

Abstract
Helicase-Dependent Amplification (HDA) is an isothermal in vitro DNA amplification method based upon the coordinated actions of helicases to separate double-stranded DNA and DNA polymerases to synthesize DNA. Previously, a mesophilic form of HDA (mHDA) utilizing the E. coli UvrD helicase, DNA polymerase I Klenow Fragment, two accessory proteins, MutL and single stranded DNA binding protein (SSB), was developed (1). In an effort to improve the specificity and performance of HDA, we have cloned and purified a thermostable UvrD helicase (Tte-UvrD) and the mutL homolog (Tte-MutL) from Thermoanaerobacter tengcongensis. Characterization of the Tte-UvrD helicase shows that it is stable and active from 45°C to 65°C. We have found that the Tte-UvrD helicase unwinds blunt-ended DNA duplexes as well as substrates possessing 3′ or 5′ ssDNA tails. Tte-UvrD was used to develop a thermophilic Helicase-Dependent Amplification (tHDA) system to selectively amplify target sequences at 60° – 65°C. The tHDA system is more efficient than mHDA, displaying heightened amplification sensitivity without the need for the MutL and SSB accessory proteins. Amplification independent of MutL corresponds with studies demonstrating that maximal Tte-UvrD helicase activity does not require the mutL homolog. The tHDA system allows for rapid amplification and detection of targets present in genomic DNA. The expeditious nature and simplistic design of the tHDA platform makes the technology ideal for use in diagnostic applications requiring rapid identification of organisms at the point-of-need.
The abbreviations used are: tHDA, thermophilic Helicase Dependent Amplification; mHDA, mesophilic Helicase dependent amplification; SSB, single-stranded binding protein; EDTA, ethylene-diamine tetra-acetic acid; DTT, dithiothreiotol; PCR, polymerase chain reaction; dNTP, deoxynucleoside triphosphate; dATP, deoxyadenoside triphosphate; LMP, low melting point; dTTP, deoxythymidine triphosphate
DNA helicases use the energy generated by the hydrolysis of nucleoside triphosphate to break the hydrogen bonds linking the two strands together in duplex DNA (2). Helicases are involved in a myriad of cellular processes requiring the manipulation of DNA, including replication, repair and recombination, because of the ability of these enzymes to allow access to the buried nucleotide sequence (3). The well-studied E. coli UvrD helicase (helicase II) unwinds DNA in a 3′ to 5′ direction (4). Unlike many other helicases, the UvrD helicase is capable of melting fully duplex molecules (DNA fragment with blunt-ends) as well as nicked circular DNA molecules (5). In E. coli cells, UvrD is recruited to a site containing an erroneously incorporated nucleotide to unwind the DNA for corrections to be made during methyl-directed mismatch repair (6) and UvrABC-mediated nucleotide excision repair (7). Direct physical interactions between UvrD and MutL, the master coordinator of the mismatch repair pathway, have shown that MutL dramatically stimulates UvrD helicase activity (8,9,10).
Although several homologs of the E. coli uvrD helicase have been identified and characterized from many bacteria, relatively few uvrD homologs have been described from thermophilic organisms. Expression of the Thermus thermophilus uvrD homolog has been shown to partially compensate for the repair function of E. coli UvrD, suggesting that the function of the helicase is evolutionarily conserved (11). Characterization of this protein indicates that the T. thermophilus UvrD possesses a 3′–5′ DNA helicase activity similar to the E. coli UvrD (12). In addition, another homolog of the uvrD helicase has been purified from Bacillus stearothermophilus (13). It was named Bst pcrA because of its sequence homology to the pcrA helicase in Staphylococcus aureus (13). Studies of two accessory proteins, ribosomal protein L3 and replication initiator protein RepD, have been shown to enhance Bst PcrA unwinding activity, much like the MutL stimulation of UvrD helicase activity in E. coli. (14,15). The stimulatory effect of MutL on UvrD has not been demonstrated to be a universal feature or species specific with uvrD homologs.
Here we report the cloning and characterization of the UvrD and MutL proteins from a fully sequenced thermophilic bacterium, Thermoanaerobacter tengcongensis (16). The helicase activity of the Tte-UvrD is described, as are the effects of the Tte-MutL protein on unwinding reactions catalyzed by Tte-UvrD helicase. Previously, we have developed isothermal DNA amplification method using the UvrD helicase from E. coli (1). Unlike the Polymerase Chain Reaction (PCR) that is dependent on heat to separate dsDNA, dsDNA in Helicase-Dependent Amplification (HDA) reactions are separated into two single strands by a helicase and, thus, enables the amplification reaction to be performed at a constant temperature. In this study, we have tested the ability of thermostable helicases to support thermophilic HDA reactions at higher temperatures, to simplify the reaction components and improve detection sensitivity.
MATERIALS AND METHODS
Bacterial strains, plasmids and enzymes
[α-32P] dTTP was obtained from PerkinElmer (Boston, MA). E. coli ER2502, E. coli ER2566, plasmid pTYB1 and chitin beads were from New England Biolabs Inc. (Beverly, MA). Plasmid pCR2.1-TOPO was from Invitrogen (Carlsbad, CA). HiTrap Heparin HP column and adenosine 5′-triphosphate (ATP) were obtained from Amersham Pharmacia (Piscataway, NJ). QIAquick Nucleotide Removal Kit was obtained from Qiagen (Valencia, CA). Bacterial genomic DNA was obtained from ATCC (Manassas, VA). Deep Vent DNA polymerase, Klenow Fragment (3′–5′ exo−), Quick T4 DNA Ligase, ThermoPol Buffer, and ThermoPol II Buffer were all from New England Biolabs (Beverly, MA).
