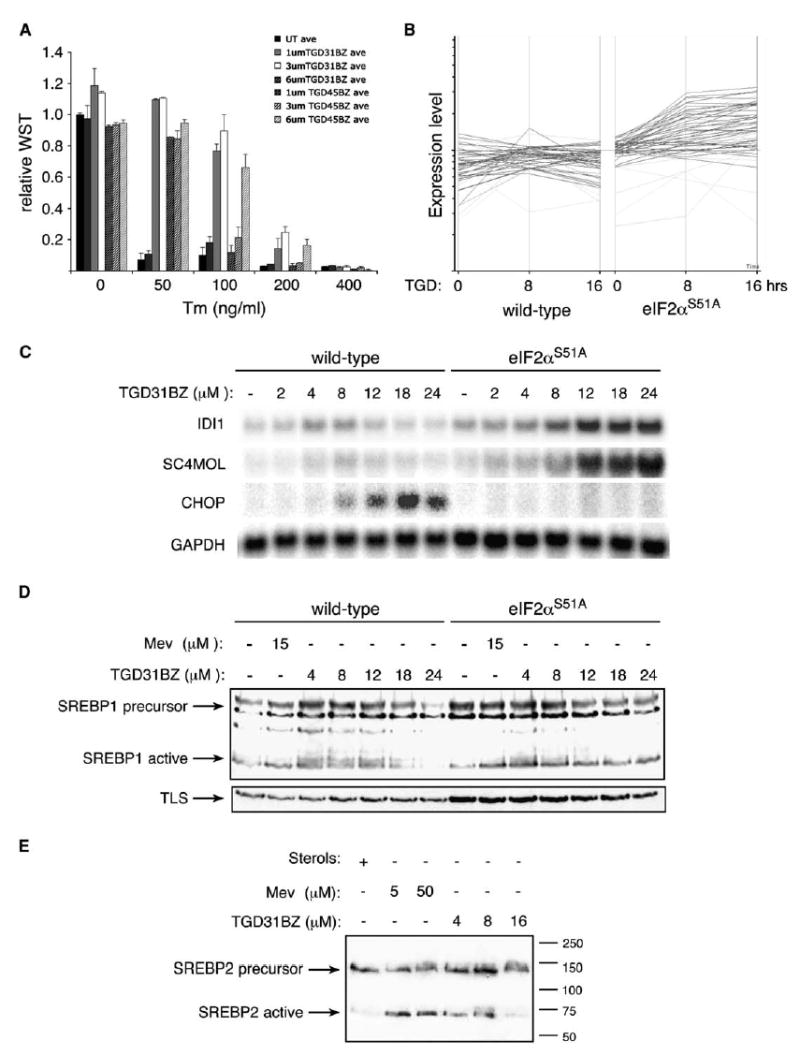
TGD31BZ and TGD45BZ protect cells against tunicamycin and induce SREBP target genes independently of eIF2α phosphorylation
A) Survival and growth of eIF2α S51A mutant fibroblasts pretreated for 14 hr with the indicated dose of TGD31BZ or TGD45BZ followed by subsequent challenge with 0, 50, 100, 200, or 400 ng/ml tunicamycin (Tm) for 24 hr. Survival is expressed as relative amount of WST reduced by cells compared to the untreated (first column, which is set arbitrarily at 1). Values shown are the mean ± range from a representative experiment performed in duplicate and reproduced twice.
B) Relative expression levels of genes previously identified as SREBP target genes (Horton et al., 2003) in wild-type and eIF2α S51A cells following 8 or 16 hr of TGD45BZ treatment.
C) Northern blot analysis of the isopentenyl-diphosphate delta-isomerase1 (IDI1), sterol-C4-methyl oxidase-like (SC4MOL), CHOP, and GAPDH genes in cells with the indicated genotypes treated for 19 hr with compound.
D) Immunoblot of SREBP1 from sterol-replete cells or cells treated with the indicated concentration of mevastatin (a known inhibitor of cholesterol biosynthesis) or TGD31BZ. The position of the inactive transmembrane SREBP1 precursor and the active, cleaved amino-terminal fragment of SREBP1 are indicated. The TLS immunoblot serves as loading control.
E) Immunoblot of SREBP2 from wild-type cells treated as in (D).
Figure 4.
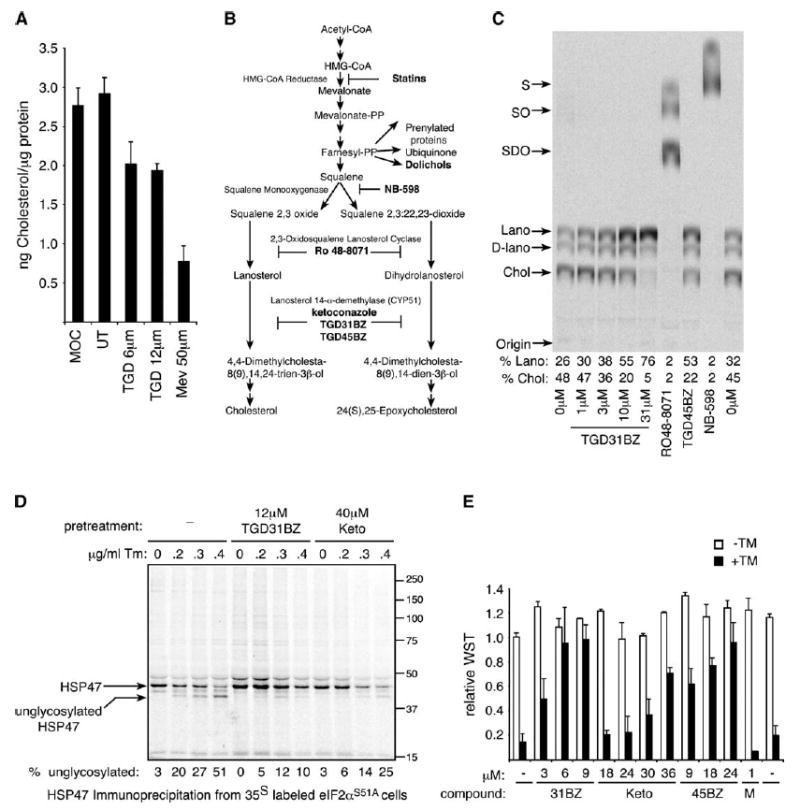
TGD31BZ and TGD45BZ inhibit cholesterol synthesis at the level of lanosterol demethylation and attenuate the inhibitory effects of tunicamycin on protein glycosylation
A) Total cholesterol levels in nonsaponifiable lipid extracts of cells cultured in lipoprotein-deficient serum with no additions (UT), supplemented with sterols and sterol precursors (MOC = mevalonate + oleic acid + cholesterol), or treated with 6 or 12 μM TGD31BZ or 50 μM mevastatin for 24 hr. The mean ± range of duplicate samples is displayed.
B) Diagram of the lipid and sterol biosynthetic pathway (Gardner et al., 2001) and the site of action of inhibitors used in this study.
C) Autoradiogram of a thin-layer chromatograph of nonsaponifiable lipids isolated from cells labeled with [14C]mevalonate for 5 hr following pretreatment for 1 hr with the indicated concentration of TGD31BZ, 31 μM RO 48-8071, 31 μM TGD45BZ, or 62 μM NB-598. The migration of the various cholesterol precursors and their metabolites is indicated to the left of the autoradiogram: squalene (S), squalene 2,3-oxide (SO), squalene 2,3:22,23-dioxide (SDO), lanosterol (Lano), dihydrolanosterol (D-lano), and cholesterol (Chol). The fraction of the label incorporated into lanosterol and cholesterol in each lane is presented below the autoradiogram.
D) Autoradiogram of 35S-labeled HSP47 immunoprecipitated from untreated mutant eIF2α S51A cells and following pretreatment with 12 μM TGD31BZ or 40 μM ketoconazole (Keto) for 17 hr prior to the addition of the indicated concentration of tunicamycin (for 6 hr) followed by a 60 min pulse of [35S]methionine-cysteine. E) Survival and growth of mutant eIF2α S51A fibroblasts pretreated for 14 hr with the indicated concentration of TGD31BZ, ketoconazole (Keto), TGD45BZ, or mevastatin (M) followed by subsequent challenge with 300 ng/ml tunicamycin (+TM, filled bars) or vehicle (−TM, open bars). Survival is expressed as relative amount of WST reduced by cells compared to the untreated (first column, which is set arbitrarily at 1). Values shown are the mean ± range from a representative experiment performed in duplicate and reproduced twice.
Faced with this evidence for ISR-independent effects of the compounds, we scrutinized more closely the gene-expression profile of compound-treated mutant cells. As expected, no ISR target genes were induced by TGD31BZ in the mutant cells (Figure 2B and Table S1). However, the compound coordinately induced a set of genes involved in cholesterol and fatty-acid biosynthesis. A significant overlap was noted between this ISR-independent set of compound target genes and genes previously identified as targets of sterol-activated transcription factors (SREBPs; sterol regulatory element binding proteins) (Horton et al., 2003) (Tables S2 and S3). Furthermore, this class of genes was expressed at lower levels in compound-treated wild-type compared to eIF2α S51A mutant cells (Figure 3B). Northern blot analysis of two representative SREBP target genes (iso-pentenyl-diphosphate delta-isomerase 1, IDI1, and sterol-C4-methyl oxidase-like, SC4MOL) revealed that both were regulated in a biphasic manner by compound in wild-type cells, whereas their mRNAs increased monotonically with compound dose in the eIF2α S51A mutant cells (Figure 3C).
SREBPs are activated by a well-characterized posttranslational proteolytic processing step triggered by cholesterol depletion (Hampton, 2002; Rawson, 2002). To determine whether TGD31BZ accesses this canonical pathway, we followed SREBP1 and SREBP2 processing by immunoblot, which distinguishes the larger unprocessed ER-membrane-associated precursor from the smaller, processed, active form. In cholesterol-replete cells, most of the SREBP1 and SREBP2 was found in the inactive precursor form. Both mevastatin (an inhibitor of cholesterol biosynthesis) and TGD31BZ increased the level of activated SREBPs (Figures 3D and 3E). The level of SREBP precursor also increased in treated cells, which is expected as SREBPs positively regulate their own expression (Horton et al., 2003). Furthermore, SREBP activation in compound-treated wild-type cells was also biphasic, decreasing with the higher concentrations of compound that are associated with ISR activation. This decrease was not observed in eIF2α S51A mutant cells (Figure 3D). These observations implicate the SREBPs in compound-mediated gene activation and suggest that inhibition of IDI and SC4MOL gene expression at higher doses, observed only in the wild-type cells, could be due to ISR-dependent repression of SREBP activation (Figures 3C–3E).
The similarities in SREBP activation noted above in mevastatin- and compound-treated cells suggested that the latter, too, might inhibit one of the enzymes involved in cholesterol biosynthesis. In support of this idea, we found that activation of SREBP1 and SREBP2 by TGD31BZ (and mevastatin) correlated with diminished cellular cholesterol content (Figure 4A). To map a possible site for inhibition of cholesterol biosynthesis, we labeled cells with 14C-mevalonate and used thin-layer chromatography to follow the accumulation of its metabolites in cells treated with various compounds. Cholesterol and its immediate precursors dihydrolanosterol and lanosterol were the only labeled species detectable by this assay in untreated cells, with most of the label accumulating in the final product, cholesterol (Figures 4B and 4C). Treatment with TGD31BZ led to a dose-dependent increase in the fraction of label retained in the lanosterol precursor, suggesting that the compounds block cholesterol biosynthesis at the level of CYP51, the enzyme that de-methylates lanosterol. Qualitatively similar observations were made in cells treated with TGD45BZ. As controls, we treated cells with established inhibitors of cholesterol biosynthesis, RO 48-8071 or NB-598, and found accumulation of intermediates predicted by their known sites of inhibition (Figures 4B and 4C).