Cloning and purification of Tte-UvrD and Tte-MutL
Oligonucleotide primers were designed to amplify the Tte uvrD gene from Thermoanaerobacter tengcongensis according to the published sequence (16). The forward primer TUF (5′-ATACATATGATTGGAGTGAAAAAG ATGAA-3′) included an NdeI site and the reverse primer TUR (5′-AAATAAGCTCTTCAGCAAG AAATTGCCTTAATAGGAG-3′) included a SapI site. The PCR product was first cloned into the pCR2.1-TOPO vector. After screening and sequencing, the correct construct was digested with NdeI and SapI, inserted into the pTYB1 expression vector and transformed into E. coli ER2502 cells (17). The sequence of the pTYB1-Tte-UvrD construct was verified and then transformed into E. coli ER2566 cells for over expression.
E. coli cells harboring the pTYB1-Tte-UvrD expression plasmid were grown at 37°C until OD600 reached 0.5 – 0.8. Tte-UvrD protein expression was induced using 0.4 mM isopropyl-ß-D-thiogalactopyranosid (IPTG) at 15°C for 12 ~ 14 hrs. All subsequent procedures were performed at 4°C except when noted. 20 grams of cells were resuspended in Buffer C (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 1 mM EDTA) andlysed by sonication. The clarified lysate was applied to a 15-ml column of chitin beads pre-equilibrated with buffer C at a flow rate of approximately 0.5–1 ml/min. The column was washed with 10 column bed volumes of buffer C and then washed with 3 bed volumes of buffer C containing 50 mM DTT at a flow rate of 2 ml/min. Following incubation for 16 hrs at 16°C, the cleaved Tte-UvrD protein was eluted with buffer C and 1 ml fractions were collected. Fractions containing protein were pooled and dialyzed against buffer A (20 mM K-Phosphate pH 6.9, 0.1 mM EDTA, 6 mM 2-mercaptoethanol, 5% v/v glycerol) supplemented with 50 mM NaCl. The dialyzed Tte-UvrD was further purified using the HiTrap Heparin HP column pre-equilibrated with buffer A supplemented with 50 mM NaCl. Protein was eluted from the column with a linear gradient of 0.05–1 M NaCl in buffer A. Protein eluted at a conductivity equivalent to that of buffer A + 0.5 M NaCl. Fractions containing the Tte-UvrD protein were analyzed by SDS-PAGE, stained with Coomassie Blue, and scanned and quantified using Quantity One 4.4.1 software to verify the purity of the protein. The purified Tte-UvrD was dialyzed against a storage buffer (20 mM Tris-HCl pH 7.4, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 50% v/v glycerol) and aliquots were stored at −80°C.
The Tte-MutL was amplified from genomic DNA by PCR using the forward primer TMF (5′-ATACATATGAATAAAATTCATCTTCTCGAC GA-3′) and the reverse primer TMR (5′-AAATATGCTCTTCAGCACTTGATTCGTTTA ACATCTTCTCAA–3′). The restriction enzyme sites, NdeI and SapI, were engineered into the forward and reverse primers, respectively, to clone the Tte-MutL gene into the expression plasmid pTYB1. The pTYB1-Tte-MutL construct was expressed in ER2566 cells and purified using the procedure described above for Tte-UvrD.
ATPase assays
The hydrolysis of ATP was detected by measuring the release of inorganic phosphate with acidic ammonium molybdate and malachite green (19). Reactions were performed using 100 ng/μl BSA, 3 mM ATP, 6.25 ng/μl M13ssDNA, 5 mM Mg(OAc)2 with varying amounts of Tte-UvrD protein at the indicated temperatures. Reactions were performed in ThermoPol buffer (20 mM Tris-HCl pH 8.8, 10 mM KCl, 10 mM (NH4) 2SO4, 2 mM MgSO4, 0.1% Triton X-100), and pre-incubated at the indicated temperatures for 2 min prior to the addition of the helicase. A parallel reaction was performed without helicase as a negative control. Incubation was continued to each specified time, wherein10 μl of the reaction mixture was removed and added to 800 μl of the ammonium molybdate (1.05% w/v)/malachite green (0.3% w/v) reagent. Each sample was incubated with the ammonium molybdate/malachite green reagent for 1 min at room temperature and then 100 μl of 34% w/v sodium citrate solution was added. After 20 min of continual incubation at room temperature the absorbance at 660 nm was measured.
Helicase assays
The DNA substrates used in this study were composed of four oligodeoxynucleotides: a fixed 23-mer top strand (5′- GCCCTGCTGCCGACCA ACGAAGG -3′) and three different bottom strands to produce either the 3′-ssDNA tailed (5′-CCTTCGTTGGTCGGCAGCAGGGC-(dT)40 –3′), the blunt-ended (5′- ACCTTCGTTGGTCG GCAGCAGGGC –3′), or the 5′-ssDNA tailed (5′-(dT)40 –ACCTTCGTTGGTCGGCAGCAGGGC –3′) duplex as depicted in Table 1. The ssDNA regions, either the 3′ or 5′ tails, were composed entirely of oligodeoxythymidylates to avoid intra-molecular base pairing within the single-stranded region (20). The top and bottom oligonucleotides were mixed in ThermoPol Buffer at a 1:1.5 molar ratio. The annealing mixture was heated at 95°C for 2 min, and then slowly cooled to 25°C over a period of 70 min. The annealed duplex was radioactivity labeled by filling one nucleotide in at the 3′ end of top strand with [α-32P]dTTP using Klenow Fragment (3′-5′ exo−). The labeled duplexes were then purified by QIAquick Nucleotide Removal Kit to remove any unincorporated nucleotides.
Table 1.