The block in cholesterol biosynthesis explains the activation of sterol-regulated signaling pathways by the novel compounds and also suggests how they might protect eIF2α S51A mutant cells, specifically against tunicamycin: Dolichol, the lipid anchor on which carbohydrate chains are assembled before their transfer to asparagine residues on glycoproteins, is synthesized from farnesyl pyrophosphate, an intermediate in the cholesterol biosynthetic pathway (Faust et al., 1980; Goldstein and Brown, 1990) (Figure 4B). Furthermore, mevalonate is limiting for dolichol synthesis and subsequent glycosylation (Carlberg et al., 1996). Thus, interrupting the flow of precursors to cholesterol downstream of farnesyl pyrophosphate might redirect them to the production of dolichol, which would counteract the inhibitory effects of tunicamycin on N-linked glycosylation by increased substrate availability. To test this hypothesis, we compared tunicamycin-mediated inhibition of glycosylation of an endogenous model glycoprotein, HSP47 (Nagata et al., 1988), in untreated and compound-treated cells. At a given concentration of tunicamycin, less underglycosylated HSP47 was noted in cells treated with either TGD31BZ or ketoconazole (a structurally unrelated azole inhibitor of CYP51; Ito et al., 1994) (Figure 4D). These effects on glycosylation were mirrored by ketoconazole’s ability to protect eIF2α S51A mutant cells against the lethal effects of tunicamycin (but not thapsigargin, which induces ER stress by the unrelated mechanism of calcium depletion; Figure 4E and Figures S2D and S2E). By contrast, mevastatin, which inhibits cholesterol biosynthesis upstream of farnesyl pyrophosphate, had no protective effect (Figure 4E). These results suggest that the compound protects cells against tunicamycin by diverting precursors to dolichol synthesis rather than by cholesterol depletion or SREBP activation.
Interactions between sterols, ER stress, and the ISR
To determine whether activation of the ISR by the compounds was mimicked by known inhibitors acting at the same step in cholesterol biosynthesis, we treated cells with the CYP51 inhibitor ketoconazole. As expected, the SREBP target genes IDI1 and SC4MOL were induced by ketoconazole. Importantly, the ISR target gene CHOP was strongly induced in wild-type cells and to much lower levels in the eIF2α S51A mutants. Activation of the SREBP target genes declined at concentrations of ketoconazole that induced CHOP, mirroring the profile of TGD31BZ (Figures 5A and 3C). This reciprocal relationship of the ISR and sterol-regulated gene expression was dependent on eIF2α phosphorylation as it was not observed in eIF2α S51A mutant cells (Figures 5A and 3C). Furthermore, like our compounds, ketoconazole also activated PERK (Figure 5B) but did not induce XBP-1 processing (Figure 5C), indicating equivalence in their biological effects in this system.
Figure 5.

Ketoconazole mimics the effect of TGD compounds
A) Northern blot analysis of the sterol-regulated genes isopentenyl-diphosphate delta-isomerase1 (IDI1) and sterol-C4-methyl oxidase-like (SC4MOL), the ISR marker CHOP, and the loading control GAPDH in total RNA from cells with the indicated eIF2α genotypes that were incubated in the presence of sterols (c) or oleic acid (o) or treated for 24 hr with the indicated concentration of ketoconazole.
B) Immunoblot of PERK, immunoprecipitated from untreated cells or cells treated with ketoconazole or tunicamycin (2.5 μg/ml for 4 hr as a positive control). The phosphorylated (P-PERK) and nonphosphorylated forms are indicated.
C) Ethidium-stained gel of XBP-1 RT-PCR of RNA isolated from untreated cells or cells treated with 2 μg/ml tunicamycin or the indicated dose of ketoconazole for 24 hr. The position of the cDNAs derived from the unprocessed (XBP-1u) or processed (XBP-1p) mRNAs is indicated.
To directly examine the role of eIF2α phosphorylation in inhibition of SREBP target genes, we exploited an experimental system that uncouples eIF2α phosphorylation from ER stress and changes in cellular calcium metabolism. The system consists of cells stably expressing a ligand-activatable chimeric eIF2α kinase, Fv2E-PERK, in which kinase activity is subordinate to an otherwise inert, small, cell-penetrant dimerizer drug, AP20187 (Figure 6A) (Lu et al., 2004b). Addition of AP20187 ligand activated the chimeric Fv2E-PERK kinase and its ISR target gene CHOP in a dose-dependent manner, as expected (Lu et al., 2004b). However, basal levels of the SREBP target mRNAs, ID1, and SC4MOL declined progressively, exhibiting an inverse relationship to ISR activity (Figure 6B). The Fv2E-PERK-mediated ISR antagonized not only the basal expression of SREBP target genes but also their mevastatin-induced activation (Figure 6C). This antagonism correlates with an ISR-mediated inhibition of SREBP activity as reflected by lower levels of processed SREBP1 in AP20187-treated cells (Figure 6D). Furthermore, this antagonism at the level of SREBP activation depended on eIF2α phosphorylation, as it was not evident in Fv2E-PERK-expressing eIF2α S51A mutant cells (Figure S3).
Figure 6.

ISR activation inhibits sterol-regulated gene expression by inhibiting SREBP activation
A) Cartoon depicting the ligand-activated Fv2E-PERK. The AP20187 ligand binding domain is fused to PERK’s eIF2α kinase domain that is activated by addition of the ligand to the culture media.
B) Northern blot analysis of samples from untreated Fv2E-PERK-expressing CHO cells or cells in which the ISR was activated by AP20187 (10 nM) for 1, 2, 4, or 8 hr. Mevastatin (25 μM) serves as a positive control.
C) As in (B) except that the cells were treated with mevastatin (25 μM) for 16 hr with or without AP20187 ligand (at 1, 2, 4, 8 nM).
D) Immunoblot of SREBP1 and phosphorylated Fv2E-PERK or total Fv2E-PERK from sterol-replete cells or cells treated with 25 μM mevastatin alone or with the indicated concentrations of AP20187. The inactive SREBP1 precursor and the active amino-terminal fragment from the same gel are shown in separate panels. Total eIF2α was used as a loading control.
E) Total cholesterol in nonsaponifiable lipid extracts of Fv2E-PERK-expressing CHO cells cultured in lipo-protein-deficient serum with no additions, supplemented with lipid-containing whole serum, or treated with the indicated concentrations of AP20187 or TGD31BZ for 24 hr. The mean and SEM of triplicate samples is displayed.
F) Comparison of the effects of ISR activation on cell growth in media containing complete serum or serum lacking lipoproteins (LPDS). Plotted is the ratio of Fv2E-PERK-expressing CHO cells following 96 hr of culture in the presence of the indicated concentration of AP20187 compared with the untreated cells (set at 100%). The inset panel shows the growth of parental CHO cells lacking Fv2E-PERK under the same conditions. The mean and SEM of triplicate samples is displayed.
ISR-mediated inhibition of SREBP target genes was also associated with a dose-dependent decrease in cellular cholesterol content (Figure 6E). Furthermore, induction of the ISR preferentially compromised cell growth in lipoprotein-deficient serum, compared to lipoprotein-containing whole serum (Figure 6F). Together, these observations indicate that the ISR inhibits the sterol-regulated gene expression program at the level of SREBP activation and that this limits the ability of cells to produce cholesterol and grow in cholesterol-deficient medium.
Discussion
Here we have described a screen for small molecule activators of the integrated stress response (ISR), which led to the discovery of hitherto unsuspected interactions between sterol biosynthetic pathways and the unfolded protein response: Blocking cholesterol biosynthesis at the level of lanosterol demethylation activates the eIF2α -phosphorylation-dependent arm of the UPR (the ISR), and the ISR inhibits sterol-regulated gene expression by blocking SREBP activation. These findings are important to our understanding of lipids’ role in ER homeostasis and the role of ER stress signaling in lipid metabolism.
Two related ISR activators, TGD31 and TGD45, were found to inhibit lanosterol demethylation, which is carried out by the P450 enzyme CYP51. ISR activation is likely to be linked mechanistically to inhibition of this step in cholesterol biosynthesis as a structurally unrelated compound, ketoconazole, which targets the same enzyme, also activates the ISR. A significant role for apoptosis in compound-mediated activation of SREBPs (Wang et al., 1996) is disfavored by the observation that, at the doses required to activate SREBPs, the compounds do not promote cell death (compare Figures 3D and 3E with Figure S4).
Interfering with the flux of metabolites to cholesterol has the predicted consequence of depleting the end product and results in lower cholesterol content of cells growing in lipoprotein-deficient media. However, our study also suggests that interference with CYP51 leads to significant accumulation of sterol-pathway precursors: Both compounds, and ketoconazole, markedly protected cells against the glycosylation inhibitor tunicamycin, an effect that was more striking in the eIF2α S51A cells that are hypersensitive to ER stress (Scheuner et al., 2001). The compounds achieved this by attenuating the toxin’s ability to block N-linked glycosylation (Lehrman et al., 1988; Zhu et al., 1992). These observations are consistent with diversion of cholesterol precursors accumulating upstream of the block to the synthesis of dolichol and an increased pool of lipid-anchored oligosaccharides in compound-treated cells. It is unclear if competition for cholesterol precursors ever limits the capacity for ER glycosylation in physiological circumstances, but our study points to an equilibrium in the partitioning of cholesterol precursors between different fates that can be perturbed to affect protein glycosylation and perhaps other processes (Figure 7).
Figure 7.
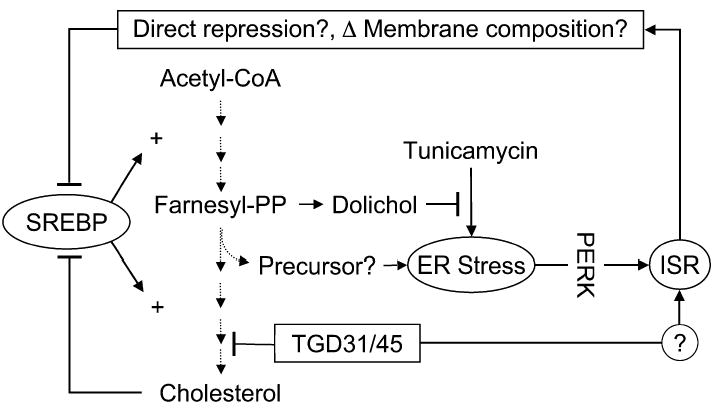
Cartoon depicting relationships between TGD31/45, cholesterol biosynthesis, and the ISR
TGD31/45 block lanosterol demethylation, a late step in cholesterol biosynthesis, and thereby activate the ISR, perhaps through the accumulation of a toxic precursor. The ISR inhibits SREBP activation by an unknown mechanism that likely requires ISR target-gene activation. These could encode a direct protein repressor of SREBP activation or could repress SREBP indirectly—for example, by altering membrane lipid composition. At the same time, the block in cholesterol biosynthesis diverts precursors to dolichol and thereby attenuates tunicamycin-mediated ER stress. By an unknown mechanism, TGD31/45 also activate the ISR independently of their ability to cause ER stress and activate PERK. We speculate that the ISR-mediated inhibition of SREBP activation fine tunes sterol biosynthesis to the circumstances of ER-stressed cells.