DNA unwin ding substrates.
| Substrate | Structure |
|---|---|
| 3′-tailed duplex |
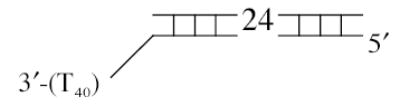
|
| blunt-ended duplex |
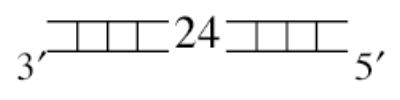
|
| 5′-tailed duplex |
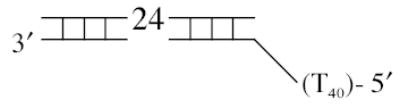
|
Helicase assay reactions were performed by mixing 0.25 nM radiolabeled DNA duplex, 3mM ATP and differing amounts of Tte-UvrD in ThermoPol buffer in a total volume of 20 μl. Reactions were preincubated at 55°C for 2 minutes and were initiated by the addition of ATP and incubated at 55°C for the indicated length of time. Reactions were terminated by the addition of 5 μl of the stop solution (0.25% Bromphenol Blue, 25% glycerol, 1% SDS, 100 mM EDTA). A positive control assay was performed by incubation of the DNA substrate at 95°C for 15 minutes in the absence of helicase to completely denature the DNA substrate. A negative control containing intact DNA substrate in the absence of helicase was performed in parallel with the sample reactions. Samples were resolved in a 20% TBE gel and radioactively labeled bands were quantified using Molecular Imager FX (Bio-Rad) and Quantity One 4.4.1 software.
HDA assays
HDA assays were performed by creating two reaction mixes, mixture A and mixture B. Mixture A contains the DNA substrate (various concentrations of either plasmid DNA or bacterial genomic DNA), primers (25–50 nM of each primer), 3.5 mM MgSO4, 1X ThermoPol II buffer, and ddH2O to a volume of 25 μl. Mixture B contains 1X ThermoPol II buffer, 200 μM dNTPs, 3 mM dATP, 20 U Bst Polymerase, 100 ng Tte-UvrD helicase, +/− 200 ng of Tte-MutL and ddH2O to a volume of 25 μl. Mixture A is heated to 95°C for 3–5 min then cooled to 65°C for 3 min when needed. Mixture B is added to mixture A and the reactions were incubated at 65°C for various amounts of time. Reactions were terminated by the addition of 12.5 μl of stop buffer (0.1% sodium dodecyl-sulphate, 50 mM Na2EDTA, 15% Ficoll and 0.2% orange G), and 10–15 μl aliquots of the reactions were separated on a 2% GPG LMP agarose gel containing ethidium bromide. The plasmid used in this study was pAH1, derived from the pUC19 plasmid. The primers for the plasmid system were as follows: forward primer: 5′-GTGAGCGGATAACAATTTCACACAGGA-3′ and reverse primer: 5′-CGCCAGGGTTTTCCCAGTCACGAC-3′. Primers for bacterial genomic DNA amplification were as follows: Neisseria gonorrhoeae pivNg forward primer: 5′-GCAAAGTTTGACAACGATTCAAAAGGTTT-3′ and reverse primer: 5′-TTGCCTCCATGCAGATATGCAGATTCT-3′.
RESULTS
Cloning and purification of Tte-UvrD and Tte-MutL
Analysis of the T. tengcongensis genome sequence revealed the presence of E. coli uvrD and mutL homologs (16). Tte-UvrD shares 42.9% sequence identity with E. coli UvrD at the amino acid level; however, the Tte-MutL shares a lower (29.5%) but still significant, amino acid sequence identity with MutL in E. coli. The corresponding genes encoding Tte-UvrD and Tte-MutL were amplified from T. tengcongensis by PCR and inserted into the expression vector pTYB1 under the control of the T7 promoter. The proteins were purified using an intein-fusion method that allows for self-cleavage on chitin columns (17). The proteins were further purified over a heparin column. Tte-UvrD and Tte-MutL proteins were found to be about 90% pure based on polyacrylamide gel electrophoresis and had an apparent molecular mass of 83 kDa and 67 kDa, respectively, which is in agreement with the calculated molecular weights of 82,800 (Tte-UvrD) and 67,200 (Tte-MutL) (Fig. 1).
Figure 1.
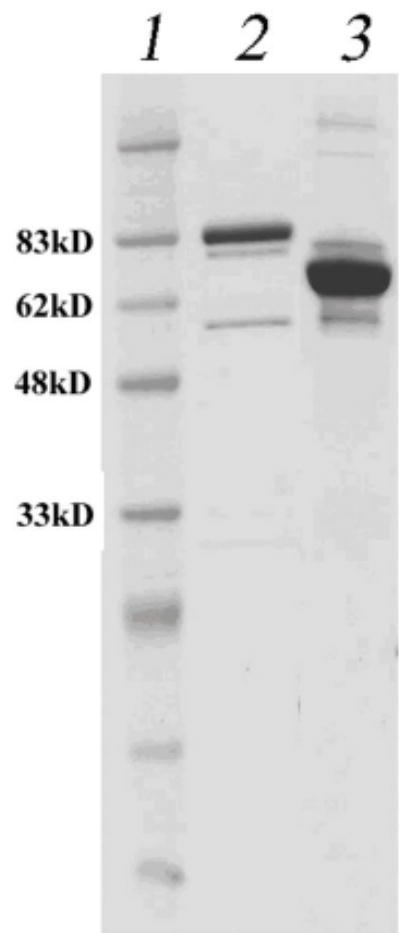
SDS-PAGE of purified Tte-UvrD and Tte-MutL proteins. Proteins were purified as described in Materials and Methods. Lane 1: Prestained protein marker; lane 2: Tte-UvrD protein, 82kDa; lane 3: Tte-MutL protein, 67kDa.