Recent evidence suggests that abnormally high free-cholesterol content of the ER membrane negatively affects organelle homeostasis (Feng et al., 2003; Li et al., 2004). We found that the TGD compounds that deplete cellular cholesterol stores and activate the ISR also do so by promoting ER stress (at least in part, as reflected by PERK activation). Thus it appears that the protein folding environment in the ER is sensitive to both extremes in cholesterol content of the cell. It is also possible that an accumulating precursor or product (or products) of a parallel pathway (perhaps dolichol itself) contributes to the development of ER stress in TGD- or ketoconazole-treated cells (Figure 7). A prominent role for a perturbing precursor, rather than cholesterol depletion, is supported by the observation that mevastatin, which potently activates the SREBPs by blocking cholesterol biosynthesis at an upstream step, did not significantly activate the ISR (Figures 6B and 6C), whereas ketoconazole and RO 48-8071, inhibitors that act downstream of the farnesyl-pyrophosphate branch point, did induce an ISR (Figure 5A and data not shown). Furthermore, we were unable to suppress TGD-mediated ISR activation by supplementing the culture media with cholesterol. However, we cannot be sure that newly synthesized cholesterol and that taken up from the media equilibrate identically through cellular compartments, and it is thus possible that exogenous cholesterol is unable to fully rescue a deficiency at a relevant intracellular site. Furthermore, the toxicity of mevastatin, which precluded high doses and lengthy treatment, limits our ability to draw conclusions regarding the role of cholesterol depletion itself in ISR activation. Either way, a perturbation of the ER likely plays a role in ISR activation following inhibition of CYP51. Surprisingly, deletion of the only known ER-stress-inducible eIF2α kinase, PERK, did not abolish eIF2α phosphorylation in compound-treated cells, indicating that ISR activation is not due exclusively to PERK activity. However, none of the other three known eIF2α kinases could be implicated in the process, suggesting that the compound may also act through additional mechanisms that remain to be worked out.
This inquiry led to the identification of a second unsuspected feature of the relationship between the ISR and sterol metabolism: In wild-type cells, sterol-regulated target-gene induction decreased at higher compound concentrations, reciprocating the induction of the ISR. This biphasic profile was not observed in mutant eIF2α S51A cells with a blocked ISR; in these cells, the induction of sterol-regulated target genes increased monotonically with compound dose. This reciprocal relationship of the ISR and sterol-regulated target-gene expression was reflected by changes in SREBP activation, which was attenuated at compound doses that activate the ISR. Furthermore, artificial activation of the ISR by a ligand-controlled eIF2α kinase directly attenuated gene expression and SREBP activation, independently of other cell stress pathways.
Phosphorylation of eIF2α attenuates protein synthesis, and the ISR may deplete cells of a labile factor required for sterol-regulated gene expression. However, SREBP inhibition is observed at rather modest reductions in global protein synthesis (data not shown); therefore, we regard it as more likely that the interaction of the two pathways is mediated by an ISR target gene (or genes). These may encode direct repressors of SREBP; alternatively, the ISR may affect SREBP activation indirectly—by altering ER membrane lipid composition, for example (Figure 7). It is notable in this regard that, whereas membrane cholesterol content is the primary regulator of SREBP’s activity in mammalian cells, fatty acids can contribute to sterol repression (Thewke et al., 1998; Hannah et al., 2001), and several genes involved in fatty-acid biosynthesis are targets of the ISR (Harding et al., 2003). The relatively long latency of ISR-mediated repression of SREBP favors, in our opinion, a metabolite-based mechanism.
In addition to coupling client-protein load to the functionality of the ER’s protein-handling machinery, the UPR also has an important role in regulating the cell’s lipid composition. This is reflected in the inositol auxotrophy of IRE1 mutations in yeast (Cox et al., 1997) and in the regulation of phospholipid biosynthesis in mammalian cells by XBP-1 (Sriburi et al., 2004). Furthermore, the enzymes that process and activate the SREBPs are also utilized for activating ATF6 in response to an unfolded client-protein load (Haze et al., 1999; Ye et al., 2000). Thus, the lipid-based and protein-based ER-centered stress responses likely diverged from a common ancestral pathway. Given the influence of cholesterol metabolism on ER function, it is tempting to speculate that ISR-mediated attenuation of SREBP fine tunes the flux of metabolites through the cholesterol biosynthetic pathway in response to the needs of ER-stressed cells.
It has recently been found that chronic ER stress in liver and fat accompanies the metabolic syndrome of obesity and insulin resistance (Ozcan et al., 2004). Therefore, the inhibitory effect of the ISR on SREBP-mediated gene expression could affect cholesterol and lipid metabolism in this disorder. It is interesting to note in this regard that, when placed on a high-fat diet, heterozygous eIF2α S51A/+ mice have elevated serum cholesterol levels, consistent with our finding of defective SREBP repression by the ISR (Scheuner et al., 2005). The pathophysiological consequences of SREBP activation by the ISR thus need to be explored in further detail.