Temperature optima and thermal stability
The temperature range of the hot spring that T. tengcongensis was isolated from fluctuates from 50° to 80°C, therefore, the Tte-UvrD helicase was expected to be stable and active at elevated temperatures (16). To determine the optimal temperature for maximal Tte-UvrD activity, we utilized a colorimetric assay to directly measure the amount of inorganic phosphate released by the ATPase activity of Tte-UvrD over a range of temperatures. Helicase activity was assessed by incubating reactions at temperatures ranging from 40° to 75°C for 8 minutes and measuring the absorbance of each sample at 660 nm. The absorbance at each temperature was expressed as a percentage of remaining activity relative to the highest absorbance in each assay group. Analysis of the data from the ATPase assay showed that ATP hydrolysis occurred over a broad temperature range (from 45°C to 65°C) and reached a maximum at 55°C (Fig. 2A). The ATPase assay was repeated using several different incubation times (2, 4, or 10 min), and similar results were obtained from each assay (data not shown). The thermal stability of Tte-UvrD was determined by incubating the helicase at 65°C or 70°C for differing amounts of time and then assaying for ATPase activity with substrates at the experimentally determined optimal temperature of 55°C for 10 min. The percentage of remaining ATPase activity at 65°C or 70°C was plotted as a function of time and demonstrated that Tte-UvrD was relatively stable at 65°C and lost 30% of ATPase activity after 90 min of continuous incubation at 65°C. The protein was much less stable at 70°C and displayed a half-life of approximately 12 min. After incubating at 70°C for one hour, approximately 90% of Tte-UvrD activity was abolished (Fig.2B). Similar findings in the thermophilic bacterium T. thermophilus have been observed when analyzing the optimal temperature for UvrD helicase activity at temperatures above 70°C (12). Considering the natural habitat of T. tengcongensis reaches temperatures as high as 80°C, the loss of activity at 70°C is an interesting observation and suggests that the helicase may require accessory proteins to function at higher temperatures.
Figure 2A.
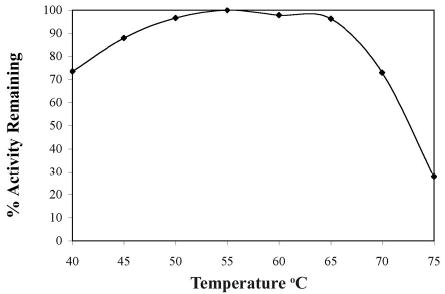
ATPase activity assays of the Tte-UvrD helicase at different temperatures. ATPase assays were performed as described in Materials and Methods. Each reaction consisted of 36 nM Tte-UvrD, incubated at the indicated temperature for 8 min. The absorbance at each temperature was expressed as a percentage of remaining activity relative to the highest absorbance in each assay group.
Figure 2B.
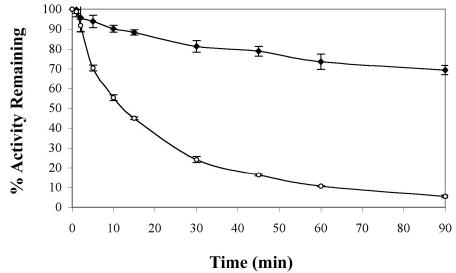
Thermal stability of Tte-UvrD helicase by ATPase assay. Tte-UvrD was diluted in ThermoPol buffer and incubated at 65°C or 70°C for the indicated time. 2 μl of protein was taken out for ATPase assays performed as described in Materials and Methods. Each reaction consisted of 36 nM Tte-UvrD, incubated at 55°C for 10 min. The absorbance at each temperature was expressed as a percentage of remaining activity relative to the highest absorbance in each assay group. Filled square, incubated at 65°C; open circle, incubated at 70°C. Each data point represents the average of at least three independent experiments. Error bars are means ± S.D.
Unwinding activity of Tte-UvrD
The unwinding reaction catalyzed by the Tte-UvrD helicase has been analyzed using an in vitro assay to measure the displacement of a radio-labeled 24-mer DNA strand from a DNA duplex in which the complementary strand is unlabeled. Three different duplex substrates were designed for the unwinding assay (Table I; 20, 21). To test the suitability of these substrates, the unwinding assay was initially performed using the well-characterized E. coli UvrD protein. Our results showed that the E. coli UvrD was capable of unwinding all three substrates, with a strong preference for the substrate containing a 3′-ssDNA tail (data not shown), in agreement with published observations (4,5). In addition, we also observed that the E. coli MutL protein significantly stimulated E. coli UvrD unwinding activity (data not shown), observations that are consistent with previous reports by Yamaguchi et al. (9) and Mechanic et al. (10).
After characterization of the substrates with the E. coli UvrD, unwinding assays were performed using a range of Tte-UvrD protein concentrations (0.25 nM – 16 nM) to unwind a fixed concentration of duplex substrate (0.25 nM). When the 3′-ssDNA tailed duplex was used, unwound single-stranded product increased as a function of increasing enzyme concentration (Fig. 3A). In parallel unwinding assays, Tte-UvrD displayed a significant unwinding activity towards blunt-ended (Fig. 3B) and 5′-ssDNA tailed duplex (Fig. 3C). Although Tte-UvrD was capable of unwinding all three substrates, slightly higher activity was displayed with the 3′-ssDNA tailed substrate as compared to either the blunt-ended or 5′-ssDNA tailed substrates. For example, in the presence of 4nM Tte-UvrD, more than 85% of the 3′-ssDNA tailed duplex was separated and less than 70% and 60% of the blunt-ended or 5′-ssDNA tailed duplexes respectively, were unwound (Fig. 3D). Unlike the E. coli UvrD, Tte-UvrD has only a marginal preference towards the 3′-ssDNA tailed duplex and is able to effectively unwind all three types of substrates (4,5). The results remained consistent over the broad range of Tte-UvrD concentrations analyzed.
Figure 3.
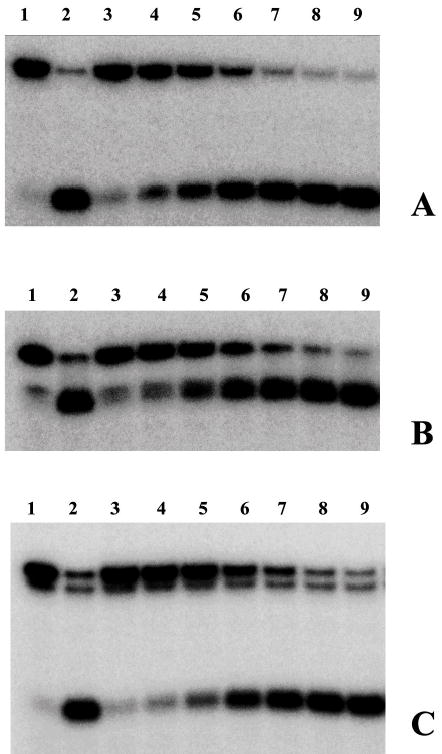
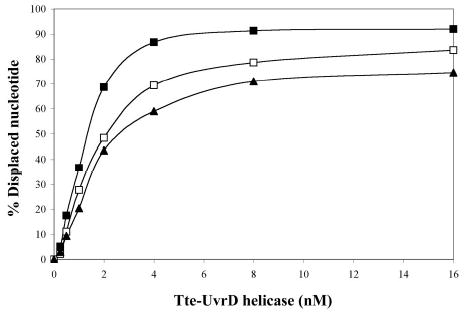
Comparison of the ability of Tte-UvrD protein to unwind DNA duplex with 3′-ssDNA tail, blunt-end, and 5′ -ssDNA tails. Helicase activity assays were performed as described in Materials and Methods. Panel A to panel C are autoradiographs showing the displacement of a 24-mer radio labeled fragment (lower band) from its duplex (upper band). The substrates in panel A were 3′-ssDNA tailed duplexes, in panel B were blunt-ended duplexes, in panel C were 5′-ssDNA tailed duplexes. The DNA concentrations were 0.25nM of duplex DNA molecules. In each panel, lane 1 was the negative control without helicase, lane 2 was the positive control (after heating at 95°C for 15 min without helicase), lanes 3–9 contained Tte-UvrD at 0.25, 0.5, 1, 2, 4, 8, and 16 nM, respectively. The oligonucleotide displacement percentage from panels A to C were calculated and shown in Panel D. Filled square, 3′-ssDNA tailed duplex; open square, blunt-ended duplex; filled triangle, 5′-ssDNA tailed duplex. Each data point represents the result of a single experiment.
In order to address the efficiency of the unwinding reaction, we also performed a time-course assay with the Tte-UvrD protein. After 4 minutes of reaction time, 66.5% of the 3′-ssDNA tailed duplex was unwound and in contrast, 46.1% of the blunt-ended and 45.2% of the 5′-ssDNA tailed duplexes were separated (Fig. 4). After 16 minutes, the unwinding assay had plateaued and increasing the incubation time did not further increase the displacement of the labeled strand. At this protein/DNA ratio (R=8; Tte-UvrD 2 nM, DNA duplex 0.25 nM), Tte-UvrD was able to maximally unwind 90% of the 3′-ssDNA tailed duplex and 71% of the blunt-ended or 5′-ssDNA tailed duplexes (Fig. 4). Although Tte-UvrD unwinds all three types of substrates, the efficiency of the reaction is higher with the 3′-ssDNA tailed duplexes. Taken together, the data from the concentration and time-course assays support the observation that the Tte-UvrD helicase is capable of unwinding all substrates analyzed, with only a marginal preference for the 3′-ssDNA tailed duplex.
Figure 4.
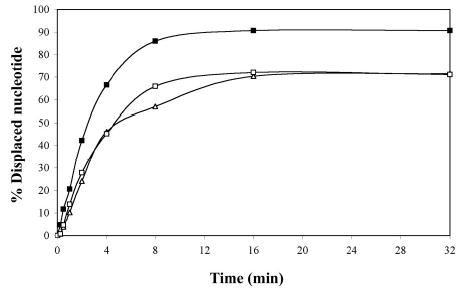
Time course of unwinding of various duplex DNA substrates by the Tte-UvrD helicase. Helicase activity assays were performed as described in Materials and Methods. Concentration of the substrate was 0.25 nM of DNA molecules, and the Tte-UvrD was 2 nM. Filled square, 3′-ssDNA tailed duplex; open square, blunt-ended duplex; filled triangle, 5′-ssDNA tailed duplex. Each data point represents the result of a single experiment.
The effect of MutL on UvrD unwinding activity
In E. coli, MutL targets and loads UvrD onto the DNA substrate and thereby enhances the unwinding activity of UvrD more than 10-fold (9, 10). To test whether Tte-MutL can similarly stimulate the unwinding reaction catalyzed by Tte-UvrD, increasing amounts of Tte-MutL was added to unwinding assays. The results indicate that the Tte-MutL did not have a significant effect on unwinding of blunt-ended and 5′-ssDNA tailed duplex substrates catalyzed by Tte-UvrD (Fig. 5). For the 3′-ssDNA tailed duplex, a low level of stimulation (<30%) was observed when the molar ratio of Tte-MutL to Tte-UvrD reached 32 or 64 (Tte-UvrD 0.5 nM, Tte-MutL 16 or 32 nM; Fig. 5). The assays were repeated with different UvrD protein to DNA substrate ratios and consistent results were observed. Stimulation of the UvrD-unwinding activity by MutL is much less in T. tengcongensis (<30%) than for E. coli (>10-fold (L. An and H. Kong, unpublished observation; 9,10), suggesting that the stimulation of UvrD unwinding activity may be specific to the mesophilic system.
Figure 5.