Experimental procedures
Cell culture
HT22 cells were cultured in standard medium (DMEM supplemented with 10% fetal clone II serum [Hyclone], penicillin-streptomycin, l-glutamine). Mouse fibroblast cell lines with defined genotypes of wild-type, Perk−/−(Harding et al., 2000b), Gcn2−/− (Harding et al., 2000a), Pkr−/− (Abraham et al., 1999), Hri−/− (Han et al., 2001), and homozygous eIF2α S51A (Scheuner et al., 2001) cells were grown in complete medium (the above medium supplemented with MEM nonessential amino acids and 55 μM β-mercaptoethanol). CHO-KI cells were grown in Hams F12 supplemented with 10% fetal clone II serum (Hyclone), penicillin-streptomycin, and l-glutamine.
Screening for ISR-activating compounds
The 3000-compound space-filling combinatorial library has been previously described (Moon et al., 2002; Khersonsky et al., 2003).
For primary and secondary screening, CHO-KI cells stably transfected with a CHOP::luciferase reporter gene (the coding sequence of CHOP::GFP [Novoa et al., 2001] was replaced by luciferase) were plated at a density of 2 × 104 cells per well in 96-well plates 24 hr before adding fresh media containing 10 μM of compound in DMSO vehicle. Sixteen hours later, cells were washed twice and lysed in 25 μl lysis buffer (25 mM glygly, 15 mM MgSO4, 4 mM EGTA, 1% Triton X-100, and 1 mM DTT [added fresh]), and luciferase activity was measured following the addition of 25 μl assay buffer (25 mM glygly, 15 mM MgSO4, 4 mM EGTA, 1% Triton X-100, 11.7 mM potassium phosphate, 1.6 mM ATP, 0.2 mg/ml coenzyme A, 500 μM luciferin, and 2 mM DTT). After screening 3000 compounds, 13 were identified as activating the reporter 1.1-fold or greater at 10 μM and 8 of these activated the reporter 1.5- to 3.6-fold at 20 μM in secondary screening. Benzoyl-modified versions of the four best compounds were synthesized, and two of these reproducibly induced the CHOP::luciferase reporter in a dose-responsive manner.
Protein detection
Cultured cells were treated for 4–16 hr with 25 μM sodium arsenite, 2 μg/ml tunicamycin, or 35 μM TGD45BZ or as indicated by the figure legends. Cytoplasmic and nuclear extracts were made as previously described (Lu et al., 2004b). To detect SREBP2, whole-cell extracts were made by collecting cells in PBS-EDTA and lysing them by three cycles of freeze thaw in whole-cell-extract buffer (10 mM HEPES [pH 8.0], 50 mM NaCl, 0.5 M sucrose, 0.1 M EDTA, 0.5% Triton X-100) supplemented just before use with 1 mM dithiothreitol (DTT), 2 mg/ml pepstatin, 4 mg/ml aprotinin, and 100 mM PMSF. The lysates were cleared by centrifugation at 425 × g (2000 rpm) for 10 min at 4° C in a tabletop centrifuge. The supernatant was transferred to a new tube, the nuclear pellet was centrifuged at 20,817 × g (14,000 rpm) for 15 min at 4° C, and the two supernatants were pooled. Equal amounts of protein were loaded per lane (40 μg of cytoplasmic extract, 20 μg of nuclear extract, or 75 μg of whole-cell extract), separated, and transferred to nitrocellulose. Immunoblotting procedures for PERK, eIF2α , ATF4, GADD34, and CHOP have previously been described (Harding et al., 2003). SREBP1 and SREBP2 were detected with monoclonal antibodies produced from the CRL-2121 and CRL-2198 hybridomas (ATCC) and HRP-conjugated goat anti-mouse secondary antibody.
Radiolabeling of endogenous HSP47 with 35S-methionine and cysteine followed by immunoprecipitation from cell lysates was as described previously (Marciniak et al., 2004).
Cell survival assay
Cells were plated at a density of 2 × 103 per well in 24-well plates. A day later, the media were replaced with standard medium containing indicated concentrations of TGD31BZ, TGD45BZ, ketoconazole, or 1 mM mevastatin. The cells were incubated with the compound for 14 hr and then exposed to the indicated concentration of glutamate, tunicamycin, or azetidine-2-carboxylic acid for an additional 24 hr (glutamate and tunicamycin) or 48 hr (azetidine-2-carboxylic acid), at which point the cells were washed and returned to standard media (HT22) or complete medium (all other cell lines). After an additional 48 hr of culture, the media were replaced with new media containing 0.05 mg/ml WST-1 (Dojindo) and 0.05 mg/ml phenazine methosulfate (Sigma), and the OD450 minus the OD650 of 100 μl medium from each well was measured after a 2–4 hr incubation period. The values are shown as relative to the untreated, which is set to a value of 1 in each graph.
Microarray, Northern blot, and RT-PCR analysis
Total cellular RNA was isolated using the acid phenol/guanidine thiocyanate method. For array analysis, wild-type or eIF2α S51A mutant cells treated for 0, 8, or 16 hr with 35 μM TGD45BZ and RNA samples were prepared in duplicate for each time point from each cell line. Fluorescently labeled RNA probes were prepared from each of the 12 samples and hybridized to Affymetrix mouse 430A high-density oligonucleotide arrays as previously described (Lockhart et al., 1996). Primary image analysis of the arrays was performed using the Genechip 3.2 software package (Affymetrix, Santa Clara, CA). The raw data from the 12 hybridization experiments were analyzed by GeneSpring software. Only genes (array spots) whose raw hybridization signal was significantly above that of the chip background in the treated wild-type cells (t = 8 and 16 hr) were evaluated (n = 11484). The raw signal strength from each gene (array spot) was normalized to the mean signal strength of all genes (array spots) from the same chip to obtain the normalized signal strength. Then, to allow visualization of all data on the same scale for subsequent analysis, the normalized signal strength of each gene (array spot) was divided by the median signal strength for that gene (array spot) among all 12 samples to obtain the expression level. One hundred and fifty-seven genes (array spots) were identified as induced 2-fold or more at 8 or 16 hr in wild-type cells, and 92 genes (array spots) were identified as induced 2-fold or more in eIF2α S51A cells. Twenty-one genes (array spots) were induced in both cell types. Fifty-three genes (array spots) that had previously been identified as being regulated by SREBP proteins were identified, and thirty-eight of these were induced 1.5-fold or more at 8 or 16 hr in eIF2α S51A cells.