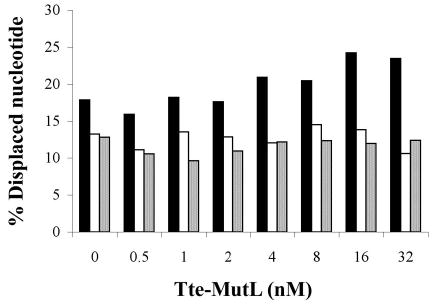
The effect of Tte-MutL protein on the unwinding activity of the Tte-UvrD helicase. Helicase activity assays were performed as described in Materials and Methods. Substrate concentration was 0.25 nM, with 0.5 nM Tte-UvrD. Solid column, 3′-ssDNA tailed duplex; open column, blunt-ended duplex; spotted column, 5′-ssDNA tailed duplex. Data points represent the result of a single experiment.
To test whether MutL from other thermophilic bacteria stimulate their corresponding UvrD’s helicase activity, we have cloned and purified a pair of MutL and UvrD homologs from the partially sequenced Bacillus stearothermophilus strain (GenBank Accession number: NC_002926). The Bst-UvrD helicase shares 91.4% amino acid sequence identity with the previously characterized Bst PcrA helicase (13). In addition, Bst-UvrD also shares 43.6% and 53.9% sequence identities with E. coli UvrD and Tte-UvrD, respectively. The corresponding Bst-MutL protein shares 32.3% and 38.9% sequence identity with E. coli MutL and with Tte-MutL, respectively. The purified Bst-UvrD with the Bst-MutL was also capable of unwinding all three duplex DNA substrates and displayed a slight preference for the 3′-ssDNA tailed substrate (Fig. 6A). A titration of Bst-MutL protein was performed in the presence of a low helicase to substrate ratio (R=4) during the unwinding assay. The results indicate that Bst-MutL had no effect on the unwinding activity of Bst-UvrD regardless of the type of DNA duplex tested (Fig. 6B), similar to the results obtained with the Tte-UvrD helicase and the corresponding MutL protein.
Figure 6.
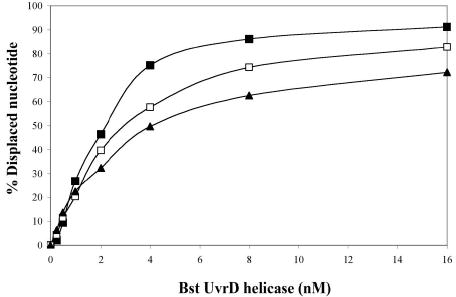
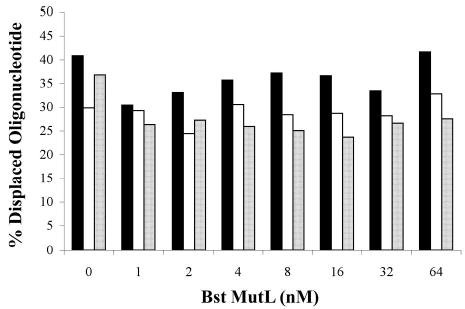
Unwinding assay of Bst-UvrD helicase and Bst-UvrD helicase with Bst-MutL. Panel A: comparison of the ability of Bst-UvrD protein to unwind DNA substrates containing either a 3′-ssDNA tail, a blunt-end, or a5′-ssDNA tail. Helicase activity assays were performed as described in Materials and Methods. Concentration of substrates was 0.25 nM. Filled square, 3′-ssDNA tailed duplex; open square, blunt-ended duplex; filled triangle, 5′-ssDNA tailed duplex. Panel B: the effect of Bst-MutL protein on the unwinding activity of Bst-UvrD helicase. Concentration of the substrate was 0.25 nM, Bst-UvrD helicase concentration was 1 nM. Solid column, 3′-ssDNA tailed duplex; open column, blunt-ended duplex; spotted column, 5′-ssDNA tailed duplex.
Thermophilic Helicase Dependent Amplification (tHDA)
To determine whether performing HDA at higher temperature would increase the specificity and efficiency of the reaction, we tested thermostable DNA helicases in HDA reaction in conjunction with various thermostable DNA polymerases. Among these thermostable helicases, Tte-UvrD helicase along with Bst DNA polymerase performed the best under our assay conditions. Studies of the tHDA reaction buffer conditions indicate that the assay is sensitive to the concentrations of magnesium and salt. Optimal reaction conditions were determined to be under low magnesium (2.5 mM to 5 mM) and salt (0 –50 mM) concentrations.
Low temperature mHDA reactions require the participation of four proteins: polymerase, UvrD helicase, MutL, and T4 gp32 SSB. The components required for tHDA reactions were determined by systematically observing the ability of the reaction to proceed in the absence of each reagent (Fig. 7). In the absence of DNA template (lane 2), primers (Fig. 7, lanes 3 & 4), or cofactor dATP (lane 5), no amplification was observed. Substitution of the dATP cofactor with ATP decreased the amplification yield (lane 6). Additional studies were carried out to test which nucleotide cofactor maximally supports the tHDA reaction and the result confirmed that dATP is better than ATP (data not shown). In terms of necessary proteins, the absence of the Tte-UvrD helicase from the reaction resulted in no amplification and confirmed that amplification is helicase dependent. Omitting both the thermostable MutL (lane 8) and SSB (lane 9) proteins had no significant effect on the DNA amplification and indicated that no accessory proteins are required for tHDA. As expected, no amplification was observed in the absence of Bst DNA polymerase large fragment (lane 10).
Figure 7.
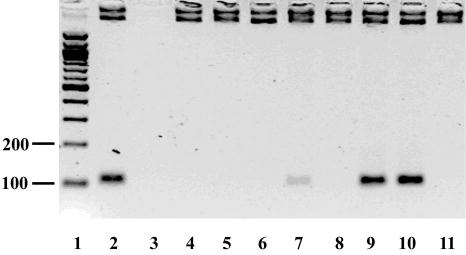
Determination of the essential components for the tHDA reaction. A pUC19 plasmid was used as a template in conjunction with forward and reverse primers, and the amplification products were analyzed by a electrophoresis on a 2% GPG LMP agarose gel. Lane 1 is the positive control containing all of the components that are required for the mesophilic HDA reaction (1), including a DNA template, primers, helicase cofactors, helicase, MutL, SSB, and DNA polymerase as described in the Materials and Methods. From lanes 2 to 10, each component is omitted respectively: lane 2 –template; lane 3 –5′ primer; lane 4 –3′ primer; lane 5 –cofactor dATP; lane 6 –substitution of dATP with ATP; lane 7 –Tte-UvrD; lane 8 –Tte-MutL; lane 9 –Bst-SSB; lane 10 –Bst Polymerase Large Fragment. Product size is 110bp.
To evaluate the sensitivity of tHDA, tHDA reactions were conducted with serial dilutions of the Neisseria gonorrhoeae template DNA. Amplification was performed using a pair of primers specific for the pivNg gene of N. gonorrhoeae, a gene that is necessary for virulence (Materials and Methods; 22). A specific DNA fragment corresponding to the 94-bp target sequence was observed with as little as 50 copies of input genome (Fig. 8). In the absence of N. gonorrhoeae DNA, only low molecular weight, non-specific product was observed that corresponded to the predicted size of primer-dimers (Fig. 8).
Figure 8.
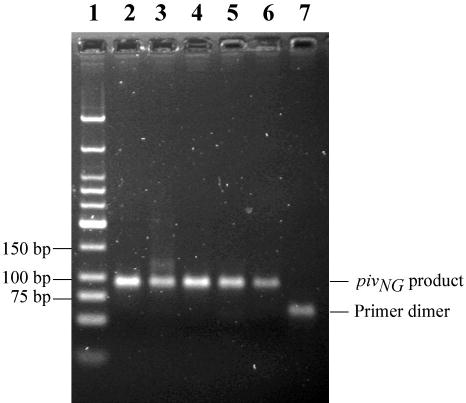
Amplification and detection of the pilin invertase homolog gene (pivNg) of Neisseria gonorrhoeae using tHDA. The tHDA reaction was carried out as described in Materials and Methods using a pair of pivNg specific primers and varying amounts of N. gonorrhoeae genomic DNA. 10 μl of the tHDA reaction was separated on a 2% agarose gel and visualized by ethidium bromide staining. The copy numbers of Neisseria gonorrhoeae genomic DNA are: lane 2: 5X105; lane 3: 5X104; lane 4: 5X103; lane 5: 5X102; lane 6: 5X101; lane 7: 0. Lane 1: 200 ng of Low Molecular Weight DNA Ladder (NEB).
DISCUSSION
We have cloned and characterized a thermostable UvrD-like helicase from T. tengcongensis. Tte-UvrD helicase is able to unwind DNA duplexes possessing a 3′ or 5′-ssDNA tails as well as blunt ended substrates. Although Tte-UvrD prefers 3′-ssDNA tailed duplex templates, the degree of the preference is much lower than that displayed by the E. coli UvrD (this study; 4, 5). Recently, a study based on single turnover DNA unwinding experiments suggests that the active form of E. coli UvrD is a dimer that is formed on 3′-ssDNA tails possessing a minimum tail-length of 12 nt (23). At high temperatures the ends of the duplex DNA may become single-stranded due to “thermal breathing” (24, 25). Since the temperature optima for Tte-UvrD is much higher than that for the E. coli UvrD, the “thermal breathing” effect is predicted to generate an increase intransient 3′-ssDNA tails. The ability of Tte-UvrD to efficiently unwind both blunt-ended and 5′-ssDNA tailed duplexes may be attributed to the transient generation of 3′-ssDNA tail during thermal breathing at higher temperatures and the subsequent capture of the tail by the helicase.
Previous work in E. coli has demonstrated that the MutL protein interacts with UvrD (8). The mechanism underlying this interaction has been shown to involve the stimulation of the UvrD unwinding activity by MutL, which is responsible for loading the UvrD helicase onto the DNA substrate (10). There is a biological relevance to the in vivo interaction between MutL and UvrD as UvrD participates in the DNA mismatch repair pathway by unwinding the strand containing the mutation (26). In addition, this effect is specific for UvrD and does not occur between the Rep helicase, which shares 40% homology with UvrD, and MutL (9).
Although sequence homologs of UvrD and MutL have been found in most of the sequenced bacterium genomes, very few biochemical and genetic studies have been reported. For example, it is not known whether the functional interaction between UvrD and MutL is universal to all bacterial systems. In this study, we have investigated whether the thermostable Tte-MutL can stimulate Tte-UvrD unwinding activity. Our results suggest that there is no significant stimulatory effect on the Tte-UvrD activity by Tte-MutL. To further verify our observations, we cloned, purified, and tested the thermostable helicase and the corresponding MutL protein from a partially sequenced Bacillus stearothermophilus strain (GenBank Accession number: NC_002926). We found that Bst-MutL also has no significant stimulation on Bst-UvrD activity (L. An and H. Kong, unpublished observation). Stimulation of UvrD activity by MutL appears to be either a specific effect observed in E. coli, or perhaps a more general mechanism necessary for efficient mesophilic DNA repair. If MutL and UvrD interact in thermophilic bacteria, the interaction may not automatically lead to the stimulation of the UvrD unwinding activity, at least in the two thermophilic bacteria reported in this study. Alternatively, UvrD may not be a necessary component of the DNA mismatch repair pathway in thermophilic bacteria and an alternative mechanism for DNA mismatch repair may exist.
In an effort to generate a thermophilic-based HDA (tHDA) platform, we evaluated the ability of the Tte-UvrD helicase and Tte-MutL accessory protein to amplify DNA in conjunction with a thermophilic polymerase. We found that Tte-UvrD was capable of efficiently amplifying target sequences from various genomes including both bacterial and human. Performing the HDA reaction at a higher temperature improves both the sensitivity and specificity of the reaction. As few as 10 copies of bacterial genomic DNA can be utilized for amplification. This is a significant advancement over the previously developed mesophilic HDA platform (mHDA), which has a detection limit of 1000 copies of bacterial genomic DNA (1).