For Northern blot analysis, total cellular RNA was separated on denaturing agarose gels, transferred to Hybond-N membranes (GE-Amersham), and hybridized to cDNA probes for mouse IDI1 (GenBank accession number 21703725, bp 233–485) or SC4MOL (GenBank accession number 13384835, bp 487–875) genes isolated by reverse transcription-PCR from fibroblast RNA or to the previously described cDNA probes for CHOP and GAPDH.
XBP-1 RT-PCR was carried out using oligo dT priming for the reverse transcriptase reaction using MLV-RT as recommended by the manufacture (Invitrogen). Primers XBP.3S (5′ -AAACAGAGTAGCAGCTCAGACTGC-3′ ) and XBP.12AS (5′ -TCCTTCTGGGTAGACCTCTGGGAG-3′ ) were used in a 25 μl PCR reaction with a single 4 min 94°C denaturation cycle followed by 30 cycles of 94°C for 10 s, 63°C for 30 s, and 72°C for 30 s followed by separation of the unprocessed (473 bp) and processed (450 bp) XBP-1 RT-PCR products on a 10 cm 2% agarose gel.
Lipid analysis
Cultured cells treated with the indicated concentrations of TGD31BZ or mevastatin for 24 hr in lipoprotein-deficient medium (complete medium made with 5% lipoprotein-deficient bovine calf serum [LPDS] instead of the usual serum and supplemented with 50 μM mevalonic acid). Total cellular lipids were extracted (Folch et al., 1957), dried under a nitrogen stream, and saponified in 90% methanol, 10% 10 M KOH for 16 hr at room temperature. After clearing by centrifugation, the soluble nonsaponifiable lipids were re-extracted and dried. Cholesterol levels were determined using enzymatic detection with a kit as directed by the manufacturer (R-Biopharm). For thin-layer chromatography analysis, [14C]mevalonic acid was prepared from [14C]mevalonic-acid lactone by alkaline cleavage: The dried compound (50 μCi) was resuspended in 340 μl 1.2N NaOH and heated for 30 min at 55°C, then neutralized by the addition of 19 μl 12N HCl and 50 μl 10× PBS. Cells growing in 10 cm dishes were switched to lipoprotein-deficient medium for 16 hr before pretreatment for 1 hr with the indicated compounds. The cells were then labeled with 1.5 μCi [14C]mevalonic acid for 5 hr, washed, and collected by scraping in PBS. The cell pellets were saponified, and the nonsaponifiable lipids were extracted, dried, and then resuspended in 50 μl hexanes:chloroform (70:30) and separated on prescored glass thin-layer chromatography plates using hexanes:diethyl ether (50:50). The migration of nonradioactive standards (Sigma) was detected by iodine vapor staining.
Supplemental Data
Supplemental Data include Supplemental References, three tables, and four figures and can be found with this article online at http://www.cellmetabolism.org/cgi/content/full/2/6/361/DC1/.
Acknowledgments
We thank Edward Fisher (NYU) and Jin Ye and Mark Lehrman (UTSW, Dallas) for scientific advice, J.J. Chen (MIT) for the Hri−/−mice, John Bell (Ottawa) for the Pkr−/− MEFs, Kazuhiro Nagata (Kyoto) for anti-HSP47 serum, Tilla Worgall and Richard Deckelbaum (Columbia University) for the anti-SREBP antibody, and ARIAD, Inc. for the Fv2E dimerization system and the AP20187 compound (http://www.ariad.com/regulationkits). This work was supported by a JDRF grant to H.P.H., NIH grant 1R01CA096912 to Y.-T.C., and NIH grants ES08681 and DK47119 to D.R.
References
- Abraham N, Stojdl DF, Duncan PI, Methot N, Ishii T, Dube M, Vanderhyden BC, Atkins HL, Gray DA, McBurney MW, et al. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J Biol Chem. 1999;274:5953–5962. doi: 10.1074/jbc.274.9.5953. [DOI] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated in-tramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Carlberg M, Dricu A, Blegen H, Wang M, Hjertman M, Zickert P, Hoog A, Larsson O. Mevalonic acid is limiting for N-linked glycosylation and translocation of the insulin-like growth factor-1 receptor to the cell surface. Evidence for a new link between 3-hydroxy-3-methylglutaryl-coenzyme a reductase and cell growth. J Biol Chem. 1996;271:17453–17462. doi: 10.1074/jbc.271.29.17453. [DOI] [PubMed] [Google Scholar]
- Chen J. (2000) Heme-regulated eIF2α kinase. In Translational Control of Gene Expression, N Sonenberg, JWB Hershey, and MB Mathews, eds (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), pp 529–546.