Unlike the mHDA system, the tHDA system does not require MutL and SSB proteins as accessory proteins and, thus, simplifies the reagent composition. In the tHDA assay, addition of the Tte-MutL protein to the reaction does not enhance product formation (Fig. 7). This is in contrast with the previously developed mHDA platform (1), which has been shown to absolutely require the presence of the corresponding MutL protein for amplification. However, the lack of enhancement observed in the tHDA assay by Tte-MutL is in agreement with the biochemical analysis of the Tte-UvrD helicase, which shows that addition of MutL to the unwinding assay did not significantly enhance unwinding activity (Fig. 5). Currently, the exact mechanism underlying the lack of an SSB requirement is not known. Perhaps at higher temperatures (65°C), the rate of rewinding of separated DNA strands is slower than that at lower temperatures (37°), eliminating the need for an SSB to stabilize the single-stranded DNA.
Acknowledgments
We thank Jun Yu and Huanming Yang from Beijing Genomics Institute, and Yuqing Tian from the Institute of Microbiology Chinese Academy for providing the genomic DNA of T. tengcongensis. We thank Myriam Vincent, Yan Xu, Lauren Higgins, and Michael Dalton for assistance in protein purification. We thank Christopher Thomas for providing us Bst-UvrD helicase (Bst-PcrA). We are very grateful to Richard Roberts, Elisabeth Raleigh, and Ann Kays for constructive discussion and critical reading of this manuscript. We thank Donald Comb for his support.
References
- 1.Vincent M, Xu Y, Kong H. EMBO Reports. 2004;5:795–800.2. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornberg,A. and Baker,T. (1992) W.H. Freeman and Company 2nd edit., New York.
- 3.Caruthers JM, McKay DB. Curr Opin Struct Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 4.Matson SW. J Biol Chem. 1986;261:10169–10175. [PubMed] [Google Scholar]
- 5.Runyon GT, Lohman TM. J Biol Chem. 1989;264:17502–17512. [PubMed] [Google Scholar]
- 6.Lahue RS, Au KG, Modrich P. Science. 1989;245:160–164. doi: 10.1126/science.2665076. [DOI] [PubMed] [Google Scholar]
- 7.Caron PR, Kushner SR, Grossman L. Proc Natl Acad Sci USA. 1985;82:4925–4929. doi: 10.1073/pnas.82.15.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall MC, Jordan JR, Matson SW. EMBO J. 1998;17:1535–1541. doi: 10.1093/emboj/17.5.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi M, Dao V, Modrich P. J Biol Chem. 1998;273:9197–9201. doi: 10.1074/jbc.273.15.9197. [DOI] [PubMed] [Google Scholar]
- 10.Mechanic LE, Frankel BA, Matson SW. J Biol Chem. 2000;275:38337–38346. doi: 10.1074/jbc.M006268200. [DOI] [PubMed] [Google Scholar]
- 11.Hiramatsu Y, Kato R, Kawaguchi S, Kuramitsu S. Gene. 1997;199:77–82. doi: 10.1016/s0378-1119(97)00349-1. [DOI] [PubMed] [Google Scholar]
- 12.Collins R, McCarthy TV. Extremophiles. 2003;7:35–41. doi: 10.1007/s00792-002-0293-4. [DOI] [PubMed] [Google Scholar]
- 13.Bird LE, Brannigan JA, Subramanya HS, Wigley DB. Nucleic Acids Res. 1998;26:2686–2693. doi: 10.1093/nar/26.11.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soultanas P, Dillingham MS, Wigley DB. Nucleic Acids Res. 1998;26:2374–2379. doi: 10.1093/nar/26.10.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soultanas P, Dillingham MS, Papadopoulos F, Phillips SE, Thomas CD, Wigley DB. Nucleic Acids Res. 1999;27:1421–1428. doi: 10.1093/nar/27.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao Q, Tian Y, Li W, Xu Z, Xuan Z, Hu S, Dong W, Yang J, Chen Y, Xue Y, et al. Genome Res. 2002;12:689–700. doi: 10.1101/gr.219302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chong S, Montello GE, Zhang A, Cantor EJ, Liao W, Xu MQ, Benner J. Nucleic Acids Res. 1998;26:5109–5115. doi: 10.1093/nar/26.22.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 19.McGlynn P, Mahdi AA, Lloyd RG. Nucleic Acids Res. 2000;28:2324–2332. doi: 10.1093/nar/28.12.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali JA, Maluf NK, Lohman TM. J Mol Biol. 1999;293:815–834. doi: 10.1006/jmbi.1999.3185. [DOI] [PubMed] [Google Scholar]
- 21.Ali JA, Lohman TM. Science. 1997;275:377–380. doi: 10.1126/science.275.5298.377. [DOI] [PubMed] [Google Scholar]
- 22.Carrick CS, Fyfe JAM, Davies JK. Gene. 1998;220:21–29. doi: 10.1016/s0378-1119(98)00424-7. [DOI] [PubMed] [Google Scholar]
- 23.Maluf NK, Fischer CJ, Lohman TM. J Mol Biol. 2003;325:913–935. doi: 10.1016/s0022-2836(02)01277-9. [DOI] [PubMed] [Google Scholar]
- 24.Putnam BF, Van Zandt LL, Prohofsky EW, Mei WN. Biophys J. 1981;35:271–287. doi: 10.1016/S0006-3495(81)84789-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roychoudhury R, Tu CP, Wu R. Nucleic Acids Res. 1979;6:1323–1333. doi: 10.1093/nar/6.4.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modrich P. J Biol Chem. 1989;264:6597–6600. [PubMed] [Google Scholar]