- Cox JS, Chapman RE, Walter P. The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol Biol Cell. 1997;8:1805–1814. doi: 10.1091/mbc.8.9.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE. Gene-specific regulation by general translation factors. Cell. 2002;108:545–556. doi: 10.1016/s0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- Faust JR, Brown MS, Goldstein JL. Synthesis of delta 2-isopentenyl tRNA from mevalonate in cultured human fibroblasts. J Biol Chem. 1980;255:6546–6548. [PubMed] [Google Scholar]
- Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, et al. The endoplasmic reticulum as the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Gardner RG, Shan H, Matsuda SP, Hampton RY. An oxysterol-derived positive signal for 3-hydroxy- 3-methylglutaryl-CoA reductase degradation in yeast. J Biol Chem. 2001;276:8681–8694. doi: 10.1074/jbc.M007888200. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Hampton RY. Proteolysis and sterol regulation. Annu Rev Cell Dev Biol. 2002;18:345–378. doi: 10.1146/annurev.cellbio.18.032002.131219. [DOI] [PubMed] [Google Scholar]
- Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, Fleming M, Leboulch P, Orkin SH, Chen JJ. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20:6909–6918. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS. Unsaturated fatty acids down-regulate srebp isoforms 1a and 1c by two mechanisms in HEK-293 cells. J Biol Chem. 2001;276:4365–4372. doi: 10.1074/jbc.M007273200. [DOI] [PubMed] [Google Scholar]
- Harding H, Zhang Y, Ron D. Translation and protein folding are coupled by an endoplasmic reticulum resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding H, Novoa I, Zhang Y, Zeng H, Wek RC, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000a;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding H, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000b;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding H, Zeng H, Zhang Y, Jungreis R, Chung P, Plesken H, Sabatini D, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in survival of secretory cells. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Harding H, Zhang Y, Zeng H, Novoa I, Lu P, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. The eIF-2 alpha kinases: regulators of protein synthesis in starvation and stress. Semin Cell Biol. 1994;5:417–426. doi: 10.1006/scel.1994.1049. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, Natarajan K. Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell. 2002;1:22–32. doi: 10.1128/EC.01.1.22-32.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Aoyama Y, Ishida K, Kudoh M, Hori K, Tsuchiya S, Yoshida Y. Selectivity of isoprenoid-containing imidazole antifungal compounds for sterol 14-demethylase P450 (P450(14)DM) and 7-ethoxycou-marin O-deethylase P450 of rat liver microsomes. Biochem Pharmacol. 1994;48:1577–1582. doi: 10.1016/0006-2952(94)90202-x. [DOI] [PubMed] [Google Scholar]
- Jousse C, Oyadomari S, Novoa I, Lu PD, Zhang Y, Harding HP, Ron D. Inhibition of a constitutive translation initiation factor 2α phosphatase, CReP, promotes survival of stressed cells. J Cell Biol. 2003;163:767–775. doi: 10.1083/jcb.200308075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman R. J. (2000) The double-stranded RNA-activated protein kinase PKR. In Translational Control of Gene Expression, N Sonenberg, JWB Hershey, and MB Mathews, eds (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), pp 503–527.
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky SM, Jung DW, Kang TW, Walsh DP, Moon HS, Jo H, Jacobson EM, Shetty V, Neubert TA, Chang YT. Facilitated forward chemical genetics using a tagged triazine library and zebrafish embryo screening. J Am Chem Soc. 2003;125:11804–11805. doi: 10.1021/ja035334d. [DOI] [PubMed] [Google Scholar]
- Lehrman MA, Zhu XY, Khounlo S. Amplification and molecular cloning of the hamster tunicamycin-sensitive N-acetylglucosamine-1-phosphate transferase gene. The hamster and yeast enzymes share a common peptide sequence. J Biol Chem. 1988;263:19796–19803. [PubMed] [Google Scholar]
- Li Y, Ge M, Ciani L, Kuriakose G, Westover EJ, Dura M, Covey DF, Freed JH, Maxfield FR, Lytton J, Tabas I. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J Biol Chem. 2004;279:37030–37039. doi: 10.1074/jbc.M405195200. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Lu PD, Harding HP, Ron D. Translation re-initiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004a;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by preemptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004b;23:169–179. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon HS, Jacobson EM, Khersonsky SM, Luzung MR, Walsh DP, Xiong W, Lee JW, Parikh PB, Lam JC, Kang TW, et al. A novel microtubule destabilizing entity from orthogonal synthesis of triazine library and zebrafish embryo screening. J Am Chem Soc. 2002;124:11608–11609. doi: 10.1021/ja026720i. [DOI] [PubMed] [Google Scholar]
- Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- Nagata K, Saga S, Yamada KM. Characterization of a novel transformation-sensitive heat-shock protein (HSP47) that binds to collagen. Biochem Biophys Res Commun. 1988;153:428–434. doi: 10.1016/s0006-291x(88)81242-7. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding H, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- Rawson RB. The SREBP pathway—insights from Insigs and insects. Nat Rev Mol Cell Biol. 2002;4:631–640. doi: 10.1038/nrm1174. [DOI] [PubMed] [Google Scholar]
- Ron D, Hampton R. Membrane biogenesis and the unfolded protein response. J Cell Biol. 2004;167:23–25. doi: 10.1083/jcb.200408117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in-vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Mierde DV, Song B, Flamez D, Creemers JW, Tsukamoto K, Ribick M, Schuit FC, Kaufman RJ. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11:757–764. doi: 10.1038/nm1259. Published online June 26, 2005. 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thewke DP, Panini SR, Sinensky M. Oleate potentiates oxysterol inhibition of transcription from sterol regulatory element-1-regulated promoters and maturation of sterol regulatory element-binding proteins. J Biol Chem. 1998;273:21402–21407. doi: 10.1074/jbc.273.33.21402. [DOI] [PubMed] [Google Scholar]
- Uttamchandani M, Walsh DP, Yao SQ, Chang YT. Small molecule microarrays: recent advances and applications. Curr Opin Chem Biol. 2005;9:4–13. doi: 10.1016/j.cbpa.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Wang X, Zelenski NG, Yang J, Sakai J, Brown MS, Goldstein JL. Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO J. 1996;15:1012–1020. [PMC free article] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zeng Y, Lehrman MA. Evidence that the hamster tunicamycin resistance gene encodes UDP-GlcNAc:dolichol phosphate N-acetylglucosamine-1-phosphate transferase. J Biol Chem. 1992;267:8895–8902. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.